
- Home
- Protocols

-

An inexpensive, fast, and robust DNA extraction method for high-quality DNA for use in genotyping and next-generation sequencing applications in plants
Last updated date: Feb 1, 2022 DOI: 10.21769/p1516 Views: 6201 Forks: 0
An inexpensive, fast, and robust DNA extraction method for high-quality DNA for use in genotyping and next-generation sequencing applications in plants
Wei Guo1# Benjamin Binstock1# Anthony Cannon1 Damon Lisch1*
1Department of Botany and Plant Pathology, Purdue University, West Lafayette, Indiana, United States of America.
#Contributed equally to this work
*For correspondence: dlisch@purdue.edu
[Abstract] Genotyping and next-generation sequencing (NGS) work can be hampered by poor DNA quality. Traditional protocols that include mortar and pestle-based homogenization procedures and toxic chemicals, such as phenol or chloroform, are useful for extracting large quantities of high quality of DNA. However, these protocols are time-consuming, environmentally harmful, and thus are inappropriate for high throughput processing or for teaching laboratories. Here, we describe a protocol that uses only common laboratory equipment and non-toxic reagents that significantly shortens the processing time to yield genomic DNA. We validated this protocol using several common plant model systems and demonstrated its utility by providing a case study of genotyping in maize. When used in combination with mortar and pestle processing, we found it suitable for yielding high-quality DNA that is ready-to-use for NGS experiments. When used with BBs, large numbers of samples can be rapidly processed to produce DNA that is suitable for PCR genotyping.
[Background] Obtaining high-quality DNA is crucial for studying molecular genetics. In plants, the initial step in the isolation of DNA is to break down cell walls to allow the release of nuclei (Allen et al., 2006). One commonly used homogenization procedure involves grinding frozen tissue in liquid nitrogen in a cold mortar and pestle (Allen et al., 2006). This ensures the full pulverization of tissue and maximizes the quantity of DNA that can be extracted. For genotyping purpose, this option is time-consuming and therefore less efficient because only a few samples can be processed at a time. Bead beater homogenizers have higher throughput capabilities than mortar and pestle methods because they significantly lower the time spent on sample homogenization and so large numbers of samples can be processed at once (Leach et al., 2016; Lunde, 2018). Furthermore, samples are directly processed in tubes from the beginning, and DNA is ready to use for PCR (no dilution is required) upon completion of extraction, this protocol can minimize the use of extra tubes and lower the risk of cross-contamination when handling large numbers of samples. However, bead beaters are expensive, costing between $8000 and $10,000, and require special tubes, adaptors, and centrifuges, making their use impractical for small laboratories or classrooms.
Here, we highlight the use of regular BBs that are much cheaper than glass beads and other disrupters commonly recommended and used from other protocols. 6000 BBs cost only around $8 and are enough to process 6000 samples, with no homogenizer required. After homogenization, cetyltrimethylammonium bromide (CTAB) based protocols are commonly used for DNA extraction (Abdel-Latif and Osman, 2017; Allen et al., 2006). CTAB is a surfactant that efficiently promotes the separation of protein from nucleic acids (Healey Adam, 2014), but it is relatively toxic, and thus must be handled with care. That protocol also requires the use of phenol and chloroform solvents to purify the nucleic acids. Chloroform and phenol are both volatile and toxic, which is not ideal when handling numerous samples or when used by undergraduate trainees or in classroom settings. Alternatively, there are a wide variety of kits that can purify plant DNA that do not use toxic chemicals. However, because of their expense (generally more than a dollar per sample), they are impractical for laboratories or schools with a need to process large numbers of samples with limited resources and are not necessary for genotyping purposes.
Instead, we recommend using a modified version of a sodium dodecyl sulfate (SDS) and potassium acetate-based protein precipitation procedure that incorporates smaller quantities of tissue and bead-based homogenization of samples (Dellaporta Stephen, 1983; Edwards K., 1991). Homogenized samples are submerged into an extraction buffer containing polyvinylpyrrolidone (PVP-40), which removes polyphenolic compounds that are often bound to and co-precipitated with nucleic acids. SDS, an anionic detergent, is added to denature and add a negative charge to proteins. The addition of potassium acetate (KOAc) results in the formation of a white SDS-protein precipitate, which is readily visualized after centrifugation. The supernatant, containing nucleic acids, is transferred to a new tube and an equal amount of isopropanol is added to precipitate the DNA. 70-75% ethanol is added to remove the salt from the DNA precipitate while minimizing DNA solubility. After air-drying the sample completely, the DNA can be dissolved in a volume of water or buffer to the desired concentration.
To demonstrate the efficacy of this protocol, we validated it in several commonly used plants, including Arabidopsis thaliana, Solanum lycopersicum, Glycine max, Sorghum bicolor, and Zea mays. We found that the quality of genomic DNA isolated from these tissues is suitable for both genotyping and NGS. We also provide a case study of genotyping work for a genetic segregation analysis using a maize line that is segregating for a transposable element insertion. These procedures, which utilize commonly available equipment and inexpensive non-toxic chemicals, can increase the throughput capabilities for genotyping purposes when working with large amounts of samples. They also work well with the mortar & pestle-based homogenization procedures that are recommended for NGS purposes.
Materials and reagents
- Leaf tissues from 2-week-old seedlings collected from Arabidopsis thaliana, tomato, soybean, sorghum, and maize.
- BBs (Daisy, catalog number: 980060-446)
- Freezer boxes (Fisher Scientific, catalog number: 03-395-465, 100/CTN)
- Eppendorf tubes (Dot Scientific, catalog number: 609-GMT; RA2000-GMT)
- Ethylenediaminetetraacetic acid (EDTA) (Fisher scientific, catalog number: BP2482100)
- Sodium chloride (NaCl) (Fisher scientific, catalog number: BP358-1)
- Sodium dodecyl sulfate (SDS) (Fisher scientific, catalog number: 28312)
- Potassium acetate (Fisher scientific, catalog number: A16321.36)
- Isopropanol (Fisher scientific, catalog number: 040983.M1)
- 100% Ethanol (Fisher scientific, catalog number: T038181000)
- DNA loading dye (NEB, catalog number: 10816015)
- 100 bp DNA ladder (GOLDBIO catalog number: D003-500)
- 2x PCR Master Mix (syd labs, catalog number: MB067-EQ2G-L)
- Liquid nitrogen
- Pipettes and tips
Extraction buffer (50 mL)
5 mL 1 M Tris, pH 8 (final conc. 100 mM)
2.5 mL 0.5 M EDTA (final conc. 25 mM)
2.5 mL 5 M NaCl (final conc. 250 mM)
0.5 g PVP-40 (final conc. 1% w/v)
40 mL Water
Equipment
- Microcentrifuge
- UV-spectrophotometer
- Qubit fluorometer
- Gel electrophoresis system
Procedure
An overview of procedures can be seen in Figure 1
Figure 1 An overview of procedures

1. Sample collection
- For genotyping, prepare 2.0 ml Eppendorf tubes corresponding to the number of samples. Add a BB to each tube and line them up on a tube rack. Leaf tips approximately ~1 cm in size can be harvested directly by hand or scissors and placed directly into each tube (Figure 2A). It is recommended to harvest tissues from younger leaves to obtain better results, but DNA quality should be adequate from older tissues. There is no need to put the tubes into liquid nitrogen or dry ice during sample collection. Samples can be stored at room temperature for a day, at -20 oC for several months, or at -80 oC for long-term storage. Regardless of how they are stored, leaf material should be avoid being frozen and then thawed prior to processing.
- For NGS experiments, collect fresh samples on a regular basis for immediate processing or store them in 50 ml falcon tubes at -80 °C.
2. Tissue homogenization
- For genotyping purposes, put all tubes (as many as 96) into a freezer box that has 4 holes in the bottom without an internal rack. Place the freezer box containing the samples into a foam box that is slightly larger than the freezer box. Pour liquid nitrogen into the box until the level of liquid nitrogen is above the tubes. Let the tubes sit for at least 5 seconds (Figure 2B). Then remove the freezer box from the foam box and let the liquid nitrogen exit through the holes at the bottom of the freezer box. Put the lid on the freezer box and vigorously shake the tubes for 5-10 seconds by hand and then put them back into the foam box. After handshaking, tissues will typically become fine powder (Figure 2C). Do not leave tubes at room temperature for too long as the BBs will pop out. Wear goggles.
- For NGS purposes, harvest tissue and place it into a pre-chilled mortar & pestle. Add enough liquid nitrogen to submerge the tissue. As the liquid nitrogen is sublimating, begin breaking up the tissue with a rocking motion. After the liquid nitrogen has sublimated, vigorously grind the tissue until it is the consistency of a fine powder. This should take no more that 10 seconds and the tissue should still be frozen after this grinding step. Transfer the fine tissue powder to 2.0 ml centrifuge tubes and proceed directly to step 3 or store it at -80 °C prior to extraction.
3. Cell lysis
After homogenization, add 650 μl extraction buffer (EB) into each tube immediately. Invert the tubes 3-6 times. If you can see that the transparent EB turns light or dark green, the cells have been sufficiently disrupted to release enough DNA for PCR (Figure 2D). After adding EB, samples are stable at room temperature (RT) for several hours. Add 90 μl 10% SDS directly and invert the tubes 3-6 times. Let the tubes sit for 20-30 minutes either at room temperature, or, if maximum yield is desired, at 65 oC, which accelerates cell lysis and thus yields more DNA.
4. Protein and DNA precipitation
To precipitate SDS complexed protein, add 250 μl KOAc. Mix well by inverting several times. A white precipitate should form immediately (Figure 2E and 2F). Spin for 5-15 minutes at 12,000 rpm at room temperature. Next, transfer 400-600 μl of the supernatant to a fresh tube. It’s recommended to spin again and move the supernatant to a fresh tube if for NGS purpose. Add an equal volume of isopropanol to the tube containing the supernatant. It’s recommended to prepare tubes filled with an equal amount of isopropanol in advance. At this point, samples can be left at -20 oC overnight. Spin samples for 5 minutes at 10,000 rpm and pour off the supernatant. At this step, DNA precipitate may not be visible with the BB-based protocol but will likely be visible as a white or opaque pellet when using samples homogenized using mortar & pestle.
5. Purification
Add 400-500 μl of 70-75% ethanol to each tube, then spin for 5 minutes at 7500 rpm at RT. Directly pour off the supernatant and air-dry samples completely. Tubes can either be left with caps opened on a rack at the bench or in a fume hood. It is recommended to put them upside-down on paper towels. This is an important measure to make sure the residual ethanol is gone as it can directly inhibit downstream enzyme activities. It is not recommended to remove liquid residuals with a pipette, as it may increase the chance of cross-contamination or loss of DNA.
6. Resuspension
For genotyping, add 100-200 μl water into each tube. There is no need to add RNase. However, for NGS purposes, it is important to add RNase, followed by one additional precipitation step, and to adjust the volume of water for each sample following quantitation to insure uniformity.
7. Genotyping
1 μl of DNA sample can be used for genotyping-PCR.
Typical genotyping PCR conditions are (Table 1)
Table 1 Overview of genotyping-PCR conditions
| Component | 12.5 μl Reaction |
| 2X Master Mix | 6.25 μl |
| 10 mM Forward Primer | 0.25 μl |
| 10 mM Reverse Primer | 0.25 μl |
| DNA | 1 μl |
| Nuclease-free water | 4.75 μl |
Thermal cycling conditions
95°C 3 minutes
95 °C 30 seconds
45-68 °C 30 seconds
70 °C 1 minute per kb
30 cycles or more
Final extension
70 °C 5 minutes
Figure 2 Demonstration of sample preparation procedures

Figure 2 Overview of sample preparation procedures. A. Leaf tissue and a BB were placed into a 2 mL Eppendorf tube. B. All tubes were submerged into liquid nitrogen in a freezer box. C. Formation of fine powder after a few seconds of handshaking. D. Extraction buffer was added into each tube. E. KOAc was added into each tube. F. Formation of protein precipitate after centrifugation.
Results
Validation of the protocol in various plant model systems
To demonstrate the usefulness of this protocol, we chose five commonly studied plants: Arabidopsis thaliana, Solanum lycopersicum, Glycine max, Sorghum bicolor, and Zea mays. Here, we focus on extracting high quality DNA while avoiding the use of expensive equipment and toxic chemicals. For each plant, one gram of two-week-old leaf tissue was collected and ground with a mortar & pestle in liquid nitrogen. The DNA was then extracted and finally dissolved into 30 μl water following the procedures described above (Figure 1). To determine DNA quality, we mixed 2 μl DNA from each sample with 10x DNA loading dye and loaded them into a gel along with the same volume of (2 μl) of DNA ladder. After electrophoresis, we visualized the gel under UV light and observed bright and intact bands corresponding to high quality high molecular weight DNA for each sample (Figure 3). The known concentration of the bands in the DNA ladder was used to estimate the concentration of the double stranded genomic DNA. For comparison, we used a Qubit fluorometer to measure for concentration (Table 2). To check for the purity of the DNA, we used a UV spectrophotometer. DNA, RNA and proteins absorb UV light at discreet wavelengths, making it possible to determine the purity of DNA samples. We examined our DNA samples using UV-spectrophotometry. The 260/230 ratio ranged from 2.0-2.2, and the 260/280 ratio was between 1.8-2.0 (Table 2), indicating that our samples were free of protein and RNA contamination.
Validation of the protocol for use in genotyping
When there are many samples to genotype, time and cost become issues. Although we have used this protocol to genotype samples for our previous work (Guo et al., 2021), here, we demonstrate the convenience of our protocol using a well-documented system called the minimal Mutator line in maize (Lisch & Freeling, 1995). In brief, mudrA encodes a transposase that can cause excision of a Mu1 transposon that transcriptionally represses a color gene, which results in pigmented spots on maize kernels. In the absence of mudrA, the kernels are uniformly pale. We genotyped for the presence or absence of mudrA in progeny seedlings from an ear derived from a test cross (mudrA/+ x +/+) segregating for spotted and pale kernels. We collected samples from one-week-old seedlings and extracted DNA using the BB-based protocol. The extracted DNA was dissolved in 200 μl of water, which is sufficient for 200 PCRs. We found that all samples collected from the spotted kernels are positive for the mudrA gene, and all of the samples from the pale kernels were negative (Figure 4). The positive control as well as the negative water control suggest that the PCR was successful and that there was no contamination.
Figure 3 Examining DNA quality in the agarose gel
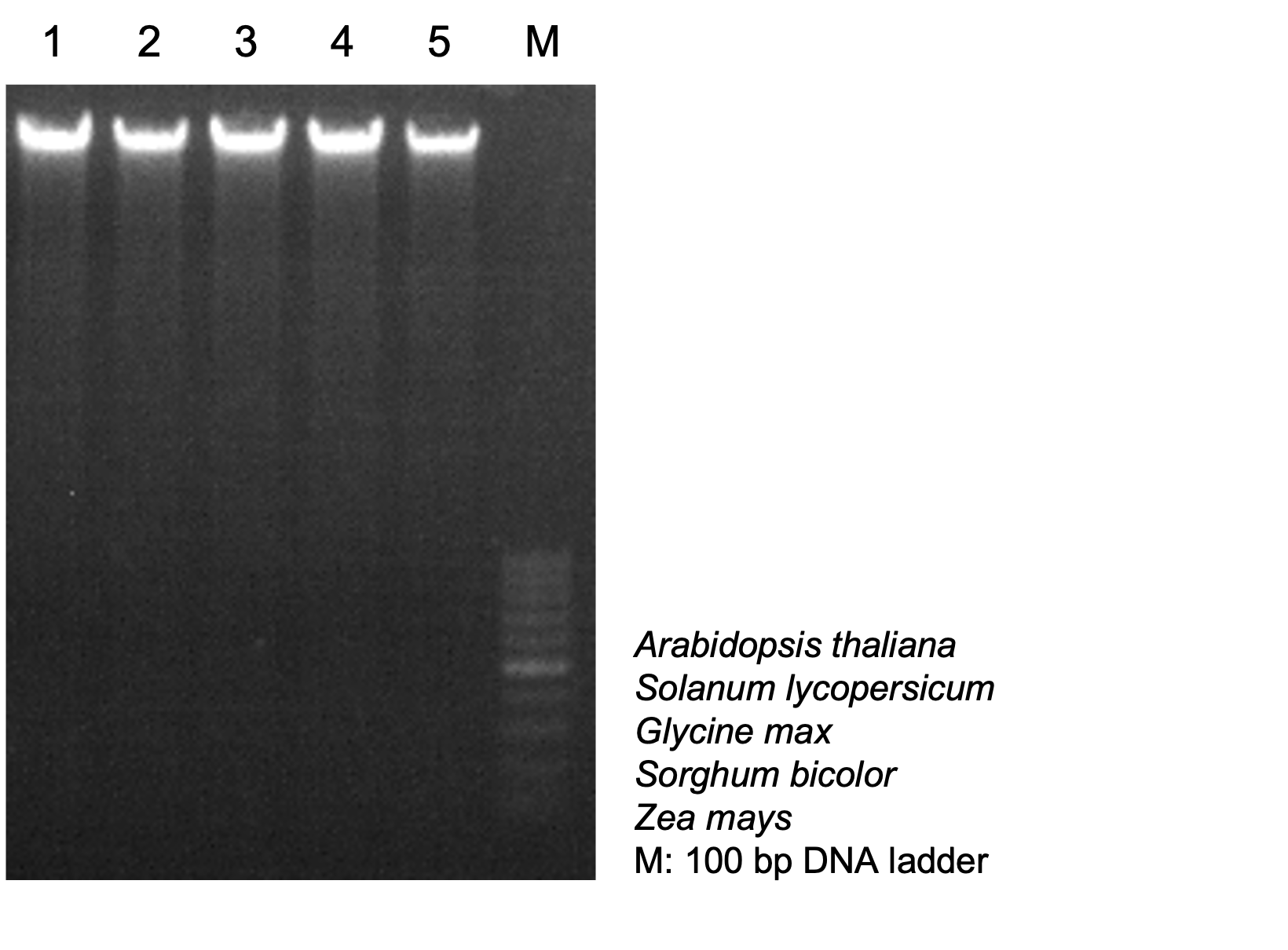
Figure 3 DNA was extracted and dissolved into 30 μl water. 1 μl RNase was added and incubated under 37 °C for 30 min to digest RNAs. 2 μl DNA from each sample was mixed with 10 x DNA loading dye, and 2 μl of 100 bp DNA ladder was loaded in 1% agarose gel. Gel was visualized under UV light. Mass of the 500 bp band from DNA ladder: 18 ng/μl
Table 2 Examining concentration and purity of genomic DNA samples
| Sample | Concentration (ng/μl, Qubit) | A260/A280 | A260/A230 |
| Arabidopsis thaliana | 152 | 1.88 | 2.12 |
| Solanum lyopersicum | 144 | 1.82 | 2.10 |
| Glycine max | 162 | 1.80 | 2.15 |
| Sorghum bicolor | 159 | 1.92 | 2.13 |
| Zea mays | 132 | 1.87 | 2.12 |
Figure 4 Genotyping result of mudrA in the maize minimal line

Figure 4 DNA was extracted from leaf tissues of ten samples of each class, either spotted or pale. PCR was performed using 2 x Master Mix. PCR products were loaded into the gel. MuDR/- was used as positive control, H2O was used as negative control.
Conclusion
We demonstrate a DNA extraction protocol that works well with multiple plant systems, and we highlight the use of inexpensive standard equipment and non-toxic chemicals that balances time, cost, and efficiency that is useful for both high-quality genotyping and NGS applications.
Acknowledgement
We thank members from Drs. Ma, Mengiste, and Kessler labs (Purdue University) for providing tissues of Glycine max, Sorghum bicolor, Solanum lycopersicum, and Arabidopsis thaliana for our project.
Competing interests
The authors declare that there is no conflict of interest.
References
Abdel-Latif, A., and Osman, G. (2017). Comparison of three genomic DNA extraction methods to obtain high DNA quality from maize Plant Methods 13, 1.
Allen, G.C., Flores-Vergara, M.A., Krasynanski, S., Kumar, S., and Thompson, W.F. (2006). A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide Nat Protoc 1, 2320-2325.
Dellaporta Stephen, W.J., Hicks James (1983). A plant DNA minipreparation: Version II Plant Mol Biol Rep 1, 19-21.
Edwards K., J.C., Thompson C. (1991). A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res 19, 1349.
Guo, W., Wang, D., and Lisch, D. (2021). RNA-directed DNA methylation prevents rapid and heritable reversal of transposon silencing under heat stress in Zea mays PLoS Genet 17, e1009326.
Healey Adam, F.A., Cooper Tal, Henry Robert (2014). Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species Plant Methods 10.
Leach, K.A., McSteen, P.C., and Braun, D.M. (2016). Genomic DNA Isolation from Maize (Zea mays) Leaves Using a Simple, High-Throughput Protocol. Curr Protoc Plant Biol 1, 15-27.
Lisch Damon, C.P., Freeling Michael (1995). Genetic characterization of the Mutator system in maize: behavior and regulation of Mu transposons in a minimal line Genetics 139, 1777-1796.
Lunde, C. (2018). Small-scale DNA Extraction Method for Maize and Other Plants Bio-Protocol 8.
- Guo, W, Binstock, B, Cannon, A and Lisch, D(2022). An inexpensive, fast, and robust DNA extraction method for high-quality DNA for use in genotyping and next-generation sequencing applications in plants. Bio-protocol Preprint. 10.21769/p1516.
Category
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.