- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Molecular Cloning and Sanger Sequencing-based Protocol for Detecting Site-specific DNA Methylation
(*contributed equally to this work) Published: Vol 12, Iss 9, May 5, 2022 DOI: 10.21769/BioProtoc.4408 Views: 3701
Reviewed by: Amanda M. RoehrkasseRama Reddy GoluguriAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2021
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics









Abstract
DNA methylation is a conserved chemical modification, by which methyl groups are added to the cytosine of DNA molecules. Methylation can influence gene expression without changing the sequence of a particular gene. This epigenetic effect is an intriguing phenomenon that has puzzled biologists for years. By probing the temporal and spatial patterns of DNA methylation in genomes, it is possible to learn about the biological role of cytosine methylation, as well as its involvement in gene regulation and transposon silencing. Advances in whole-genome sequencing have led to the widespread adoption of methods that examine genome-wide patterns of DNA methylation. Achieving sufficient sequencing depth in these types of experiments is costly, particularly for pilot studies in organisms with large genome sizes, or incomplete reference genomes. To overcome this issue, assays to determine site-specific DNA methylation can be used. Although often used, these assays are rarely described in detail. Here, we describe a pipeline that applies traditional TA cloning, Sanger sequencing, and online tools to examine DNA methylation. We provide an example of how to use this protocol to examine the pattern of DNA methylation at a specific transposable element in maize.
Keywords: EpigeneticsBackground
DNA methylation is a conserved chemical modification at the 5’ position in the deoxyribose ring of cytosine that is catalyzed by methyltransferases (Figure 2A and Schultz et al., 2012). In plants, DNA methylation occurs in three sequence contexts: CG, CHG, and CHH, where H represents A, T, or C (Zhang et al., 2018). DNA methylation plays an important role in regulating both genes and transposable elements (TEs) (Schultz et al., 2012; Zhang et al., 2018; Guo et al., 2021). Using a maize MuDR reporter system to study TE silencing, we have demonstrated that DNA methylation at a terminal inverted region (TIR) in one of the MuDR elements, mudrA, is associated with silencing (Woodhouse et al., 2006; Guo et al., 2021).
Bisulfite sequencing involves bisulfite treatment of genomic DNA, resulting in the conversion of unmethylated cytosines into uracil, while methylated cytosines remain unchanged. After treatment, the region of interest is amplified, cloned, and sequenced. Investigating DNA methylation at a gene or a TE requires high-quality genomic DNA, and the conversion of unmethylated cytosines into uracil, while retaining methylated cytosines using sodium bisulfite treatment (Gruntman et al., 2008; Zhang et al., 2018). After this treatment, the region of interest is amplified via polymerase chain reaction (PCR), which is followed by TA-cloning and Sanger sequencing (Foerster, 2010). Because PCR amplification of the converted cytosines will replace uracil with thymine, determining the methylation profile at a region of interest can be achieved by comparing the sequence of the bisulfite-treated DNA with that of untreated DNA sequences (Gruntman et al., 2008). To minimize the noise created by incomplete conversion of unmethylated cytosine to uracil, examining DNA methylation using an internal control gene, and assaying multiple individual colonies of PCR products are needed.
Here, we provide a case study with step-by-step procedures, to examine the pattern of DNA methylation at a silenced MuDR element in maize, which is a well-documented system to validate our protocol. We described a site-specific DNA methylation pipeline, using traditional TA cloning and Sanger sequencing and provide procedures for sample preparation, treatment, cloning work, and data analysis. We highlight the applicability of this traditional DNA methylation pipeline as an inexpensive method for pilot studies, important for decision-making, or for use when an organism’s genome is not fully sequenced. Additionally, this pipeline describes the basis of molecular cloning and epigenetic analysis, and how it can be used as a method for teaching.
Materials and Reagents
Eppendorf tubes (Dot Scientific, catalog number: RA1700-GMT)
Glass beads (MO-SCI, catalog number: GL0191B4/38-53)
Axygen 96-well PCR Microplates (Fisher Scientific, catalog number: 14-222-326)
Tissue Culture Treated Petri Dishes (dot scientific, catalog number: 667621)
Competent E. coli cells (Invitrogen, catalog number: 18265017)
Minimal MuDR line (Damon Lisch lab, Purdue University)
Ethylenediaminetetraacetic acid (EDTA) (Fisher Scientific, catalog number: BP2482100)
Sodium chloride (NaCl) (Fisher Scientific, catalog number: BP358-1)
Sodium dodecyl sulfate (SDS) (Fisher Scientific, catalog number: 28312)
Potassium acetate (Fisher Scientific, catalog number: A16321.36)
Isopropanol (Fisher Scientific, catalog number: 040983.M1)
100% Ethanol (Fisher Scientific, catalog number: T038181000)
DNA loading dye 10× (NEB, catalog number: 10816015)
2× PCR Master Mix (Syd Labs, catalog number: MB067-EQ2G-L)
EpiMark Hot Start Taq DNA Polymerase (NEB, catalog number: M0490S)
Bisulfite conversion kit (ZYMO RESEARCH, catalog number: D5005)
Lysogeny broth (LB) medium (Sigma-Aldrich, catalog number: L2897)
T4 ligase (NEB, catalog number: M0202S)
TA cloning vector (Fisher Scientific, catalog number: K1231)
RNase A (Fisher Scientific, catalog number: EN0531)
8-strip PCR tubes and Caps (dot scientific, catalog number: 503-8PCR-A)
Zymoclean Gel DNA recovery Kits (Zymo catalog number: D4007)
ZymoPURE Plasmid Miniprep Kit (Zymo catalog number: D4210)
Equipment
Pipettes (Fisher Scientific, catalog number: EPPR4331)
Microcentrifuge (Fisher Scientific, catalog number: 5424)
Incubator (VWR, catalog number: 13259-36)
Qubit fluorometer (Fisher Scientific, catalog number: Qubit 2.0)
Gel electrophoresis system (NeoSCI, catalog number: 55-1094)
Low speed orbital shaker (Corning LSE, catalog number: 6780-FP)
PCR machine (Bio-Rad, catalog number: 1861096)
Procedure
An overview of procedures can be seen in Figure 1.

Figure 1. An overview of procedures of DNA extraction protocol
Sample collection and DNA extraction
1. Collect samples and store them at -80°C, or immediately perform DNA extraction using the protocol described previously (Guo et al., 2022).
2. Digest genomic DNA with RNase.
Normally, 1 μL of RNase is added to each DNA sample, and incubated at 37°C for 30 min.
3. Evaluate the quality of DNA.
The quality of genomic DNA should be evaluated by running gel electrophoresis, and using a Qubit fluorometer or UV spectrophotometer. A large, clear, intact band on top indicates the presence of high quality genomic DNA. In addition to high-quality DNA, a successful bisulfite conversion reaction requires a quantity of DNA suggested by the commercial kit being used.
Primer design
Primers can be designed using a variety of online tools, such as Bisulfite Primer Seeker (https://www.zymoresearch.com/pages/bisulfite-primer-seeker).
Note: It is recommended that primers be designed before the bisulfite treatment steps. Primers for an internal control gene should also be designed. Ideally, this should be a gene that has already been determined to be entirely or largely unmethylated.
Bisulfite conversion
Readers should follow the instructions from the commercial kit being used for bisulfite conversion.
Note: Converted DNA can be stable at 4°C or -20°C for a short period of time, but it is best to check the manual from the kit you are using. It is recommended to PCR-amplify the region of interest immediately after the conversion is done to ensure the best results.
TA cloning of the PCR amplicons
Typical cloning PCR conditions are (Table 1):
Table 1. Overview of cloning PCR conditions
Component 50 μL Reaction 5× EpiMark Hot Start Taq Reaction buffer
10 μL 10 mM dNTPs 1 μL 10 μM Forward Primer 1 μL 10 μM Reverse Primer 1 μL EpiMark Hot Start
Taq Polymerase
0.25 μL Converted DNA 1 μL/variable Nuclease-free water To 50 μL Thermal cycling conditions:
95°C, 30 s
35–40 cycles of:
95°C, 15–30 s
45–68°C, 15–60 s
68°C, 1 min per kb
Final extension:
68°C, 5 min
Note: Because genomic DNA is treated and converted by the bisulfite reagent, a regular Taq DNA polymerase may not tolerate uracil-containing DNA and high AT targets. Therefore, it is necessary to use the appropriate DNA polymerase to ensure high fidelity, low bias, and sufficient yield. We recommend EpiMark Hot Start Taq DNA Polymerase from the New England Lab (NEB), which generates the PCR products containing dA overhangs at the 3’ end. This allows for a ligation to vector with dT/dU-overhangs. To clone the region of interest using PCR, it’s recommended to run a PCR with a larger volume, to ensure sufficient DNA can be recovered. Usually, 50 μL or more gives better results. One can directly purify the PCR product using a commercial kit. Alternatively, the PCR product can be recovered from the gel slice. If using this method, it’s important to use fresh TAE buffer and a clean razor blade, when performing electrophoresis and recovery.
Ligation. Typical ligation conditions are (Table 2):
Table 2. Overview of ligation conditions
Component 20 μL Reaction T4 DNA Ligase Buffer (10×) 2 μL Vector DNA (3 kbp) 50 ng Insert DNA (300 bp) 15 ng T4 DNA Ligase 1 μL Nuclease-free Water To 20 μL Note: After PCR amplification and gel recovery, incubate the PCR products with the cloning vector, following the instructions below (Table 2). Our previous work used the cloning kit from Thermo Fisher (Guo et al., 2021). Ligation can be completed in 5 min at room temperature, but it is recommended to extend this time to 30 min or longer for any PCR products larger than 3,000 bp.
Transformation
Readers should follow the instructions from the commercial competent cells being used for transformation.
Note: Once ligation is complete, samples are ready to undergo vector transformation using E. coli competent cells. Note the antibiotic-resistant gene carried by the vector. Typically, the Top10 strain grows faster than DH 5-alpha strain, but there is no significant difference in using either for a regular cloning experiment. After transformation, cells can be spread directly onto an LB medium containing appropriate antibiotics. Be sure to use either sterilized glass beads or a spreader. Incubate the LB plates at 37°C for 10–16 h.
To determine the colonies that carry a positive amplicon, propagate the cells from individual colonies in liquid LB medium, and run a colony-PCR to select the positive colonies that will undergo sequencing. Typically,
Prepare 2.0-mL tubes on a tube rack, and fill them with 2 mL of liquid LB medium containing antibiotics.
Inoculate each tube with an individual colony from each plate, using a sterilized toothpick or pipette tip.
Tubes are then placed onto a shaker set at 200 rpm and 37°C for 10–14 h.
Note: Due to some unsuccessful ligation and propagation, and depending on the size of the PCR products, it is recommended to inoculate at least two-fold more colonies than the actual number you want to send for Sanger sequencing.
Once transparent liquid LB medium becomes turbid, run a colony-PCR directly, using 1 mL of liquid LB medium containing cells as the DNA template.
Note: Since the size of the vector is usually small, and this is a direct PCR amplification from the plasmid from E. coli, a strong positive amplification typically results after as few as 20 PCR cycles.
Typical colony-PCR conditions are (Table 3):
Table 3. Overview of colony-PCR conditions
Component 12.5 μL Reaction 2× Master Mix 6.25 μL 10 μM Forward Primer 0.25 μL 10 μM Reverse Primer 0.25 μL DNA 1 μL Nuclease-free Water 4.75 μL Thermal cycling conditions
95°C, 3 min
20 cycles or more of:
95°C, 30 s
50°C, 30 s
70°C, 1 min per kb
Final extension:
70°C, 5 min
Once positive colonies are identified, plasmids can be extracted from the remaining liquid culture. Extraction can be done using traditional methods, or a commercial kit. Once plasmids are ready, samples can be sent for Sanger sequencing. It’s recommended to send a few to test, before multiple samples are sequenced. Distinct sequences from ten colonies from each genotype or treatment should be obtained for analysis.
Sequences can be trimmed using SnapGene viewer or any available software. Reference sequences and samples sequences are placed in a separate text file in the FASTA format. Be sure that all sequences are in the correct orientation relative to the reference sequence, and primer sequences are excluded. The two files are uploaded into Kismeth (http://katahdin.mssm.edu/kismeth/revpage.pl), which provides quantitative data, as well as images for result visualization.
Results
Examining DNA methylation patterns at MuDR elements in the maize minimal MuDR line
Using this protocol, we have demonstrated that DNA methylation at TIRB, the terminal inverted repeats (TIR) adjacent to the mudrB gene in MuDR, is not associated with transcriptional activity of mudrB, and that mudrA activity is associated with DNA hypomethylation at TIRA (Li et al., 2010; Guo et al., 2021). Here, we describe how to examine DNA methylation in a step-by-step manner. First, we collected leaf tissue from 2-week-old seedlings from active and silenced MuDR lines, and then extracted genomic DNA from those tissues. Gel examination indicated that the DNA was of good quality, as indicated by the intact bright band at the top of the gel (Figure 2B). We then measured the concentration of DNA, to determine the right volume of DNA for the bisulfite conversion reaction (Table 4). Next, we performed bisulfite conversion using a commercial kit, and PCR to amplify short fragments from TIRA and TIRB in two separate amplifications, using two different pairs of primers (Figure 2C). Previous experiments demonstrated that TIRA lacks DNA methylation, which can serve as an internal control in the active MuDR/- line, whereas DNA methylation is present at TIRB (Woodhouse et al., 2006; Guo et al., 2021).
After we harvested the PCR products directly from the gel slices using a commercial kit, we performed ligation and E. coli transformation, following the procedures described above. We obtained plates with several hundred colonies (Figure 2D). To determine the positive colonies and extract plasmids from those for Sanger sequencing, we propagated cell clones in liquid medium, and performed colony-PCR. We found that most colonies carried inserts of the expected size, suggesting that the cloning work was successful. We then sent the plasmids for Sanger sequencing. Reads were trimmed and pasted into a text file and uploaded to Kismeth (http://katahdin.mssm.edu/kismeth/revpage.pl).
Table 4. Examining concentration and purity of genomic DNA samples
| Sample | Concentration (Qubit) | A260/A280 | A260/A230 |
|---|---|---|---|
| MuDR/- | 75 ng/μL | 1.82 | 2.12 |
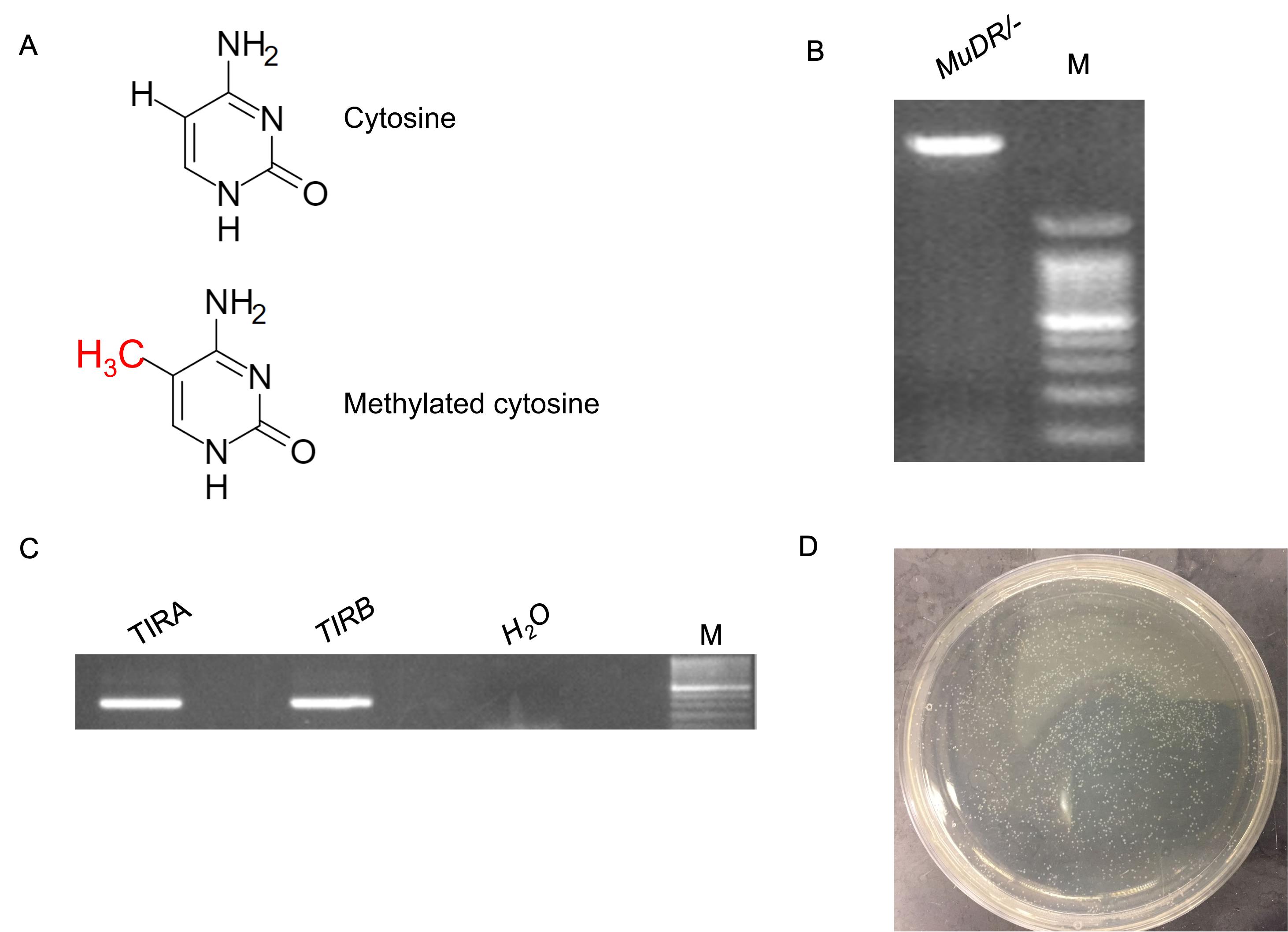
Figure 2. TA cloning of TIRA and TIRB in MuDR/-. A. An illustration of methylated and unmethylated cytosine. B. Genomic DNA of MuDR/-. A total volume of 2 mL of DNA mixed with 10× DNA loading dye was loaded into 1% agarose gel, together with 5 mL of the 100 bp DNA ladder that was loaded into the 2nd lane. M denotes the 100-bp DNA ladder. C. PCR amplification of TIRA and TIRB from MuDR/-. M denotes the 100-bp DNA ladder. A negative control of water as DNA template was included. D. Colonies on LB agar plate containing 50 μg/mL ampicillin.
A dot plot with three colors, indicating DNA methylation occurring in three sequence contexts, was generated and visualized for TIRA and TIRB, respectively (Figure 3A). To get data for the ratio of DNA methylation and show an individual value for each colony, each sequence from each plasmid was uploaded, and data were recorded. A histogram plot with a dot standing for each colony was generated (Figure 3B and 3C), of which the results were consistent, and supported the results obtained using the dot plots.
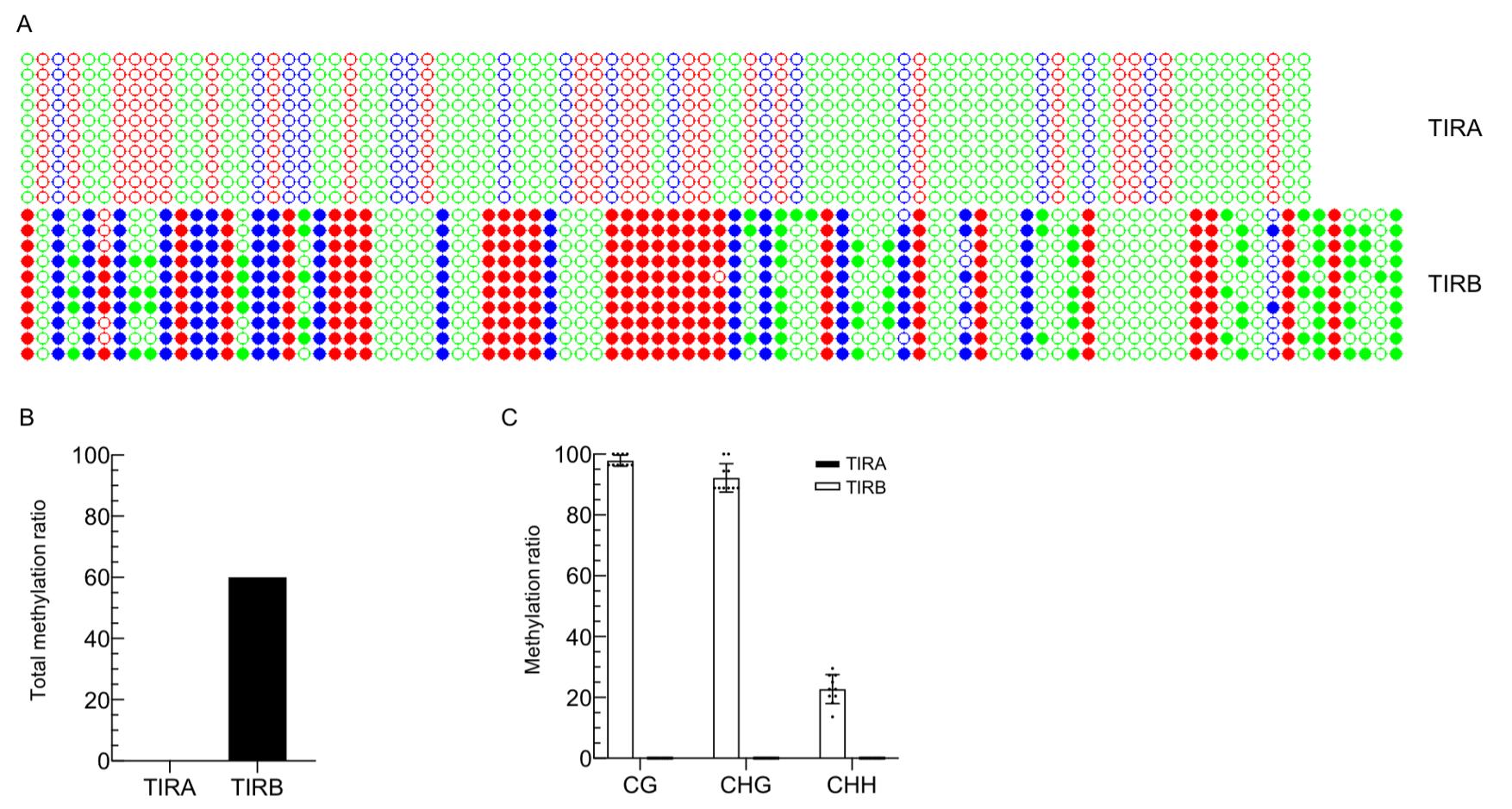
Figure 3. DNA methylation pattern at TIRA and TIRB in the MuDR line. A. Dot plot of DNA methylation pattern at TIRA and TIRB. B. Total DNA methylation ratio at TIRA and TIRB. C. DNA methylation ratio for cytosines in each sequence context at TIRA and TIRB. Ten individual clones were sequenced from PCR products of bisulfite-treated samples. The cytosines in different sequence contexts are represented by three colors (red: CG, blue: CHG, green: CHH; where H =A, C, or T). Filled cycles stand for methylated cytosines.
Acknowledgments
This work was funded by a NSF grant to DL (IOS-1237931).
Competing interests
The Authors declare that there is no conflict of interest.
References
- Foerster, A. S. M. O. (2010). Analysis of DNA Methylation in Plants by Bisulfite Sequencing. Methods Mol Biol 631: 1-11.
- Gruntman, E., Qi, Y., Slotkin, R. K., Roeder, T., Martienssen, R. A. and Sachidanandam, R. (2008). Kismeth: analyzer of plant methylation states through bisulfite sequencing. BMC Bioinformatics 9: 371.
- Guo, W., Wang, D. and Lisch, D. (2021). RNA-directed DNA methylation prevents rapid and heritable reversal of transposon silencing under heat stress in Zea mays. PLoS Genet 17(6): e1009326.
- Guo, W., Binstock, B., Cannon, A. and Lisch, D. (2022). An inexpensive, fast, and robust DNA extraction method for high-quality DNA for use in genotyping and next-generation sequencing applications in plants. Bio-protocol 10.21769/p1516.
- Li, H., Freeling, M. and Lisch, D. (2010). Epigenetic reprogramming during vegetative phase change in maize. Proc Natl Acad Sci U S A 107(51): 22184-22189.
- Schultz, M. D., Schmitz, R. J. and Ecker, J. R. (2012). 'Leveling' the playing field for analyses of single-base resolution DNA methylomes. Trends Genet 28(12): 583-585.
- Woodhouse, M. R., Freeling, M. and Lisch, D. (2006). The mop1 (mediator of paramutation1) mutant progressively reactivates one of the two genes encoded by the MuDR transposon in maize. Genetics 172(1): 579-592.
- Zhang, H., Lang, Z. and Zhu, J. K. (2018). Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19(8): 489-506.
Article Information
Copyright
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Guo, W., Cannon, A. and Lisch, D. (2022). A Molecular Cloning and Sanger Sequencing-based Protocol for Detecting Site-specific DNA Methylation. Bio-protocol 12(9): e4408. DOI: 10.21769/BioProtoc.4408.
Category
Systems Biology > Epigenomics > DNA methylation
Microbiology > Microbial genetics > DNA
Molecular Biology > DNA
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.