- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Most Read
Most viewed in the last three months

A Guide to Basic RNA Sequencing Data Processing and Transcriptomic Analysis
RNA sequencing (RNA-Seq) has transformed transcriptomic research, enabling researchers to perform large-scale inspection of mRNA levels in living cells. With the growing applicability of this technique to many scientific investigations, the analysis of next-generation sequencing (NGS) data becomes an important yet challenging task, especially for researchers without a bioinformatics background. This protocol offers a beginner-friendly step-by-step guide to analyze NGS data (starting from raw .fastq files), providing the required codes with an explanation of the different steps and software used. We outline a computational workflow that includes quality control, trimming of reads, read alignment to the genome, and gene quantification, ultimately enabling researchers to identify differentially expressed genes and gain insights on mRNA levels. Multiple approaches to visualize this data using statistical and graphical tools in R are also described, allowing the generation of heatmaps and volcano plots to represent genes and gene sets of interest.
Cycloheximide (CHX) Chase Assay to Examine Protein Half-life
Cycloheximide (CHX) is a small molecule derived from Streptomyces griseus that acts as fungicide. As a ribosome inhibitor, CHX can restrict the translation elongation of eukaryotic protein synthesis. Once protein synthesis is inhibited by CHX, the level of intracellular proteins decreases by degradation through the proteasome or lysosome system. Thus, the CHX chase assay is widely recognized and used to observe intracellular protein degradation and to determine the half-life of a given protein in eukaryotes. Here, we present a complete experimental procedure of the CHX chase assay.Graphical overview

sc3D: A Comprehensive Tool for 3D Spatial Transcriptomic Analysis
Serial spatial omics technologies capture genome-wide gene expression patterns in thin tissue sections but lose spatial continuity along the third dimension. Reconstructing these two-dimensional measurements into coherent three-dimensional volumes is necessary to relate molecular domains, gradients, and tissue architecture within whole organs or embryos. sc3D is an open-source Python framework that registers consecutive spatial transcriptomic sections, interpolates bead coordinates in three dimensions, and stores the result in an AnnData object compatible with Scanpy. The workflow performs slice alignment, 3D reconstruction, optional downsampling, and interactive visualization in a napari-sc3D-viewer, enabling virtual in situ hybridization and spatial differential gene expression analysis. We tested sc3D on Slide-seq and Stereo-seq datasets, including E8.5 and E16.5 mouse embryos, recovering continuous tissue morphologies, cardiac anatomical markers, and the expected anterior–posterior gradients of Hox gene expression. These results show that sc3D allows reproducible reconstruction and analysis of volumetric spatial omics data across different samples and experimental platforms.
Generation of Human Induced Pluripotent Stem Cell (hiPSC)-Derived Astrocytes for Amyotrophic Lateral Sclerosis and Other Neurodegenerative Disease Studies
Astrocytes are increasingly recognized for their important role in neurodegenerative diseases like amyotrophic lateral sclerosis (ALS). In ALS, astrocytes shift from their primary function of providing neuronal homeostatic support towards a reactive and toxic role, which overall contributes to neuronal toxicity and cell death. Currently, our knowledge on these processes is incomplete, and time-efficient and reproducible model systems in a human context are therefore required to understand and therapeutically modulate the toxic astrocytic response for future treatment options. Here, we present an efficient and straightforward protocol to generate human induced pluripotent stem cell (hiPSC)-derived astrocytes implementing a differentiation scheme based on small molecules. Through an initial 25 days, hiPSCs are differentiated into astrocytes, which are matured for 4+ weeks. The hiPSC-derived astrocytes can be cryopreserved at every passage during differentiation and maturation. This provides convenient pauses in the protocol as well as cell banking opportunities, thereby limiting the need to continuously start from hiPSCs. The protocol has already proven valuable in ALS research but can be adapted to any desired research field where astrocytes are of interest.Key features• This protocol requires preexisting experience in hiPSC culturing for a successful outcome.• The protocol relies on a small molecule differentiation scheme and an easy-to-follow methodology, which can be paused at several time points.• The protocol generates >50 × 106 astrocytes per differentiation, which can be cryopreserved at every passage, ensuring a large-scale experimental output.Graphical overview
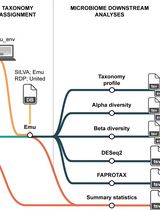
Reproducible Emu-Based Workflow for High-Fidelity Soil and Plant Microbiome Profiling on HPC Clusters
Accurate profiling of soil and root-associated bacterial communities is essential for understanding ecosystem functions and improving sustainable agricultural practices. Here, a comprehensive, modular workflow is presented for the analysis of full-length 16S rRNA gene amplicons generated with Oxford Nanopore long-read sequencing. The protocol integrates four standardized steps: (i) quality assessment and filtering of raw reads with NanoPlot and NanoFilt, (ii) removal of plant organelle contamination using a curated Viridiplantae Kraken2 database, (iii) species-level taxonomic assignment with Emu, and (iv) downstream ecological analyses, including rarefaction, diversity metrics, and functional inference. Leveraging high-performance computing resources, the workflow enables parallel processing of large datasets, rigorous contamination control, and reproducible execution across environments. The pipeline’s efficiency is demonstrated on full-length 16S rRNA gene datasets from yellow pea rhizosphere and root samples, with high post-filter read retention and high-resolution community profiles. Automated SLURM scripts and detailed documentation are provided in a public GitHub repository (https://github.com/henrimdias/emu-microbiome-HPC; release v1.0.2, emu-pipeline-revised) and archived on Zenodo (DOI: 10.5281/zenodo.17764933).
A Simple, Reproducible Procedure for Chemiluminescent Western Blot Quantification
Western blotting is a universally used technique to identify specific proteins from a heterogeneous and complex mixture. However, there is no clear and common procedure to quantify the results obtained, resulting in variations due to the different software and protocols used in each laboratory. Here, we have developed a procedure based on the increase in chemiluminescent signal to obtain a representative value for each band to be quantified. Images were processed with ImageJ and subsequently compared using R software. The result is a linear regression model in which we use the slope of the signal increase within the combined linear range of detection to compare between samples. This approach allows to quantify and compare protein levels from different conditions in a simple and reproducible way. Graphical overview
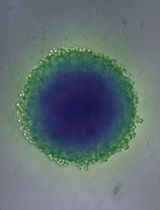
A Simple and Cost-Effective Method for Generating Spheroids From Triple-Negative Breast Cancer Cell Line (MDA-MB-231)


Breast cancer (BC) is the most frequently diagnosed malignancy in women and a leading cause of cancer-related mortality worldwide. Current clinical management relies on molecular classification—based on estrogen receptor (ER), progesterone receptor (PR), HER2, and Ki67 expression—to guide prognosis and therapy. Triple-negative breast cancer (TNBC), which lacks ER, PR, and HER2 expression, represents 15%–20% of cases and is characterized by aggressive behavior, early recurrence, and a paucity of targeted treatment options. These challenges underscore the urgent need for improved preclinical models that better recapitulate tumor biology to accelerate therapeutic discovery. While conventional monolayer (2D) cultures have contributed significantly to cancer research, they fail to mimic critical features of the three-dimensional (3D) tumor microenvironment (TME), thereby limiting clinical translation. To address this gap, 3D spheroid models have emerged as a powerful intermediary, more accurately replicating in vivo conditions such as cell–cell and cell–matrix interactions, nutrient and oxygen gradients, and the development of hypoxic cores. These features make spheroids a physiologically relevant platform for studying complex processes like metastasis, drug resistance, and treatment response. Here, we present a robust, simple, and cost-effective protocol for generating uniform 3D spheroids. Our method enables consistent monitoring of spheroid formation and growth over time, with quantitative, image-based size analysis to ensure reproducibility and scalability. Designed for flexibility, the protocol is broadly applicable across diverse cell types, effectively bridging the gap between traditional 2D cultures and complex in vivo studies. By providing an accessible and reliable model of the 3D TME, this protocol opens new avenues for high-throughput drug screening, mechanistic studies of tumor progression, and the advancement of personalized medicine strategies in breast cancer and beyond.
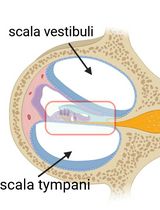
Cochlear Organ Dissection, Immunostaining, and Confocal Imaging in Mice
The organ of Corti, located in the inner ear, is the primary organ responsible for animal hearing. Each hair cell has a V-shaped or U-shaped hair bundle composed of actin-filled stereocilia and a kinocilium supported by true transport microtubules. Damage to these structures due to noise exposure, drug toxicity, aging, or environmental factors can lead to hearing loss and other disorders. The challenge when examining auditory organs is their location within the bony labyrinth and their small and fragile nature. This protocol describes the dissection procedure for the cochlear organ, followed by confocal imaging of immunostained endogenous and fluorescent proteins. This approach can be used to understand hair cell physiology and the molecular mechanisms required for normal hearing.
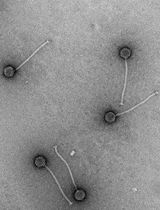
Quick and Cheap: Optimized Purification and Concentration of Bacteriophages Produced in Rich Culture Media

This protocol describes an easy, quick, cheap, and effective method for the purification and concentration of bacteriophages (phages) produced in rich culture media, meeting the quality criteria required for structural analyses. It is based on a tube dialysis system that replaces the classical but expensive and tedious density gradient ultracentrifugation step. We developed this protocol for the Oenococcus oeni bacteriophage OE33PA from its amplification to imaging by negative stain electron microscopy (NS-EM). The host bacterium, O. oeni, is a lactic acid bacterium that lives in harsh oenological ecosystems and grows only in rich and complex media such as Man–Rogosa–Sharpe (MRS) or fruit juice-based media in laboratory conditions. This raises experimental challenges in pure and concentrated phage preparations for further uses such as structure-function studies.
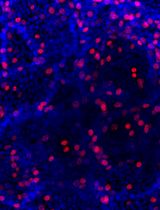
An Optimized Protocol for Simultaneous Propagation of Patient-derived Organoids and Matching CAFs
Recurrent hormone receptor-positive (HR+) breast cancer is a leading cause of cancer mortality in women. Recurrence and resistance to targeted therapies have been difficult to study due to the long clinical course of the disease, the complex nature of resistance, and the lack of clinically relevant model systems. Existing models are limited to a few HR+ cell lines, organoid models, and patient-derived xenograft models, all lacking components of the human tumor microenvironment. Furthermore, the low take rate and loss of estrogen receptor (ER) expression in patient-derived organoids (PDOs) has been challenging. Our protocol allows simultaneous isolation of PDOs and matching cancer-associated fibroblasts (CAFs) from primary and metastatic HR+ breast cancers. Importantly, our protocol has a higher take rate and enables long-term culturing of PDOs that retain ER expression. Our matching PDOs and CAFs will provide researchers with a new resource to study the influence of the tumor microenvironment on various aspects of cancer biology such as cell growth and drug resistance in HR+ breast cancer.

How to Train Custom Cell Segmentation Models Using Cell-APP
The deep learning revolution has accelerated discovery in cell biology by allowing researchers to outsource their microscopy analyses to a new class of tools called cell segmentation models. The performance of these models, however, is often constrained by the limited availability of annotated data for them to train on. This limitation is a consequence of the time cost associated with annotating training data by hand. To address this bottleneck, we developed Cell-APP (cellular annotation and perception pipeline), a tool that automates the annotation of high-quality training data for transmitted-light (TL) cell segmentation. Cell-APP uses two inputs—paired TL and fluorescence images—and operates in two main steps. First, it extracts each cell’s location from the fluorescence images. Then, it provides these locations to the promptable deep learning model μSAM, which generates cell masks in the TL images. Users may also employ Cell-APP to classify each annotated cell; in this case, Cell-APP extracts user-specified, single-cell features from the fluorescence images, which can then be used for unsupervised classification. These annotations and optional classifications comprise training data for cell segmentation model development. Here, we provide a step-by-step protocol for using Cell-APP to annotate training data and train custom cell segmentation models. This protocol has been used to train deep learning models that simultaneously segment and assign cell-cycle labels to HeLa, U2OS, HT1080, and RPE-1 cells.
Cloning a Chloroplast Genome in Saccharomyces cerevisiae and Escherichia coli
Chloroplast genomes present an alternative strategy for large-scale engineering of photosynthetic eukaryotes. Prior to our work, the chloroplast genomes of Chlamydomonas reinhardtii (204 kb) and Zea mays (140 kb) had been cloned using bacterial and yeast artificial chromosome (BAC/YAC) libraries, respectively. These methods lack design flexibility as they are reliant upon the random capture of genomic fragments during BAC/YAC library creation; additionally, both demonstrated a low efficiency (≤ 10%) for correct assembly of the genome in yeast. With this in mind, we sought to create a highly flexible and efficient approach for assembling the 117 kb chloroplast genome of Phaeodactylum tricornutum, a photosynthetic marine diatom. Our original article demonstrated a PCR-based approach for cloning the P. tricornutum chloroplast genome that had 90%–100% efficiency when screening as few as 10 yeast colonies following assembly. In this article, we will discuss this approach in greater depth as we believe this technique could be extrapolated to other species, particularly those with a similar chloroplast genome size and architecture.
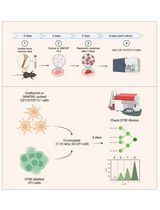
In Vitro Bone Marrow–Derived Dendritic Cells (BMDC) Generation for Antigen Presentation Assay

Dendritic cells (DC) are sentinel cells of the immune system that process and present antigens to activate T cells, thus serving to bridge the innate and adaptive immune systems. DCs are particularly efficient at cross-presentation whereby exogenously acquired antigens are processed and presented in context with MHCI molecules to activate CD8+ T cells. Assaying antigen presentation by DCs is a critical parameter in assessing immune functionality. However, the low abundance of bona fide DCs within the lymphoid compartments limits the utility of such assays. An alternative approach employing the culturing of bone marrow cells in the presence of factors needed for DC lineage commitment can result in the differentiation of bone marrow dendritic cells (BMDCs). This protocol details the process of in vitro generation of BMDCs and demonstrates their subsequent utility in antigen presentation assays. The protocol described can be adapted to various conditions and antigens.
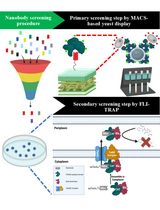
Isolation of Antigen-Specific Nanobodies From Synthetic Libraries Using a Protein Selection Strategy That Combines MACS-Based Screening of YSD and FLI-TRAP
Although protein–protein interactions (PPIs) are central to nearly all biological processes, identifying and engineering high-affinity intracellular binders remains a significant challenge due to the complexity of the cellular environment and the folding constraints of proteins. Here, we present a two-stage complementary platform that combines magnetic-activated cell sorting (MACS)-based yeast surface display with functional ligand-binding identification by twin-arginine translocation (Tat)-based recognition of associating proteins (FLI-TRAP), a bacterial genetic selection system for efficient screening, validation, and optimization of PPIs. In the first stage, MACS-based yeast display enables the rapid high-throughput identification of candidate binders for a target antigen from a large synthetic-yeast display library through extracellular interaction screening. In the second stage, an antigen-focused library is subcloned into the FLI-TRAP system, which exploits the hitchhiker export process of the Escherichia coli Tat pathway to evaluate binder–antigen binding in the cytoplasm. This stage is achieved by co-expressing a Tat signal peptide–tagged protein of interest with a β-lactamase-tagged antigen target, such that only binder–antigen pairs with sufficient affinity are co-translocated into the periplasm, thus rendering the bacterium β-lactam antibiotic resistant. Because Tat-dependent export requires fully folded and soluble proteins, FLI-TRAP further serves as a stringent in vivo filter for intracellular compatibility, folding, and stability. Therefore, this approach provides a powerful and cost-effective pipeline for discovering and engineering intracellular protein binders with high affinity, specificity, and functional expression in bacterial systems. This workflow holds promise for several applications, including synthetic biology and screening of theragnostic proteins and PPI inhibitors.

Deaminase-Assisted Sequencing for the Identification of 5-glyceryl-methylcytosine
DNA epigenetic modifications play crucial roles in regulating gene expression and cellular function across diverse organisms. Among them, 5-glyceryl-methylcytosine (5gmC), a unique DNA modification first discovered in Chlamydomonas reinhardtii, represents a novel link between redox metabolism and epigenetic regulation. Accurate genome-wide detection of 5gmC is essential for investigating its biological functions, yet no streamlined method has been available. Here, we present deaminase-assisted sequencing (DEA-seq), a simple and robust approach for base-resolution mapping of 5gmC. DEA-seq employs a single DNA deaminase that efficiently converts unmodified cytosines (C) and 5-methylcytosine (5mC) into uracils or thymines, while leaving 5gmC intact. This selective resistance generates a clear sequence signature that enables precise identification of 5gmC sites across the genome. The method operates under mild reaction conditions and is compatible with low-input DNA, minimizing sample loss and improving detection sensitivity. Overall, DEA-seq provides an accessible, efficient, and highly accurate protocol for profiling 5gmC, offering clear advantages in workflow simplicity, DNA integrity, and analytical performance.
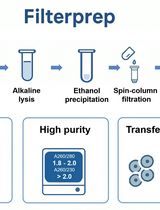
Plasmid DNA Purification Using Filterprep With an Optional Endotoxin Removal Step
This protocol presents a modified version of the Filterprep method originally reported in New Biotechnology, adding an optional step to reduce endotoxin levels. Filterprep is a simple, rapid, and cost-effective approach to plasmid DNA purification that couples ethanol precipitation with a single spin-column filtration step, eliminating chaotropic salts and silica binding. The formulations and parameters are fully transparent and do not rely on proprietary buffers, using only standard laboratory reagents and widely available miniprep columns. Under matched conditions, the method recovers high-purity plasmid DNA with yields up to fivefold higher than those obtained with representative commercial midiprep kits. The workflow is readily adoptable in most molecular biology laboratories and, under routine conditions, can be completed in approximately 40 min. The resulting DNA is suitable for molecular cloning, PCR, sequencing, and other downstream biochemical applications. Endotoxin is a lipopolysaccharide (LPS) found in the outer membrane of Gram-negative bacteria and may carry over during plasmid preparation. For experiments requiring lower endotoxin input, an optional modification resuspends the DNA pellet in a Triton X-114 wash buffer before column loading to decrease lipopolysaccharide carryover. The method is modular and extensible, allowing adjustment of precipitation and wash conditions, variation in the number of washes, selection of alternative column formats, and integration of endotoxin-reduction modules without altering the core principle. These features facilitate troubleshooting and quality control, enable scaling from routine batches to larger culture volumes and higher throughput, and allow seamless integration with existing workflows.
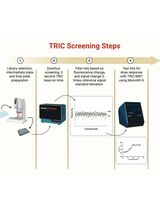
Orthogonal Temperature-Related Intensity Change and Time-Resolved Förster Resonance Energy Transfer High-Throughput Screening Platform for the Discovery of SLIT2 Binders
SLIT2 is a secreted glycoprotein implicated in axon guidance, immune modulation, and tumor biology, whose extracellular and glycosylated nature can complicate conventional biophysical screening workflows. Here, we provide a complete, step-by-step protocol for an orthogonal high-throughput discovery pipeline that integrates temperature-related intensity change (TRIC) as a solution-based primary binding screen with time-resolved Förster resonance energy transfer (TR-FRET, homogeneous time-resolved fluorescence format) as a functional assay for inhibition of the SLIT2–ROBO1 interaction. The workflow is designed to be fast and convenient, uses low reaction volumes and low nanomolar protein concentrations to minimize material use, and includes built-in quality control steps to support reproducible hit triage. In TRIC (NanoTemper Dianthus), binding is detected as temperature-dependent fluorescence intensity changes of a labeled target protein under an infrared (IR)-mediated thermal gradient, enabling immobilization-free detection of small-molecule interactions and instrument-assisted filtering of autofluorescent, quenching, or aggregating compounds. Candidate binders are advanced to multi-point TRIC/microscale thermophoresis (MST) measurements on Monolith X to determine binding affinity (Kd). In TR-FRET, disruption of SLIT2–ROBO1 association is quantified by changes in the ratiometric 665/620 nm emission readout, measured with a time delay to suppress short-lived background fluorescence, enabling concentration-response analysis and reporting of relative IC50 values (including partial inhibition behavior where applicable). Although presented using the SLIT2–ROBO1 extracellular interaction as a representative model system, this orthogonal screening strategy is designed to be adaptable to other extracellular protein-protein interactions where minimizing immobilization artifacts and fluorescence interference is critical.

A Bioinformatics Workflow to Identify eccDNA Using ECCFP From Long-Read Nanopore Sequencing Data
Extrachromosomal circular DNA (eccDNA) is a type of circular DNA that exists independently of chromosomes and has garnered significant attention in various fields, particularly in the context of smaller eccDNAs, which have considerable roles in gene regulation through various mechanisms. Current methods such as Circle-Seq and 3SEP can enrich small eccDNAs during sample preparation, but most bioinformatics pipelines remain challenging, exhibiting low accuracy and efficiency. This protocol describes the detailed workflow of a newly developed bioinformatics analysis pipeline, named EccDNA Caller based on Consecutive Full Pass (ECCFP), to accurately identify eccDNA from long-read Nanopore sequencing data. Compared to other pipelines, ECCFP significantly improves detection sensitivity, accuracy, and runtime efficiency. The process includes raw data quality control, trimming of adapters and barcodes, alignment to a reference genome, and identification of eccDNA, with detailed results encompassing accurate positioning of eccDNA, consensus sequences, and variants of individual eccDNA.
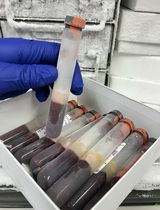
Nuclei Isolation Methods on Frozen Clotted Blood Samples
It is common practice for laboratories to discard clotted blood or freeze it for future DNA extraction after extracting serum from a serum-separating tube. If freezing for DNA extraction, the blood clot is not usually cryopreserved, which leads to cell membrane fragility. In this protocol, we describe steps to isolate high-quality nuclei from leukocytes derived from whole blood samples frozen without a cryoprotective medium. Nuclei isolated from this protocol were able to undergo ATAC (assay for transposase-accessible chromatin) sequencing to obtain chromatin accessibility data. We successfully characterized and isolated B cells and T cells from leukocytes isolated from previously frozen blood clot using Miltenyi’s gentleMACS Octo Dissociator coupled with flow sorting. Nuclei showed round, intact nuclear envelopes suitable for downstream applications, including bulk sequencing of nuclei or single-cell nuclei sequencing. We validated this protocol by performing bulk ATAC-seq.
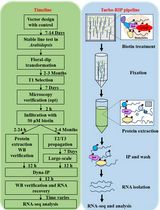
Turbo-RIP: A Protocol for TurboID-based RNA Immunopurification to Map RNA Landscapes in Plant Biomolecular Condensates
Biomolecular condensates organize cellular processes through liquid–liquid phase separation, creating membrane-less compartments enriched in specific proteins and RNAs. Understanding their RNA composition is essential for elucidating plant stress responses, yet capturing these transiently associated RNAs remains technically challenging. We present Turbo-RIP (TurboID-based proximity labeling with RNA immunopurification), a comprehensive protocol for identifying condensate-associated RNAs in plants. Turbo-RIP employs the biotin ligase TurboID to label proximal proteins at 22 °C, followed by formaldehyde crosslinking and streptavidin-based capture of protein–RNA complexes. We provide detailed procedures for three cloning strategies, transformation of Nicotiana benthamiana and Arabidopsis thaliana, validation of TurboID activity, and RNA recovery. The protocol successfully captured processing body–associated RNAs with minimal background. Turbo-RIP enables systematic mapping of RNA populations within plant condensates under diverse conditions. The protocol requires 3–5 days from sample preparation to RNA isolation, with construct validation taking 2–4 weeks. All procedures use standard laboratory equipment, making Turbo-RIP accessible for plant molecular biology laboratories.

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics





