- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Micrococcal Nuclease (MNase) Assay of Arabidopsis thaliana Nuclei


Published: Vol 3, Iss 7, Apr 5, 2013 DOI: 10.21769/BioProtoc.455 Views: 24597
Original research article
The authors used this protocol in:

Aug 2012
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Micrococcal nuclease (MNase) is able to produce double-strand breaks within nucleosome linker regions. The efficiency of MNase digestion depends on the degree of chromatin compaction, being more easily digested the regions of less compacted chromatin. The MNase protocol described here can be used to asses changes in the chromatin structure of nuclei extracted from Arabidopsis seedlings.
Keywords: Arabidopsis thalianaMaterials and Reagents
- Micrococcal Nuclease (MNase) (Roche Applied Science, catalog number: 10107921001 , 15,000 U)
- Proteinase K (Roche Applied Science, catalog number: 03115836001 )
- PIPES (Sigma-Aldrich, catalog number: P-9291 )
- Liquid nitrogen
- Sucrose
- KCl
- MgCl2
- CaCl2
- Triton X-100
- 1 mM PMSF (freshly added)
- Tris/HCl (pH 7.8)
- EDTA
- SDS
- Ethidium bromide
- Nuclei extraction buffer A, B, C (see Recipes)
- MNase buffer (see Recipes)
- 2x stop buffer (see Recipes)
- 10x Proteinase K buffer (see Recipes)
Equipment
- Pestle and mortar
- Centrifuges
- 37 °C oven
- 70 μm Nylon mesh
- 50 ml Falcon tubes
- Centrifuge tubes
- 2 ml microtubes
- Nylon mesh
Procedure
- Grind frozen Arabidopsis plantlets (2 g) with a pestle and a mortar (previously cooled with liquid nitrogen) under liquid nitrogen.
Note: Perform the following steps on ice. - Add the homogenized plant material to 10 ml of nuclei extraction buffer A in a 50 ml Falcon tube. Mix it well by vortexing.
- Filter twice the plant homogenate obtained in step 2, using a 70 μm nylon mesh placed in a funnel.
Note: perform the second filtering step in the centrifuge tubes needed for step 4. - Centrifuge at 10,000 x g for 20 min at 4 °C.
- Discard the supernatant by decantation.
Note: Discard all the supernatant by pipetting. - Resuspend the pellet in 200-500 μl of nuclei extraction buffer B (this volume can be adjusted depending on the size of the pellet).
- Pipet 200-500 μl of nuclei extraction buffer C (it must be the same volume as the one used in step 6) and place it into an empty 2 ml Eppendorf tube.
Note: Nuclei extraction buffer C is viscous because of its high sucrose content. You should perform this step slowly in order to avoid the formation of bubbles. - Add the resuspended pellet from step 6 onto the layer of buffer C obtained in step 7.
Note: Pay attention in not disturbing the layer of Buffer C when you add the resuspended pellet. - Centrifuge at 12,000 x g for 1 h at 4 °C.
- Discard all the supernatant by pipetting.
- Resuspend the pellet in 250 μl of MNase buffer (this volume can be adjusted depending on the size of the pellet).
Note: At this point you can add 35% glycerol to the nuclei samples, submerge them into liquid nitrogen, and keep frozen at -80 °C until MNase digestion is going to be performed. At that moment, nuclei are defrozen and centrifuged at 16,000 x g for 20 min at 4 °C in order to remove the glycerol. Finally, the nuclei are resuspended in MNase buffer. - Quantify the DNA concentration by measuring the absorbance at 260 nm. Usually a dilution 1:20 should be used.
Optional: Analyze the DNA integrity before performing the MNase digestion. For this purpose, mix equal volumes of nuclei and 2x stop buffer, and centrifuge at 16,000 x g for 10 min at 4 °C. Analyze the supernatant on a 1.2% agarose gel stained with ethidium bromide.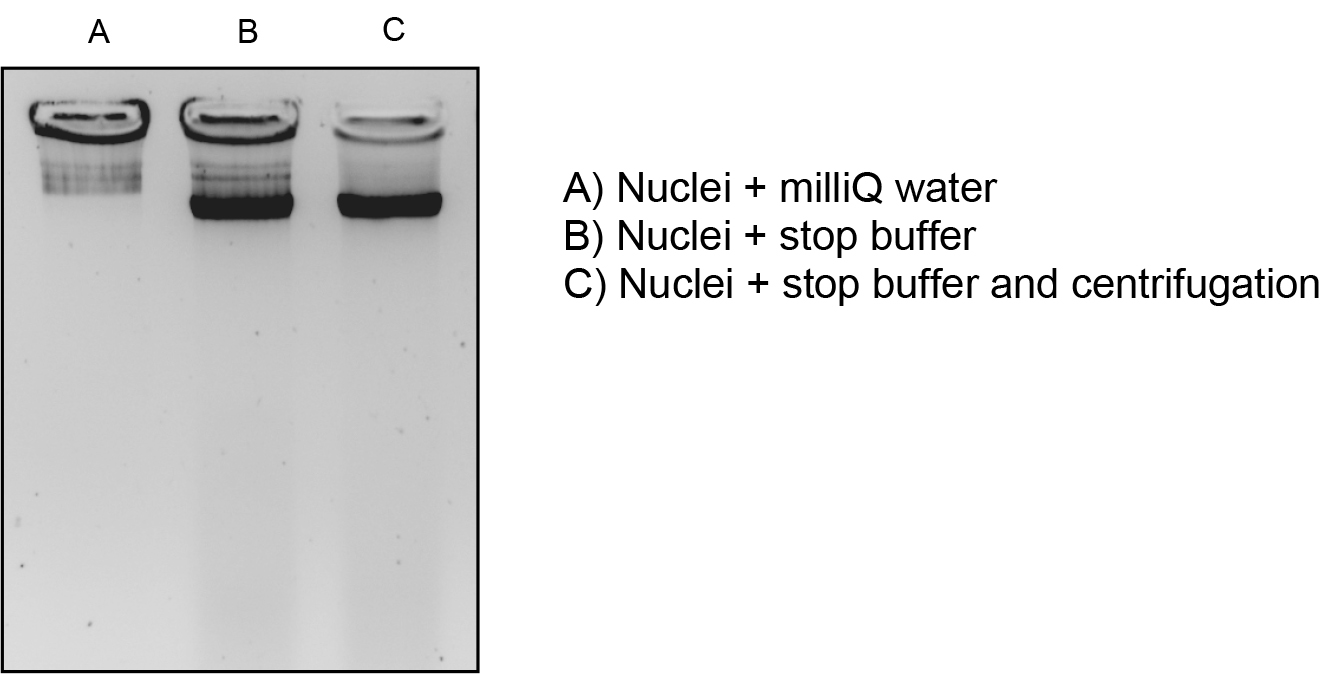
Figure 1. Analysis of DNA integrity before performing the MNase digestion. Several procedures were used for this purpose: (A) mixing equal volums of nuclei and milliQ water; (B) mixing equal volumes of nuclei and 2x stop buffer and, (C) mixing equal volumes of nuclei and 2x stop buffer, and centrifugation as described in the protocol. The procedure in (C) gave the best results and was used routinely. - Perform this step if you want to compare different samples, if not, you can directly proceed to MNase digestion (step 14). Dilute the sample(s) with MNase buffer in order to obtain the same DNA concentration in all of them (usually a range of concentrations between 300 ng/μl and 600 ng/μl).
Note: The final volume for all the samples must be the same and it must be adjusted depending on the number of MNase digestions you want to perform. As a rule, use between 20 μl and 40 μl per reaction.
To study chromatin sensitivity to MNase, two complementary protocols can be used: Digestion with different concentrations (Units) of MNase (step 14) and/or digestion with a desired MNase concentration during different incubation times (step 15). - Incubate the suspension of nuclei from step 13 with different Units of MNase, at 37 °C for 15 min. For instance, you can use 1, 2.5, 5, 10, 20, 40 U/ml or higher concentrations, until you observe total degradation of the high molecular weight DNA band (Figure 2). For this purpose, prepare the MNase solutions by making serial dilutions from the enzyme stock (for example 10,000 U/ml), in order to add the same volume of MNase to each individual reaction. For example, aliquot 30 μl of nuclei and add 10 μl of MNase at the desired concentration. To stop the reaction, add 40 μl of 2x stop buffer. Finally, add Proteinase K 1x buffer and 1 μl of Proteinase K (stock 10 mg/ml), and incubate overnight at 37 °C.
- Incubate the diluted nuclei with the MNase at 4 °C (*) for different periods of time, in order to perform time-course studies at a desired concentration of MNase. At each time point, transfer 20 μl of the reaction to an eppendorf tube containing 20 μl of stop buffer 2x and mix. Add Proteinase K 1x buffer and 1 μl of Proteinase K (stock 10 mg/ml), and incubate overnight at 37 °C.
(*) The optimal digestion temperature of MNase is 37 °C. At 4 °C MNase digestion is slower than at 37 °C, so you can use longer incubation time of digestion at 4 °C than at 37 °C.
Note: RNase treatment can be performed at the end of step 14 and step 15 if you observe RNA contamination in your samples. - Add DNA loading buffer to the sample obtained in step 14 and step 15 and visualize the results by electrophoresis on 1.2% agarose gels stained with ethidium bromide.
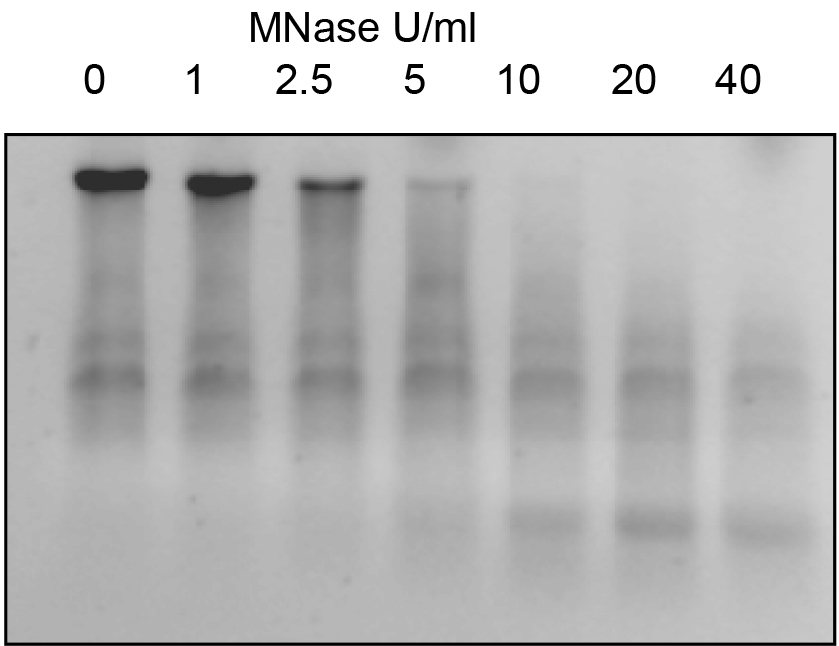
Figure 2. Example of MNase digestion using diferent concentrations of MNase. Same amounts of nuclei were digested with increasing concentrations of MNase at 37 ºC for 15 min and analysed in an 1.2% agarose gel.
Recipes
- Nuclei extraction buffer A
0.25 M sucrose
60 mM KCl
15 mM MgCl2
1 mM CaCl2
15 mM PIPES (pH 6.8)
0.8% Triton X-100
1 mM PMSF (freshly added) - Nuclei extraction buffer B
0.25 M sucrose
10 mM Tris/HCl (pH 8.0)
10 mM MgCl2
1% v/v Triton X-100
5 mM β-mercaptoethanol (freshly added)
1 mM PMSF (freshly added) - Nuclei extraction buffer C
1.7 M sucrose
10 mM Tris/HCl (pH 8.0)
10 mM MgCl2
0.5% Triton X-100
5 mM β -mercaptoethanol
1 mM PMSF - MNase buffer
0.3 M sucrose
20 mM Tris/HCl (pH 7.5)
3 mM CaCl2 - 2x stop buffer
50 mM EDTA
1% SDS - 10x Proteinase K buffer
100 mM Tris/HCl (pH 7.8)
50 mM EDTA
5% SDS
Acknowledgments
This protocol was developed for the work previously published in Plant Journal (Moreno-Romero et al., 2012). This work was supported by grants BFU2007-60569, BFU2010-15090 and Consolider Ingenio 2010 CSD2007-00036 from the Ministerio de Educación y Ciencia, (Spain) and grants 2005SGR-00112 and 2009SGR-795 from the Generalitat de Catalunya, Catalunya (Spain). L.A. and J.M.-R. were recipients of fellowships from the Ministerio de Educación y Ciencia (Spain) and the Universitat Autònoma de Barcelona respectively.
References
- Moreno-Romero, J., Armengot, L., Mar Marques-Bueno, M., Britt, A. and Carmen Martinez, M. (2012). CK2-defective Arabidopsis plants exhibit enhanced double-strand break repair rates and reduced survival after exposure to ionizing radiation. Plant J 71(4): 627-638.
Article Information
Copyright
© 2013 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Armengot, L. and Moreno-Romero, J. (2013). Micrococcal Nuclease (MNase) Assay of Arabidopsis thaliana Nuclei. Bio-protocol 3(7): e455. DOI: 10.21769/BioProtoc.455.
Category
Cell Biology > Organelle isolation > Nuclei
Molecular Biology > DNA > DNA structure
Plant Science > Plant molecular biology > DNA > DNA structure
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.