Visualizing Diverse RNA Functions in Living Cells With SpinachTM Family of Fluorogenic Aptamers
利用SpinachTM 系列荧光适配体可视化活细胞中多种RNA功能
Vendor protocol
0 Favorites
1840 Views
Jun 20, 2026
Vendor protocol
0 Favorites
1840 Views
Jun 20, 2026
0 Favorites
1840 Views
Jun 20, 2026
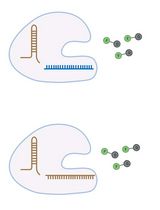
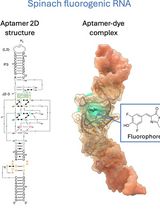
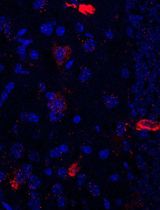
RNA is now recognized as a highly diverse and dynamic class of molecules whose localization, processing, and turnover are central to cell function and disease. Live-cell RNA imaging is therefore essential for linking RNA behavior to mechanism. Existing approaches include quenched hybridization probes that directly target endogenous transcripts but face delivery and sequestration issues, protein-recruitment tags such as MS2/PP7 that add large payloads and can perturb localization or decay, and CRISPR–dCas13 imaging that requires substantial protein cargo and careful control of background and off-target effects. Here, we present a protocol for live-cell RNA imaging using the SpinachTM family of fluorogenic RNA aptamers. The method details the design and cloning of SpinachTM-tagged RNA constructs, selection and handling of cognate small-molecule fluorophores, expression in mammalian cell lines, dye loading, and image acquisition on standard fluorescence microscopes, followed by quantitative analysis of localization and dynamics. We include controls to verify aptamer expression and signal specificity, guidance for multiplexing with related variants (e.g., Broccoli, Corn, Squash, Beetroot), and troubleshooting for dye permeability and signal optimization. Application examples illustrate use in tracking cellular delivery of mRNA therapeutics, monitoring transcription and decay in response to perturbations, and the forming of toxic RNA aggregates. Compared with prior methods, SpinachTM tags are compact, genetically encodable, and fluorogenic, providing high-contrast imaging in both the nucleus and cytoplasm with single-vector simplicity and multiplexing capability. The protocol standardizes key steps to improve robustness and reproducibility across cell types and laboratories.