材料与试剂
- 0.2 ml和1.5 ml微量离心管 (BBI Life Sciences,常温储存)
- 200 μl八联排管 (BBI Life Sciences,常温储存)
- 各种型号的枪头 (10 μl,200 μl,1 ml,BBI Life Sciences,常温储存)
- 基因组DNA提取试剂盒 (北京天根生化科技有限公司,常温储存,catalog number: DP304-03)
- 无水乙醇 (分析纯) (广州化学试剂厂,常温储存)
- AMPure XP beads (Beckman Coulter,4 °C储存,catalog number: A63881)
- Dynabeads MyOne Streptavidin C1 (Life Technologies,4 °C储存,catalog number: 65001)
- FastDigest MluI (Thermo Fisher Scientific,-20 °C储存,catalog number: FD0564)
- FastDigest SbfI (Thermo Fisher Scientific,-20 °C储存,catalog number: FD1194)
- T4 DNA ligase (5 U/μl) (Thermo Fisher Scientific,-20 °C储存,catalog number: EL0014)
- Human Cot-1 DNA (1 µg/µl) (Thermo Fisher Scientific,-20 °C储存,catalog number: 15279011)
- NEBNext Ultra DNA Library Prep Kit for Illumina (New England Biolabs,-20 °C储存,catalog number: E7645L)
- NEBNext dsDNA Fragmentase (New England Biolabs,-20 °C储存,catalog number: M0348L)
- TransTaq DNA Polymerase High Fidelity (HiFi) (北京全式金生物技术有限公司,-20 °C储存,catalog number: AP131-03)
- 100 mM ATP (上海生工生物工程股份有限公司,-20 °C储存,catalog number: A600311)
- 10 mM dNTPs (上海生工,-20 °C储存,catalog number: A610056)
- 1X TE buffer (上海生工,4 °C储存,catalog number: B548106)
- 20X SSPE buffer (上海生工,4 °C储存,catalog number: B548111)
- 50X Denhardt’s Solution (上海生工,-20 °C储存,catalog number: B548209)
- 10 % SDS Solution (上海生工,常温储存,catalog number: B548118)
- 20X SSC buffer (上海生工,4 °C储存,catalog number: B548109)
- 5 M NaCl (上海生工,4 °C储存,catalog number: B548121)
- 1 M Tris-HCl Solution,pH 8.0 (上海生工,4 °C储存,catalog number: B548127)
- 0.5 M EDTA,pH8.0 (上海生工,4 °C储存,catalog number: B300599)
- 吐温20 (上海生工,常温储存,catalog number: A600560)
- 矿物油 (上海生工,常温储存,catalog number: A630217)
- 琼脂糖 (Invitrogen,常温储存,catalog number: 75510-019)
- 双蒸水 (广州誉维生物科技仪器有限公司Unique超纯水机制)
- 吸附缓冲液 (见配方,4 °C储存)
- 洗涤缓冲液1 (见配方,4 °C储存)
- 洗涤缓冲液2 (见配方,4 °C储存)
仪器设备
- 0.5-5 μl,2-20 μl,20-200 μl,100-1000μl移液器 (Thermo Fisher Scientific F2系列)
- Gene Pro PCR扩增仪 (杭州博日科技股份技术有限公司,model: TC-E-96G)
- Vortex Genie2漩涡混合器 (Scientific Industries,model: G560E)
- Cubee迷你离心机 (广州美津生物技术有限公司,model: aqbd-i)
- 振荡型恒温金属浴 (杭州奥盛仪器有限公司,model: MS-100)
- DynaMag-2 Magnet磁力架 (Thermo Fisher Scientific,model: 12321D)
- 超微量分光光度仪 (Thermo Fisher Scientific,model: NanoDrop2000)
- 通用电泳仪 (北京百晶生物技术有限公司,model: BC-power6001)
- 高压蒸汽灭菌锅 (Kagoshima Seisakusyo Inc.,model: SX-500)
- 服务器 (Linux系统,32 G以上的内存,500 G的存储)
软件
- Seqtk_demultiplex https://github.com/jameslz/fastx-utils/blob/master/seqtk_demultiplex/
- SPAdes https://github.com/ablab/spades/
- CD-HIT https://github.com/weizhongli/cdhit/
- BLAST 2.6.0 ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.6.0/
- Python 2.7.9 https://www.python.org/ftp/python/2.7.9/
- Biopython 1.68 https://pypi.org/project/biopython/1.68/
- PASTA https://github.com/smirarab/pasta
- FastTree http://www.microbesonline.org/fasttree/#Install
- Phylopypruner https://gitlab.com/fethalen/phylopypruner/-/wikis/Installation
- RAxML https://github.com/stamatak/standard-RAxML
- ASTRAL 5.7.4 https://github.com/smirarab/ASTRAL
实验步骤
AFLP基因组捕获技术的实验流程,可分为三个步骤: AFLP探针制备,基因组文库构建以及杂交捕获,其工作流程如图1所示。

图1. AFLP基因组捕获技术的工作流程图
一、AFLP探针制备 (总耗时: 约7 h)
读者需要挑选一个与研究类群相关的探针制备物种,并将它的基因组DNA作为起始模板,按照以下四个步骤完成AFLP探针的制备 (见图1):(1) 限制性内切酶消化;(2) 接头连接和片段筛选;(3) 预扩增;(4) 选择性扩增。其中,限制性内切酶消化和选择性扩增是影响AFLP探针制备结果的关键步骤,因为它们决定了AFLP探针的长度范围和数量等。需要指出的是,探针制备物种的基因组DNA质量对AFLP探针制备的实验结果至关重要。在利用限制性内切酶消化基因组DNA的过程中,如果基因组DNA的质量较差 (如DNA无主带或DNA有主带,但同时存在明显的弥散条带),大量的限制性酶切识别位点会因DNA降解而丢失,这将导致酶切片段总数量的减少,从而导致AFLP探针数量的减少。因此,建议读者优先挑选基因组DNA质量较好的样本作为探针制备物种,选择标准为:在基因组DNA的电泳检测结果中,基因组DNA条带的长度大于20 Kb,且无明显的弥散条带。
- 限制性内切酶消化 (耗时:约1 h)
- 接头连接和片段筛选 (耗时:约1.5 h)
- 预扩增 (耗时:约2 h)
- 选择性扩增 (耗时:约2.5 h)
本方法提供了两条上游的选择性扩增引物和两条下游的选择性扩增引物,共四对组合 (引物序列见附录,组合见表5)。每一条引物的3’ 末端都添加了单个选择性核苷酸 (见图1的AFLP探针制备),目的是为了控制AFLP片段数目。例如,使用表5中任意一对引物组合进行选择性扩增,理论上能够获得已回收酶切片段数目的1/16 (1/4 × 1/4)。此外,两条下游的选择性扩增引物的5’ 末端都添加了生物素标记 (biotin),目的是通过PCR扩增的方法,将生物素标记添加到AFLP片段中,以获得可直接用于杂交捕获实验的AFLP探针 (见图1的AFLP探针制备)。考虑到每对引物组合扩增出的AFLP探针不同,读者可以根据自身对目标基因数目的需求量仅使用其中某一组探针,或者将不同组的探针混合在一起使用。根据本方法在臭蛙属 (隶属于两栖纲,蛙科) 中的测试结果 (Li et al., 2019),当混合使用四组AFLP探针 (表5) 进行序列捕获时,最终能够在属内种间水平上得到511个直系同源序列组 (orthologous groups,OGs)。这511个OGs的长度分布范围为203-7779 bp,与AFLP探针的长度范围相近 (200-5000 bp)。其中有一些OG的长度大于5000 bp,这证明了该方法不仅可以捕获目标区域,还可以捕获目标区域两侧的侧翼序列 (flanking sequence),获得这些额外的侧翼序列可能会增强系统发育分析的信号。本方法提供的引物具有通用性,适用于不同的生物类群。若读者需要更多的选择性扩增引物,可参考上述设计思路自行设计。值得一提的是,选择性核苷酸的数目与研究对象的基因组大小有关,若基因组小于5 Gb,建议在上游和下游引物中添加单个选择性核苷酸 (与本方法提供的引物设计思路一致),若基因组大于5 Gb,建议在上游或下游引物中添加两个及两个以上的选择性核苷酸。
表5. 选择性扩增引物组合信息表

二、基因组文库构建 (总耗时:约5 h)
本方法对高通量测序平台的测序读长没有严格要求,所以它适用于所有的二代测序平台,如Illumina,Ion Torrent,BGISEQ等。读者可根据自身对测序平台的熟悉程度,购买与测序平台相匹配的基因组文库构建试剂盒进行文库构建。本方法以NEBNext Ultra DNA Library Prep Kit for Illumina试剂盒为例,简要地介绍了Illumina基因组文库构建的方法。为了保证基因组文库具有较高的质量和良好的多样性,建议尽量选择DNA质量好 (> 4 Kb) 的样本用于文库构建,DNA初始投入量在200 ng-1 μg,1 μg最优。
- DNA片段化 (耗时:约1 h)
该步骤的目的是为了获得符合Illumina测序平台的要求的,具有一定长度范围的DNA片段。以HiSeq X ten测序平台为例,该平台期望的插入DNA片段大小在200-400 bp之间,所以片段化后的DNA片段最好集中在这个区域。片段化的方法有超声处理,酶处理等,对于新手而言,推荐使用后者,因为该过程不依赖特殊仪器 (超声波DNA打断仪),且入手简单。本方法以NEBNext DNA双链片段化酶为例,介绍了DNA片段化的方法,具体操作步骤和注意事项可参阅产品说明书:
https://international.neb.com/protocols/0001/01/01/digestion-with-nebnext-dsdna-fragmentase-m0348 - 补平加A和接头连接 (耗时:约2.5 h)
这些步骤的主要目的是为了在DNA片段的两端添加Illumina测序接头。此外,为了区分不同的样本,测序接头上还应包含样本特异性标签 (barcode或index)。具体操作步骤请参阅文库构建试剂盒说明书
https://international.neb.com/protocols/2015/01/14/protocol-for-use-with-nebnext-ffpe-dna-repair-mix-m6630-and-ultra-dna-library-prep-kit-for-illumina-e7370 - 片段筛选及PCR扩增 (耗时:约1.5 h)
三、杂交捕获 (总耗时:约41 h)
在系统发育研究中,捕获样本往往与探针制备样本不同。为了从捕获样本的基因组文库中杂交捕获到与探针同源的基因组区域,特别是从亲缘关系较远的样本中,通常需要根据捕获样本与探针制备样本之间的序列相似度,对杂交体系中的盐离子浓度,杂交反应温度等条件进行优化和调整。在本实验方法中,杂交反应体系中盐离子的终浓度为5X,且杂交反应的温度呈梯度下降 (Touch-down),即杂交温度从65 °C起始,每隔一段时间下降3°C,最终下降至50°C。在此条件下,与探针的核酸序列相似度在65 %以上的单链DNA文库将被捕获。这种反应条件适合于大多数的系统发育研究。
- 杂交 (耗时:约37 h)
杂交反应体系共50 μl,其中包括18.5 μl的杂交缓冲液体系和31.5 μl的探针文库混合体系,在杂交反应开始前,两个子体系需要进行不同的预处理 (具体操作见下文)。为了降低实验成本,提高实验效率,本方法建议使用混合文库进行杂交实验。在准备混合文库时,读者首先需要根据样本间亲缘关系的远近, 基因组文库的质量 (指浓度和片段范围) 和文库特异性标签对捕获样本进行分组,将亲缘关系相近,基因组文库质量相近且不具有相同特异性标签的样本分为一组,每组的样本数量为2-7个,4个最优 (Portik et al. 2016)。随后将同一组的基因组文库等质量混合,以用于后续的杂交实验。 - 链霉亲和素磁珠吸附 (耗时:约1 h)
链霉亲和素磁珠的用量与探针投入量存在对应关系,磁珠用量过多会造成浪费,过少可能会影响吸附效果。以Dynabeads Myone streptavidin C1磁珠为例,1 μl (50 μg) 磁珠可以吸附约200 ng的DNA片段。根据该对应关系,当杂交反应的AFLP探针投入量为200 ng时,建议使用3ul磁珠进行吸附。增加磁珠用量的原因是在吸附反应开始前,本实验方法会对链霉亲和素磁珠进行洗涤,部分磁力较弱的珠子将被丢弃,这有助于提高捕获的效率。 - 洗涤 (耗时:约1.5 h)
该步骤的主要目的是利用不同盐离子浓度的缓冲液对杂交产物进行洗涤,以去除非目标捕获 (non-target) 和非特异性杂交的单链DNA文库,从而提高杂交捕获的效率。在本实验方法中,洗涤条件先宽松后严格,首先使用高离子浓度的洗涤缓冲液1 (1X SSC,0.1 % SDS) 对杂交产物进行洗涤,以去除非目标捕获的单链DNA文库,然后使用 50 °C (最终杂交温度) 预热的低离子浓度的洗涤缓冲液2 (0.1X SSC,0.1 % SDS) 对杂交产物进行洗涤,以去除非特异性杂交的单链DNA文库。 - PCR扩增及高通量测序 (耗时:约1.5 h)
该步骤的目的是通过PCR扩增的方式将杂交捕获到的单链DNA文库变为双链DNA文库。对于探针和捕获文库的投入量分别为200 ng和500 ng的杂交反应,本方法推荐PCR扩增的模板投入量为3 μl,扩增循环数为10-12。PCR扩增反应体系的其余成分 (如引物和dNTPs等) 以及扩增反应程序,均与基因组文库制备流程中的PCR扩增步骤一致,详细信息可参阅试剂盒说明书。需要注意的是,在添加PCR扩增的模板时,务必保证带有磁珠的杂交产物溶液是充分悬浮的状态。
结果与分析
AFLP基因组捕获技术可以产生匿名基因序列数据和SNP数据。其中,匿名基因序列数据可以用于属内种间水平上的系统发育分析,而SNP数据可以用于种群水平上的系统地理学分析。读者需要根据自身的研究需求进行数据的提取。目前,SNP数据提取与分析的流程已在现有文献中被详细描述 (McKenna et al. 2010; Depristo et al. 2011; Van der Auwera et al. 2013),读者可参考上述文献对SNP数据进行提取与分析。本文将重点介绍从AFLP捕获数据中获取匿名基因序列数据的生物信息学分析流程,其主要流程可概括为: 数据分选与组装,直系同源序列的鉴定与提取,序列比对,数据质量控制以及系统发育分析。
一、数据分选与组装
- 使用Trimmomatic (v 0.39) 对测序得到的原始数据 (raw data) 进行质量控制和筛选,去除reads中的测序接头序列,去除平均碱基质量值低于20的reads,去除reads首端和末端碱基质量小于3或N的碱基,得到高质量的测序数据 (clean reads)。
以处理双端测序数据为例:
$ java -jar trimmomatic-0.39.jar PE RawReads_R1.fq.gz RawReads _R2.fq.gz RawReads_R1_paired.fq.gz RawReads_R1_unpaired.fq.gz RawReads_R2_paired.fq.gz RawReads_R2_unpaired.fq.gz ILLUMI-NACLIP:TruSeq3-PE.fa:2:30:10:2:keepBothReads LEADING:3 AVGQUAL:20 TRAILING:3 MINLEN:36 - 依据各个样本的特异性标签 (barcode),使用fastq-multx将clean reads分选到对应的实验样本中。
以分选双端测序数据为例:
$ fastq-multx -B barcode.txt -b CleanReads_R1_paired.fq.gz Clean-Reads_R2_paired.fq.gz -o %_R1_clean.fq %_R2_clean.fq
注:“-B”用于提供特异性标签文件,%指的是输出文件名与barcode文件中的样本名称相对应。详细使用方法可参阅https://github.com/brwnj/fastq-multx. - 使用基因组拼接软件SPAdes对每个样本的clean data进行从头组装 (de novo assembly)。
以组装样本A的clean reads为例:
$ python spades.py –t 4 –m 20 -1 taxaA_R1_clean.fq -2 taxaA _R2_clean.fq –o taxaA_outdir
注:“-t”用于设定线程数,“-m”用于设定内存大小 (Gb),“-o”用于指定输出文件夹的路径和名称。 - 使用cd-hit-est软件去除contigs中的冗余序列。
以处理样本A的contigs为例:
$cd-hit-est –i taxaA_contig.fasta –o taxaA_contig095.fasta –c 0.95 –n 10 –M 16000 –T 4
注:“-c”用于设定冗余序列相似度阈值,“-M”用于设定内存大小 (Mb),“-T”用于设定线程数。
二、直系同源序列的鉴定与提取
使用同一套AFLP探针从不同样本的基因组文库中进行序列捕获时,不同样本间被捕获到的序列具有同源性。所以,本方法直接采取双向BLASTN策略从所有样本的contigs中鉴定直系同源序列组 (orthologous groups,OGs) (如图3中的步骤一)。做法是:首先从所有样本拼接的contigs中选择拼接总长度最长的contigs作为参考序列,然后将其他样本的contigs分别与参考序列进行双向BLASTN。若1号样本的A序列在参考序列的第一个BLAST Hit是B序列,反过来参考序列的B序列在1号样本中的第一个BLAST Hit 还是A序列,则A序列与B序列为1号样本与参考序列的直系同源序列。考虑到在一个OG中,样本间contigs的长度有所差异,这使它们很难进行序列比对。为了解决这个问题,本方法提出了一种修剪OG的策略,即在原始OG中寻找一个最佳区域,以降低比对的难度 (如图3中的步骤二)。上述所有过程可通过本实验室开发的“SearchOGs.py”脚本实现 (下载地址:https://figshare.com/s/5a4eee383e2dc9afba53)。
$ python SearchOG.py -c ./contigDir -r ./contigDir/reference.fasta -e 1E-10 -L 200 -S 0.7 -B 2 -C 0.5 -N 2 -o outdir
注:“-c”用于提供包含所有样本contigs文件在内的文件夹路径,“-r”用于提供参考序列文件的路径,“-e”用于设置BLAST比对的E-value阈值, “-L”用于设置BLAST Hit长度阈值,“-S”用于设置BLAST Hit的序列相似度阈值,“-B”用于设置最佳比对区域中上下限包含样本数目的阈值,“-C”用于设置最佳比对区域中修剪后的contigs长度与参考序列长度比例的阈值,“-N”用于设置直系同源序列组中所包含样本数目的阈值,“-o”用于指定输出文件夹的路径和名称。运行环境要求和参数详解请参阅该脚本的说明文件。

图3. 鉴定直系同源序列组的流程图
三、序列比对
常用的多序列比对的软件有Muscle (http://www.drive5.com/muscle),MAFFT (https://mafft.cbrc.jp/alignment/software/),Bali-Phy (http://www.bali-phy.org/),SATé (http://phylo.bio.ku.edu/software/sate/),PASTA (https://github.com/smirarab/pasta)等。考虑到大多数被AFLP探针捕获的匿名基因组序列都是快速进化的非编码序列,因此,为了提高序列比对的准确性,推荐使用针对高变序列比对专门设计的程序如SATé和PASTA。
例如,使用PASTA处理一个OG文件:
$ python run_pasta.py –i OG1.fasta
注:PASTA具体使用方法请参阅 https://github.com/smirarab/pasta/blob/master/pasta-doc/pasta-tutorial.md#step-2-inspecting-the-output-of-pasta
四、数据质量控制
为了进一步提高数据质量,确保所鉴定的直系同源基因数据集的准确性,本方法对每个alignment 构建单基因树。通过分析基因树中各分枝的长度和判断基因树的拓扑结构,将枝长异常的序列,旁系同源序列以及污染序列从数据集中剔除 (如图4)。以臭蛙属为例,本方法能够从11个臭蛙属物种和2个外类群物种中鉴定出511个OGs,共4142条序列,碱基缺失率为35.6% (包含外类群物种在内)。经过数据质量控制之后,有11条不合格的序列被剔除,碱基缺失率变为35.9%。虽然数据集的碱基缺失率有所增加,但剔除枝长异常的序列,旁系同源序列以及污染序列将大大提高最终数据集的可靠性。

图4. 基于单基因树构建的方法去除长枝,旁系同源序列以及污染序列的示意图 (Kapli et al. 2020)。
以处理一个OG为例:
- 单基因树构建:
$ FastTree –gtr –nt OG1_align.fasta > OG1_align.tree - 借助Phylopypruner去除长枝 (含有长枝的基因树拓扑结构可参照图3中的拓扑结构A),例如,删除某一枝长大于树中所有分枝标准差3倍的序列:
$ phylopypruner --trim-lb 3 OG1_align.fasta OG1_align.tree
注:Phylopypruner具体使用方法请参阅 https://gitlab.com/fethalen/phylopypruner/-/wikis/methods#paralogy-pruning- - 去除旁系同源序列或污染序列,其标准是观察单基因树拓扑结构中是否存在聚类关系怪异的分支,可参照图4中基因树拓扑结构B和C同正确的基因树拓扑结构之间的差异。
- 使用PASTA对数据质量控制后的alignment再次进行比对。
$ python run_pasta.py –i OG1_realign.fasta - 使用Gblocks剔除比对模糊的区域。
$ Gblock OG1_realign.fasta –b5=a
注:Gblock软件的详细参数介绍可参阅http://molevol.cmima.csic.es/castresana/Gblocks/Gblocks_documentation.html
五、系统发育分析
- 串联树构建
将所有的OG比对矩阵进行串联处理,生成一个大型的数据矩阵集,并使用RAxML构建ML树:
$ raxmlHPC-PTHREADS-AVX –T 4 -f a -x 12347 -p 12845 -# 500 -m GTRGAMMA –s CombineOGs.fasta –n CombineOGs_MLtree_500boostrap
注:“-f”用于选择RAxML运算的算法,a表示执行快速Bootstrap分析并搜索最佳得分的 ML树,“-x”用于指定一个随机数作为随机种子,以启动快速Bootstrap算法,“-#”用于提供Bootstrap的次数,“-m”用于指定核苷酸或氨基酸的替代模型。RAxML参数的详细使用说明请参阅https://cme.h-its.org/exelixis/resource/download/NewManual.pdf - 物种树构建
溶液配方
- 10X接头淬火缓冲液配方 (表11)
表11. 10X接头淬火缓冲液体系

- 10 μM MluI adapter制备方法
在0.2 ml 离心管中,建立双链MluI接头淬火体系 (表12)。
表12. 双链MluI接头淬火体系

充分涡旋混匀,利用下述条件进行淬火反应,形成局部互补的DNA双链接头:95 °C 5 min;95 °C降到12 °C,每秒0.1 °C;保持在12 °C。反应结束后,将退火后的接头溶液放置于-20 °C保存
- 10 μM SbfI adapter制备方法
在0.2 ml 离心管中,建立双链SbfI接头淬火的体系 (表13)。
表13. 双链SbfI接头淬火体系

充分涡旋混匀,利用下述条件进行淬火反应,形成局部互补的DNA双链接头: 95 °C 5 min;95 °C降到12 °C,每秒0.1 °C;保持在12 °C。反应结束后,将退火后的接头溶液放置于-20 °C保存。
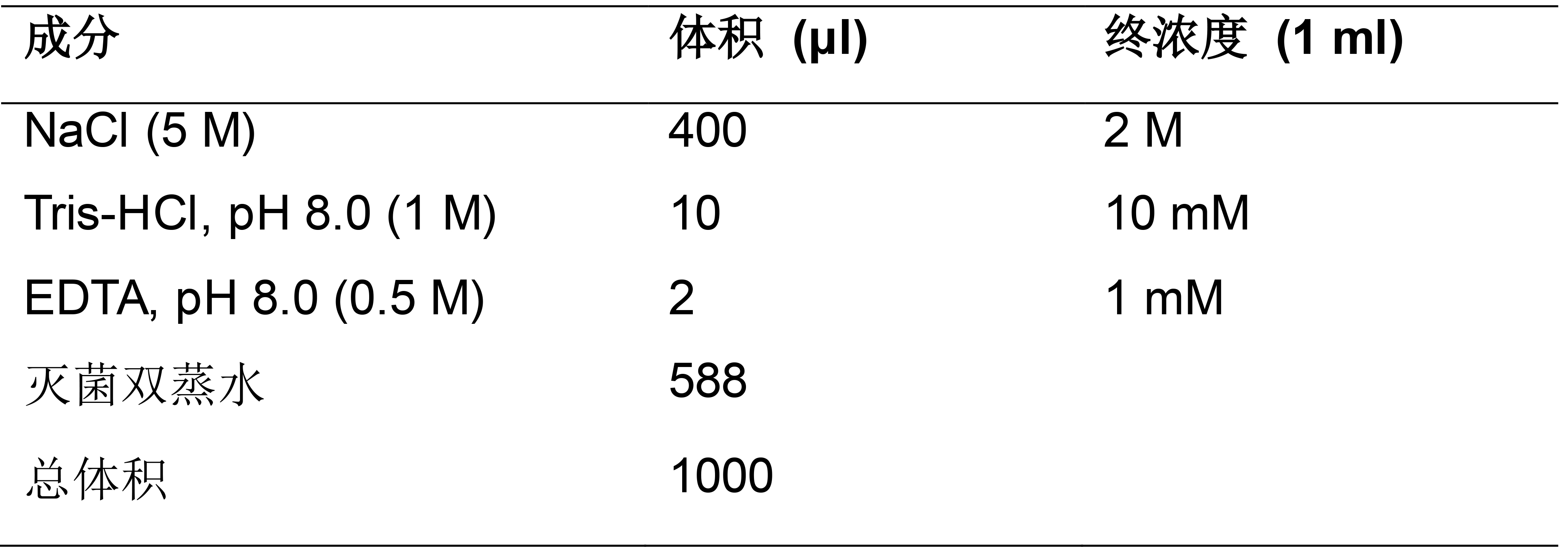
- 吸附缓冲液配方 (表14)
表14. 吸附缓冲液体系
- 洗涤缓冲液1 (表15)
表15. 洗涤缓冲液1体系

- 洗涤缓冲液2 (表16)
表16. 洗涤缓冲液2体系

附件
附表1 附表2
致谢
实验方案摘自发表的文章Jia-Xuan Li, Zhao-Chi Zeng, Ying-Yong Wang, Dan Liang, Peng Zhang*. (2019). Sequence capture using AFLP-generated baits: A cost effective method for high-throughput phylogenetic and phylogeographic analysis. Ecology and Evolution, 9(10), 5925-5937.
竞争性利益声明
作者声明没有利益冲突
参考文献
- DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C. and Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics 43(5), 491–498.
- Li,C., Hofreiter, M., Straube, N., Corrigan, S. and Naylor, G. J. (2013). Capturing protein-coding genes across highly divergent species. BioTechniques 54(6), 321-326.
- Li, J., Zeng, Z., Wang, Y., Liang, D., Zhang, P. (2019). Sequence capture using AFLP-generated baits: A cost effective method for high-throughput phylogenetic and phylogeographic analysis. Ecology and Evolution 9(10): 5925-5937.
- Kapli, P., Yang, Z. H. and Telford, M. J. (2020). Phylogenetic tree building in the genomic age. Nature Review Genetics 21(7): 428-444.
- McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A. and DePristo, M. a. (2010). The genome analysis tool‐kit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Re-search 20(9), 1297–1303.
- Portik, D. M., Smith, L. L, Bi, K. (2016). An evaluation of transcriptome-based exon capture for frog phylogenomics across multiple scales of divergence (Class: Amphibia,Order: Anura). Molecular Ecology Resources 16(5):1069-83.
- Van der Auwera, G. A., Carneiro, M. O., Hartl, C., Poplin, R., del Angel, G., Levy‐Moonshine, A. and DePristo, M. A. (2013). From fastQ data to high‐confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics 43, 11.10.1–11.10.33.
引用格式:李佳璇, 梁丹, 张鹏. (2021). AFLP基因组捕获技术: 一种通用的低阶元的系统发育分析方法.
Bio-101: e1010598. DOI:
10.21769/BioProtoc.1010598.
 How to cite:
How to cite: Li, J. X., Liang, D. and Zhang, P. (2021). AFLP Genome Capture Technique: A General Method for Shallow Scale Phylogenetic Analysis.
Bio-101: e1010598. DOI:
10.21769/BioProtoc.1010598.
