
- Home
- Protocols

-

Proteomic profiling of hypusinated protein candidates using a clickable spermidine probe
Last updated date: Apr 30, 2026 Views: 49 Forks: 0
Proteomic profiling of hypusinated protein candidates using a clickable spermidine probe
Author List: Tian Zhang1,2,4, Jianlong Li1,2,4, Xinbo Hu1,4, Zhaoyin Wang1, Yaoyang Zhang1,3, Junying Yuan1,3,6*, Bing Shan1,3,5*
Affiliations:
1Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China;
2University of Chinese Academy of Sciences, Beijing, China;
3Shanghai Key Laboratory of Aging Studies, Shanghai, China;
4These authors contribute equally
5Technical contact
6Lead contact
*Correspondence: junying_yuan@sioc.ac.cn; shanbing@sioc.ac.cn
Abstract
Spermidine has been implicated in the regulation of diverse biological processes associated with aging. It exerts its effects through a unique posttranslational protein modification known as hypusination. This process involves the transfer of an aminobutyl moiety from spermidine to specific lysine residues, catalyzed by the enzyme deoxyhypusine synthase (DHPS), followed by hydroxylation mediated by deoxyhypusine hydroxylase (DOHH). Here, we present a protocol that enables proteome-wide identification of candidate hypusinated proteins. This workflow includes the synthesis of a clickable alkynyl-spermidine probe, DHPS-dependent metabolic labeling in cells, click chemistry–mediated biotinylation, streptavidin-based enrichment, and subsequent mass spectrometry analysis of probe-labeled proteins. Additionally, we also describe the procedures for data processing and statistical analysis.
For complete details on the use and execution of this protocol, please refer to Zhang et al. (2024)1.
Graphical overview
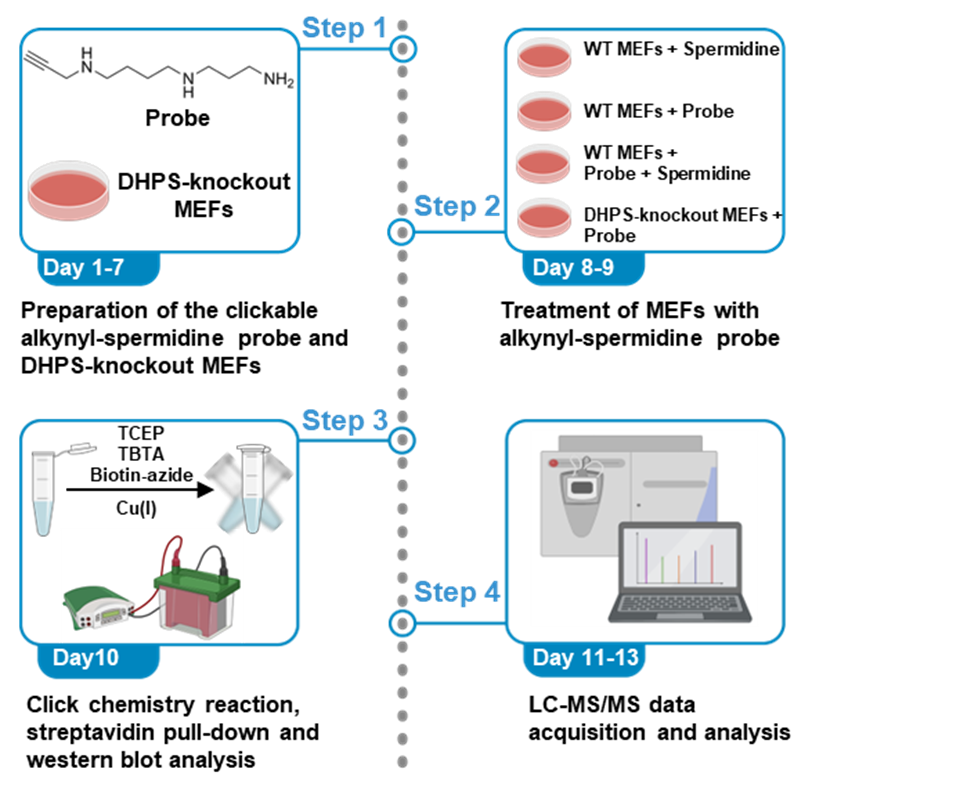
Background
Spermidine is a natural polyamine that has been widely recognized for its anti-aging properties and protective effects against age-associated pathologies, including cardiovascular, neurodegenerative, and inflammatory diseases2. However, the molecular mechanisms underlying these pleiotropic functions remain incompletely understood. Protein hypusination is a unique post-translational modification in which spermidine donates an aminobutyl moiety to a specific lysine residue in targeting proteins mediated sequentially by deoxyhypusine synthase (DHPS) and then hydroxylated by deoxyhypusine hydroxylase (DOHH)3,4. In this process, spermidine functions not only as a metabolic intermediate but also as a direct regulator of protein activity via covalent modification.
The first hypusination substrate, the eukaryotic initiation factor eukaryotic initiation factor 5A (eIF5A), was identified using two-dimensional gel electrophoresis (2-D gel electrophoresis) in the 1980s5 . Largely due to technical limitations, studies on hypusination had been restricted to eIF5A. Such limitations have hindered a mechanistic understanding of spermidine-mediated biology. This gap underscores the urgent need for unbiased, proteome-wide approaches to define the full spectrum of spermidine-dependent protein modification, which is essential for elucidating the molecular basis of spermidine’s diverse biological functions, particularly with regard to aging.
Here, we describe an innovative chemical proteomics workflow that challenges the existing paradigm by enabling proteome-wide identification of hypusinated proteins. The protocol employs a clickable alkynyl-spermidine probe for DHPS-dependent metabolic labeling, followed by Copper-catalyzed Azide–Alkyne Cycloaddition (CuAAC)-mediated biotinylation and streptavidin-based enrichment1. This strategy facilitates the capture and detection of rare, low-abundance hypusinated proteins that are typically undetectable by conventional mass spectrometry.
To ensure specificity, the workflow incorporates DHPS-knockout cell lines as negative controls and uses the polyamine oxidase inhibitor MDL-72527 to maintain the probe stability. Coupled with high-resolution LC–MS/MS, this approach enabled the identification of 1,627 candidate hypusinated proteins, including receptor-interacting serine/threonine-protein kinase 1(RIPK1) 1. By integrating synthetic chemistry with advanced proteomics, this method offers a robust platform for the systematic discovery of previously unrecognized hypusination targets.
Prior to starting the protocol, the alkyne-functionalized spermidine probe should be designed, synthesized and characterized. The synthesis of the probe is accomplished through sequential alkylation reactions followed by global deprotection step to yield the probe as its trifluoroacetate salt. The purity and identity of the intermediate and final product should be confirmed by NMR and, if possible, mass spectrometry. The probe should be dissolved in water or appropriate buffer and stored at −20°C in aliquots to avoid repeated freeze–thaw cycles.
Mouse embryonic fibroblasts (MEFs), including wild-type and DHPS knockout cells, should be maintained under standard culture conditions and confirmed to be free of mycoplasma contamination. The use of DHPS-deficient cells provides an essential control to assess labeling specificity of spermidine mediated hypusination. For optimal labeling efficiency, cells should be in a healthy proliferative state prior to treatment. The polyamine oxidase inhibitor MDL-72527 is used to stabilize intracellular polyamine levels and should be prepared freshly or stored according to the manufacturer’s instructions.
Efficient click chemistry and enrichment depend on protein quality and buffer compatibility. Cell lysis should be performed in SDS-containing buffer supplemented with protease inhibitors (cOmplete EDTA-free protease inhibitor cocktail) and N-Ethylmaleimide (NEM) to block serine/cysteine proteases and prevent post-lysis protein degradation, thereby ensure efficient solubilization and stabilization of proteins. Copper-catalyzed azide–alkyne cycloaddition (CuAAC) reagents, including azide-biotin, CuSO4, TCEP, and TBTA ligand, should be in good quality and freshly prepared to maintain reaction efficiency. Streptavidin beads should be equilibrated prior to use, and extensive washing is critical to reduce nonspecific binding.
For downstream mass spectrometry analysis, the users should ensure access to a high-resolution LC–MS/MS system and appropriate data analysis pipelines. Protein samples should be of sufficient quantity and quality, and experimental design should include biological replicates and appropriate controls to support confident identification of probe-labeled proteins.
The hit selection protocol involves the identification of bona fide hits after enrichment and with parallel controls which can be used to eliminate any non-specific hits that are not competed away by free spermidine and/or not eliminated by DHPS knockout. Potential non-specific hits such as azide-alkyne-thiol adduct formation during the bioorthogonal ligation reaction is neither DHPS-dependent nor competable by spermidine, any such non-specific products were excluded from the final hit list by the target selection scheme.
Materials and reagents
Antibodies
- Streptavidin-HRP (Beyotime, catalog number: A0305)
Chemicals
- Compound 2 (N-Boc-2-propyn-1-amine) (Accela ChemBio, catalog number: SY025866)
- Sodium hydride (60% in mineral oil) (Macklin, catalog number: S817935-100g)
- 1,4-dibromobutane (Macklin, catalog number: D665801-5g)
- Potassium carbonate (Macklin, catalog number: P816302-500g)
- tert-butyl (3-aminopropyl) carbamate (Macklin, catalog number: N803413-5g)
- CF3CO2H (Macklin, catalog number: T818781-100ml)
- Urea (MERCK, catalog number: U5128)
- Tris(2-carboxyethyl)phosphine (TCEP) (MERCK, catalog number: C4706)
- Iodoacetamide (IAA) (MERCK, catalog number: I1149)
- CaCl2 (MERCK, catalog number: 223506)
- Tris-HCl (MERCK, catalog number: T5941)
- Trypsin (Promega, catalog number: V511C)
- C18 desalting spin columns (Thermo Fisher Scientific, catalog number: 87784)
- C18 resin (1.9 μm) (Dr. Maisch GmbH, catalog number: r119.aq.0003)
- Formic acid, LC-MS grade (Thermo Fisher Scientific, catalog number: 85178)
- Acetonitrile, LC-MS grade (Fisher Chemical, catalog number: A955)
- Water, LC-MS grade (Fisher Chemical, catalog number: W6)
- TBTA (Sigma, catalog number: 678937)
- CuSO4 (Sigma, catalog number: C1297)
- Biotin-azide (Cayman, catalog number: 13040)
- MDL72527 (Sigma, catalog number: M2949)
- Spermidine (Sigma, catalog number: S2501)
- cOmplete EDTA-free protease inhibitor cocktail (Roche, catalog number: C762Q77)
- N-Ethylmaleimide (Sigma, catalog number: 34115-M)
- SDS-PAGE loading buffer (reduced) (Bioswamp, catalog number: WB9207)
- SDS (Sigma, catalog number: 436143)
- Pierce ECL western blotting substrate (Thermo Fisher Scientific, catalog number: 32209)
- Non-fat dry milk (Bio-Rad, catalog number: 1706404XTU)
- 10× Tris/Glycine/SDS running buffer (Bio-Rad, catalog number: 1610772)
- 4–15% Criterion TGX stain-free protein gels (Bio-Rad, catalog number: 5678083)
- TBST (Thermo Fisher Scientific, catalog number: 28360)
- Transfer buffer (Thermo Fisher Scientific, catalog number: J63664.K2)
- BSA (CST, catalog number: 9998)
- DMEM (Thermo Fisher Scientific, catalog number: C11995500)
- FBS (Thermo Fisher Scientific, catalog number: 10099141)
- Penicillin-Streptomycin (Thermo Fisher Scientific, catalog number: 15140122)
- PBS (Thermo Fisher Scientific, catalog number: 10010023)
Experimental models: Cell lines
- MEFs (ATCC, catalog number: CRL-2991)
- DHPS knockout MEFs (home-made)
Solutions
1. 8M urea buffer (prepare fresh)
| 8 M urea buffer (prepare fresh) | Final concentration | Amount |
| Urea | 8 M | 0.48 g |
| 500 mM Tris-HCl pH 8.5 | 100 mM | 0.2 mL |
| ddH2O | n/a | 0.44 mL |
| Total | n/a | 1 mL |
2. Protein digestion reagent
Reagent | Concentration (stock) | Dilution factor |
| TCEP | 500 mM | 100× |
| IAA | 500 mM | 50× |
| CaCl2 | 100 mM | 100× |
| Tris-HCl pH 8.5 | 100 mM | 1× |
| Trypsin | 0.5 μg/μL | n/a |
Equipment
- Thermo Scientific Nunc EasYDish 150 mm dish (Thermo Fisher Scientific, catalog number: 150462)
- Criterion Vertical Electrophoresis Cell (Bio-Rad, catalog number: 1656001)
- Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad, catalog number: 1703940)
- Digital Dry Bath/Block Heater (Thermo Fisher Scientific, catalog number: 88870010)
- Nitrocellulose membrane (Bio-Rad, catalog number: 1620112)
- X-Ray Film (Thomas Scientific, catalog number: 1148B77)
- EASY-nLC 1200 system (Thermo Fisher Scientific, catalog number: LC140)
- Q-Exactive HFX mass spectrometer (Thermo Fisher Scientific, catalog number: 0726042)
Software
- MaxQuant (Max Planck Institute for Biochemistry, version: 2.0.3.0)
Perseus (Max Planck Institute for Biochemistry, version: 2.0.7.0)
Procedure
A. Preparation of the clickable alkynyl-spermidine probe
A three-step synthesis (Figure 1) is designed to obtain the compound N1-(3-ammoniopropyl)-N4-(prop-2-yn-1-yl) butane-1,4-diaminium 2,2,2-trifluoroacetate (Compound 1). The overall process converts the readily available precursor (Compound 2) through two intermediates (Compound 3 and 4) and final removal of Boc groups with trifluoracetic acid (TFA) to afford the desired clickable probe as its trifluoroacetate salt (Compound 1). The steps below describe the detailed procedures for each stage of the synthesis.
Timing: 7 days
1. First step of synthesis of tert-butyl (4-bromobutyl) (prop-2-yn-1-yl) carbamate (compound 3). Please refer to Figure 1 and Table 1.
Weigh compound 2 in a 100 mL round-bottom flask containing a magnetic stirring bar.
Add N,N-dimethylformamide (DMF, 50 mL) to the flask using a syringe and cool the flask in an ice bath for 10 minutes.
Weigh sodium hydride (0.96 g) and add it slowly to the solution. After addition, stir the mixture in an ice bath for 30 minutes.
Weigh 1,4-dibromobutane (5.17 g) and add it to the reaction mixture via syringe. After addition, heat the reaction to 50 °C and stir for 16 h at this temperature.
Treat the reaction mixture with saturated aqueous NH4Cl solution (10 mL) and extract with EtOAc (25 mL).
Wash the organic layer with saturated aqueous NH4Cl solution (10 mL × 3) followed by brine (10 mL × 2), then dry over anhydrous Na2SO4, filter, and concentrate under reduced pressure.
Purify the residue by flash silica gel chromatography (petroleum ether as eluent) to afford compound 3 (4.17 g, 72% yield).
Characterize the title product by NMR spectroscopy. 1HNMR (400 MHz, CDCl3): δ 4.05 (s, 2H), 3.45-3.42 (m, 2H), 3.38-3.34 (m, 2H), 2.20(s, 1H), 1.89-1.84 (m, 2H), 1.76-7.71 (m, 2H), 1.47 (s, 9H).
Table 1. Reagents for synthesis of compound 3 | |||
Reagent | Formula weight | Equivalent | Amount |
Compound 2 | 155.20 Da | 1.00 | 3.10 g |
Sodium hydride (60% in mineral oil) | 24.00 Da | 1.20 | 0.96 g |
1,4-dibromobutane | 215.91 Da | 1.20 | 5.17 g |
Pause point: The purified compound 3 can be stored in a sealed flask under nitrogen at -20 °C for several days.
2. Second step of synthesis of tert-butyl (4-(3-(tert-butoxy carbonyl) amino) propyl) amino) butyl) (prop-2-yn-1-yl) carbamate (compound 4). Please refer to Figure 1 and Table 2.
Weigh Compound 3 (1 g) in a 100 mL round-bottom flask containing a magnetic stirring bar.
Add acetonitrile (20 mL) to the flask using a syringe.
Weigh potassium carbonate (2.66 g) and add it to the solution.
Weigh tert-butyl (3-aminopropyl) carbamate and add it to the mixture. After the addition, heat the reaction to 50 °C and maintain at this temperature for 15 h.
Treat the reaction mixture with saturated aqueous NH4Cl solution (10 mL) and extract with EtOAc (25 mL). Wash the organic layer with saturated aqueous NH4Cl solution (10 mL × 3) followed by brine (10 mL × 2), then dry over anhydrous Na2SO4, filter, and concentrate under reduced pressure. Purify the resulting residue by flash silica gel chromatography using a gradient of 0–5% MeOH in CH2Cl2 to afford compound 4 (405 mg, 31% yield).
Characterize the title product by NMR spectroscopy. 1HNMR (400 MHz, CDCl3): δ 5.16 (s, 1H), 4.15-4.02 (m, 2H), 3.35-3.31 (m, 2H), 3.22-3.21(m, 2H), 2.71-2.66 (m, 4H), 2.20 (s, 1H), 1.71-1.70 (m, 2H), 1.62-1.61 (m, 2H), 1.53-1.52 (m, 2H), 1.47 (s, 9H), 1.44 (s, 9H).
Table 2. Reagents for synthesis of compound 4
Reagent
Formula weight
Equivalent
Amount
Compound 3
290.20 Da
1.00
1.00 g
Potassium carbonate
138.20 Da
3.54
2.66 g
tert-butyl (3-aminopropyl) carbamate
174.24 Da
1.06
0.57 g
Pause point: The purified compound 4 can be stored in a sealed flask under nitrogen at -20 °C for several days.
3. Third step of synthesis of N1-(3-ammoniopropyl)-N4-(prop-2-yn-1-yl) butane-1,4-diaminium 2,2,2-trifluoroacetate (compound 1). Please refer to Figure 1 and Table 3.
Weigh Compound 4 (405 mg) in a 100 mL round-bottom flask containing a magnetic stirring bar.
Add CH2Cl2 (10 mL) to the flask using a syringe.
Cool the flask in an ice bath.
Add CF3CO2H (TFA, 2.0 mL) using syringe.
Stir the reaction for 1 h, then concentrate to dryness. Triturate the residue with toluene (5.0 mL) for 30 min. Collect the solid by filtration and air-dry to afford Compound 1 as the CF3CO2H salt (470 mg, 84% yield).
Table 3. Reagents for synthesis of compound 1
Reagent
Formula weight
Equivalent
Amount
Compound 4
383.53 Da
1.00
405.00 mg
CF3CO2H
114.02 Da
excess
2.00 mL
Pause point: The purified Compound 1 as its TFA salt can be stored in a sealed flask under nitrogen at -20 °C for several months.
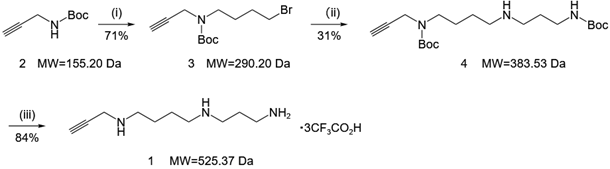
Figure 1. Three-step synthesis of the clickable alkynyl-spermidine probe (Compound 1).
(i) 1,4-dibromobutane, NaH, DMF, 50 oC, 16h; (ii) t-butyl (3-aminopropyl) carbamate, K2CO3, CH3CN, 50 oC, 15h; (iii) CF3CO2H, CH2Cl2, rt, 1h.
B. Preparation of the DHPS knockout MEFs
Timing: 7 days
1. Plasmid construction for knockout of Dhps in cells
Annealing: Phosphorylate and anneal sgDhps (or sgGfp) forward and reverse oligos.
Digestion: Digest lentiCRISPR v2 with BsmBI and gel-purify the backbone.
Ligation: Ligate the annealed oligos into the digested backbone.
Transformation: Transform the ligation product into Stbl3 competent cells, plate on LB agar containing ampicillin, and perform Sanger sequencing of individual colonies using a U6 forward primer to confirm correct sgRNA insertion. Use endotoxin-free plasmids extracted from sequence-verified clones provided by a commercial sequencing service.
Annealing reaction master mix
Reagent | Amount |
sgDhps forward oligo (5’-CACCGGCGCATGGTAATCTACGCCG-3’) (100 µM) | 1 μL |
sgDhps reverse oligo (5’-AAACCGGCGTAGATTACCATGCGCC-3’) (100 µM) | 1 μL |
10x T4 ligation buffer (contains ATP) | 1 μL |
T4 PNK (Polynucleotide Kinase) | 0.5 μL |
ddH2O | 6.5 μL |
Total volume | 10 μL |
Annealing conditions
Temperature | Time |
37 °C | 30 min |
95 °C | 5 min |
Ramp down to 25 °C at a rate of 0.1 °C per second (or -5°C every 1 minute) | |
Digestion master mix
Reagent | Amount |
lentiCRISPR v2 Plasmid | 5 μg |
BsmBI-v2 | 1 μL |
10x FastDigest/NEBuffer | 5 μL |
ddH2O | Up to 50 μL |
Digestion conditions
Temperature | Time |
55 °C | 4 hours |
Ligation master mix
Reagent | Amount |
Digested lentiCRISPR v2 | 50 ng |
Diluted Annealed Oligos (1:100) | 1 μL |
10x T4 ligation buffer | 1 μL |
T4 DNA ligase | 0.5 μL |
ddH2O | Up to 10 μL |
Ligation conditions
Temperature | Time |
16 °C | Overnight |
CRITICAL: Sanger sequencing using the U6 forward primer is the only way to ensure that the sgRNA was inserted correctly and without mutations in the scaffold.
Pause point: Endotoxin-free plasmids can be stored at 4°C for several months.
2. Lentivirus production (HEK293T)
a. Transfection: Co-transfect HEK293T cells (70-80% confluence) with lentiCRISPR-sgDhps, psPAX2, and pMD2G.
b. Media Change: Replace the medium with fresh DMEM supplemented with 10% FBS after 6-8 hours.
c. Harvest: Collect viral supernatants at 48 h and 72 h post-transfection and then filter through a 0.45 µm membrane.
Pause point: Filtered viral supernatants can be stored at -80 °C for several months or at 4 °C for several days, though a slight loss in virus titer may occur with freeze-thaw cycles.
3. MEFs transduction and selection
a. Infection: Infect low-passage MEFs with viral supernatant in the presence of Polybrene (8 µg/mL).
b. Recovery: Replace with fresh media 24 h post-infection.
c. Selection: Add Puromycin (3 µg/mL) at 48h post-infection.
d. Maintenance: Continue selection for 3–5 days until non-infected control cells are completely eliminated.
CRITICAL: Always include a non-infected control well when adding puromycin. Selection is only successful once 100% of the control cells have died.
4. Validation of knockout efficiency
a. Lyse the cells and perform western blot analysis.
b. Detect DHPS protein levels using a DHPS-specific antibody to confirm knockout efficiency.
CRITICAL: Western Blot validation of DHPS knockout is mandatory before proceeding to click chemistry reaction to ensure the biological model is valid.
Pause point: DHPS knockout MEFs can be stored in liquid nitrogen for several years.
C. Click chemistry reaction and streptavidin pull-down
Timing: 2 days
Pre-treat wild-type (WT) or DHPS knock-out (KO) mouse embryonic fibroblasts (MEFs) in 150 mm dish with 20 μM of MDL-72527 (a polyamine oxidase inhibitor) for 1 h.
Incubate the cells with either 400 μM alk-spermidine probe or spermidine for 4 h, or in the case of competition experiments, co-treat cells with 400 μM alk-spermidine and 2 mM spermidine for 4 h.
Harvest the cells and lyse by sonication in PBS buffer containing 1% SDS and a protease inhibitor cocktail supplemented with 10 mM N-Ethylmaleimide (NEM).
Clarify lysates by centrifugation at 14,000 × g for 10 min, and determine protein concentration in the supernatant using a BCA assay.
Aliquot 1 mg of protein (~1 mg/mL) and perform CuAAC click chemistry by adding azide-biotin, TBTA, CuSO₄, and TCEP to final concentrations of 200 μM, 400 μM, 1 mM, and 1 mM, respectively.
Add an additional aliquot of TCEP after 30 min to maintain reaction activity and continue incubation for a total of 1 h at room temperature.
Precipitate the proteins by adding ice-cold methanol to the reaction mixture.
Re-suspend protein pellets in 500 μL of 1% SDS by sonication.
Dilute SDS to 0.2% with PBS, then incubate proteins with streptavidin agarose beads for 2 h at room temperature.
Wash the beads sequentially with 1 mL of 1% SDS (six times), 1 mL of 0.1% SDS (six times), and 1 mL of PBS (six times) for a total of 18 washing steps,carefully removing the supernatant after each wash.
Elute the enriched proteins in 1× loading buffer at 95 °C for 10 min to prepare for western blot analysis.
CRITICAL: The order of addition and freshness of TCEP and CuSO₄ are vital. The solution should be mixed well to ensure the formation of the active Cu(I) catalyst.
CRITICAL: Pretreatment with MDL-72527 (20 μM, 1h) is required to maintain probe stability. Polyamine oxidase can promote the degradation of spermidine click probe to reduce the efficiency of probe labeling. In addition, polyamine oxidation generates reactive aldehydes and ROS, which may further compromise labeling specificity and introduce artifactual modifications6-8. Therefore, inhibition of polyamine oxidation (e.g., with MDL72527) is essential to preserve probe integrity and enable accurate identification of click probe-labeled targets, especially for low abundant protein targets.
Pause point: Denatured samples after boiling for western blotting can be stored at 4 °C for up to two days.
D. Western blot analysis of probe-labeled proteins enriched by streptavidin agarose after click chemistry
Timing: ~8 h
Mix protein samples with SDS loading buffer and denature at 95 °C for 10 minutes.
Load samples into the SDS-page gel and run at 80 V for the stacking phase, then increase to 120 V for the resolving phase until the tracking dye reaches the bottom.
Assemble the transfer sandwich and transfer the proteins from the gel to the Nitrocellulose membrane in transfer buffer at a constant current of 250 mA for 90 minutes under chilled conditions.
Block the membrane in 5% non-fat milk in TBST for 1 hour at room temperature. Wash the membrane three times for 5 minutes each with TBST.
Dilute Streptavidin-HRP at 1:2000 in 5% BSA/TBST and incubate for 2 hours at room temperature with gentle agitation.
Wash the membrane three times for 10 minutes each with TBST to remove unbound HRP.
Apply ECL chemiluminescence substrates to the membrane for 1 minute, then capture the signal using X-ray films.
E. On-beads digestion of biotin pull-down samples, and desalting
Timing: ~16 h
This section describes the preparation of peptide samples of enriched probe substrates using the on-beads digestion method for Liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
Following step 10 in the last section, resuspend the streptavidin agarose beads in 50 μL of 8 M urea solution, then add 0.5 μL of 0.5 M TCEP to a final concentration of 5 mM. Incubate at room temperature for 20 min.
Add 1 μL of 0.5 M IAA to a final concentration of 10 mM, and incubate at room temperature in the dark for 15 min.
Add 150 μL of 100 mM Tris-HCl (pH 8.5) buffer to dilute the urea concentration to 2 M.
Add 2 μL of 100 mM CaCl2 solution to a final concentration of 1 mM.
Add 2 μL of trypsin (0.5 μg/μL), then incubate at 37 °C with shaking at 800 rpm for 12 h.
Add 10 μL of formic acid (FA) to terminate the digestion.
Centrifuge the enzymatically digested sample at 20,000 × g and 4 °C for 15 min, and transfer the resulting supernatant to a new EP tube for desalting.
Insert the C18 desalting spin columns into the cap of a 1.5 mL EP tube, and add 200 μL of acetonitrile. Centrifuge the EP tube in a microcentrifuge to activate the C18 packing material, and the flow-through was discarded.
Load the C18 desalting columns with 200 μL of 0.1% formic acid (FA) solution and centrifuge it in a microcentrifuge to equilibrate the C18 desalting columns. Discard the flow-through, and repeat this step three times.
Load the supernatant sample obtained in step 25 onto the C18 desalting columns and centrifuge it in a microcentrifuge. Reload the flow-through onto the C18 desalting columns, and repeat this process three times. Discard the final flow-through.
Load the C18 desalting columns with 200 μL of 0.1% FA solution and centrifuge it in a microcentrifuge, and discard the flow-through. Repeat this step twice.
Transfer the C18 desalting columns to a new 1.5 mL EP tube, and add 100 μL of peptide elution buffer (containing 70% acetonitrile and 0.1% FA). Centrifuge the EP tube in a microcentrifuge, and collect the eluate into a new 1.5 mL EP tube. Repeat this step three times and pool all eluates.
Dry the pooled peptide eluate under vacuum at 4 °C.
Pause point: The dried sample can be stored at −20 °C until further analysis by LC-MS/MS.
F. LC-MS/MS data acquisition
Timing: ~1.5 day
Analyze the peptides processed in previous steps by LC-MS/MS.
- Dissolve the dried peptide sample in 15 µL of 0.1% FA solution, then centrifuge it at 20,000 × g and 4 °C for 15 min. Subsequently, transfer 10 µL of the supernatant to an autosampler vial for LC-MS/MS analysis.
First load peptide samples by injecting 2 µL onto a trapping column, then separate them on a C18 analytical column (150 mm × 100 μm i.d., packed in-house with 1.9 μm C18 resin from Dr. Maisch GmbH) using an EASY-nanoLC 1200 system equipped with an autosampler.
Use mobile phases consisting of (A) 0.1% FA; (B) 0.1% FA, 80% ACN.
Separate peptides using a 90-min gradient at a constant flow rate of 300 nL/min: 3% B (0 min), 8% B (4 min), 28% B (67 min), 40% B (79 min), 50% B (84 min), 100% B (87 min), hold at 100% B (90 min).
Analyze the eluting peptides online using a Q-Exactive HFX mass spectrometer via electrospray ionization under the following parameters:
Acquire the data in data-dependent acquisition mode (top-40).
For MS1, set the scan range to 300 - 1650 m/z with a resolution of 60,000. Set the automatic gain control (AGC) target to 3e6, with a maximum injection time of 20 ms.
For MS2, set the resolution to 15,000. Set the AGC target to 1e5, with a maximum injection time of 25 ms.
Set the isolation window to 1.4 m/z. Fragment precursor ions using higher-energy collisional dissociation (HCD) with a normalized collision energy of 27%.
Finally, process all acquired raw files using a uniform data analysis workflow as outlined in the following steps.
G. Data analysis and processing
Timing: ~1 day
Perform protein identification and quantification using MaxQuant (version 2.0.3.0)9 with the following parameters:
Search the tandem mass spectra against the UniProt mouse protein database (containing 17,073 entries; downloaded in May 2021).
Set Trypsin/P as the enzyme, and set the maximum number of missed cleavages to 2.
Set cysteine carbamidomethylation (+57.021 Da) as a static modification, while set methionine oxidation (+15.995 Da) and protein N-terminal acetylation (+42.010 Da) as variable modifications.
Set both the precursor and fragment mass tolerances to 20 ppm.
Control the false discovery rates at both the peptide-spectrum match and protein levels at <1%.
Use unique and razor peptides for quantification, and use the summed peptide intensities for protein quantification.
Perform data processing using Perseus (version 2.0.7.0)10 with the following parameters:
First, perform quality control by filtering the identified proteins to retain only those consistently detected in at least three independent replicates.
Then normalize the dataset using the abundance of streptavidin, which is immobilized on the agarose beads used for probe pull-down and can be digested during sample preparation for mass spectrometry.
Subsequently, impute missing values with random values drawn from a downward-shifted distribution to facilitate statistical analysis. Apply a down shift of 1.8 and a distribution width of 0.5 to simulate the abundance levels of low-abundance proteins that typically result in missing values.
Export the imputed data to an Excel spreadsheet for statistical analysis. Calculate P values (two-tailed Student’s t-test) and fold changes for comparisons between groups (e.g., probe vs. spermidine; probe vs. probe + spermidine; WT probe vs. DHPS-KO probe). Consider proteins with a fold change > 2 and p < 0.05 as significantly differentially expressed protein targets.
Define candidate hypusinated proteins as those specifically labeled by alkyne–spermidine probe in wild-type (WT) cells and dependent on DHPS activity. Probe-specific labeling in WT cells is defined as proteins significantly enriched in the presence of the probe compared with spermidine control (probe vs spermidine; fold change > 2; P < 0.05), with signal significantly reduced upon competition with excess native spermidine (probe vs probe + excess spermidine; fold change > 2; P < 0.05). In parallel, DHPS dependence is defined as proteins significantly enriched in WT cells compared with DHPS knockout (Dhps KO) cells under the probe labeling condition (WT probe vs Dhps KO probe; fold change > 2; P < 0.05). Only proteins that met all of the above criteria were defined as candidate hypusinated proteins.
Validation of the protocol
The execution of this protocol is expected to yield a high-purity alkynyl-spermidine probe, confirmed by NMR spectroscopy showing characteristic peaks for the propargyl group and the removal of Boc protecting groups. In Western blotting analysis, researchers should observe a robust pattern of protein labeling in wild-type (WT) MEFs treated with the probe, visualized via Streptavidin-HRP (Figure 2). This labeling should be significantly outcompeted by natural spermidine and markedly reduced in DHPS-knockout (KO) cells, confirming the biological specificity of the metabolic labeling.
For the proteomic workflow, the on-beads digestion and subsequent LC-MS/MS analysis are expected to identify hundreds of potential hypusination substrates. Successful data processing using MaxQuant and Perseus should reveal significantly enriched proteins, such as eIF5A and RIPK1, with a fold change >2 and p < 0.05 when comparing probe-treated groups to controls. Additionally, endogenous biotinylated proteins like PYC and ACACA11,12 should serve as reliable internal recovery markers and showed no differential enrichment between conditions.
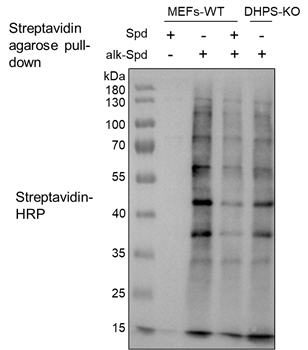
Figure 2. Validation of alkynyl-spermidine-probe-labeled proteins by western blotting analysis.
The specificity of the clickable alkynyl-spermidine probe was assessed in wild-type (WT) and DHPS knockout (DHPS-KO) mouse embryonic fibroblasts (MEFs). Cells were pretreated with 20 μM MDL-72527 for 1 h to stabilize intracellular polyamine levels, followed by treatment with 400 μM probe for 4 h, with or without 2 mM competing natural spermidine. Following cell lysis in 1% SDS containing proteinase inhibitor cocktail including NEM, the lysates were treated with CuAAC-mediated click chemistry followed by biotin-azide. Biotinylated proteins were enriched using streptavidin agarose beads, subjected to SDS-PAGE, and visualized via streptavidin-HRP blotting.
Limitations
This protocol does not directly identify hypusination sites on target proteins. Instead, it enables the identification of candidate hypusination proteins based on competitive labeling and comparison with DHPS knockout controls. As such, the approach cannot unambiguously distinguish bona fide hypusinated substrates from proteins indirectly associated with spermidine probe incorporation. Definitive site-specific validation requires additional experiments, including enrichment of individual proteins of interest followed by mass spectrometry–based site identification, in vitro hypusination assays to confirm modification sites, and the generation of site-specific antibodies for orthogonal validation. In addition, the metabolic labeling strategy may introduce biases related to probe uptake, intracellular availability, and competition with endogenous spermidine. Furthermore, transient or low-stoichiometry hypusination events may escape detection. Finally, because this workflow relies on cell-based labeling, its applicability to certain cell types or in vivo systems may be limited and require further optimization.
Troubleshooting
Problem 1:
Low yield or purity of the alkynyl-spermidine probe (Related to steps 1–3 in preparation of the clickable alkynyl-spermidine probe).
Potential solution:
Ensure all anhydrous solvents are strictly dry and reagents are added slowly in an ice bath to prevent side reactions. Verify intermediate purity by NMR before proceeding to the global deprotection step.
Problem 2:
Inconsistent knockout efficiency in MEFs (Related to step 3 in preparation of the DHPS knockout MEFs).
Potential solution:
Use low-passage MEFs for transduction and confirm DHPS depletion via western blot prior to starting the metabolic labeling experiments.
Problem 3:
Weak or absent labeling signal in western blot (Related to Steps 1-6 in click chemistry reaction and streptavidin pull-down).
Potential solution:
Check the activity of the CuAAC reagents; prepare CuSO4 and TCEP freshly for every experiment. Ensure the protein concentration is at least 1 mg/mL to facilitate efficient reaction kinetics.
Problem 4:
High non-specific background in pull-down samples (Related to Steps 7–11 in click chemistry reaction and streptavidin pull-down).
Potential solution:
Strictly adhere to the washing protocol, using 1% SDS to remove non-covalently bound proteins from the streptavidin beads.
Problem 5:
Poor peptide recovery after on-beads digestion (Related to Steps 19–31 in on-beads digestion of biotin pull-down samples, and desalting).
Potential solution:
Confirm the urea concentration is diluted to 2 M or lower before adding trypsin, as high urea concentrations inhibit enzymatic activity.
Problem 6:
Column clogging or high pressure during LC separation (Related to Steps 32–35 in LC-MS/MS data acquisition).
Potential solution:
Perform high-speed centrifugation (≥13,000 × g, 4 °C, 15 min) before injection. Carefully transfer only the clear supernatant. Optionally, filter samples using a 0.22 μm filter prior to LC loading.
Problem 7:
Poor chromatographic separation or unstable retention time (Related to Steps 32–35 in LC-MS/MS data acquisition).
Potential solution:
Ensure proper preparation and degassing of mobile phases. Include blank or wash runs between different sample groups to minimize carryover. Replace the analytical column if performance declines.
Problem 8:
Low identification rate or high missing values in MaxQuant analysis (Related to Steps 36 in Data analysis and processing).
Potential solution:
Verify mass accuracy and calibration of the instrument. Ensure correct database selection and parameter settings (enzyme specificity, modifications, FDR). Increase LC gradient length or sample loading to improve peptide coverage. Consider increasing the number of biological or technical replicates.
Problem 9:
Failure to identify candidate hypusinated proteins (Related to Steps 36–37 in Data analysis and processing).
Potential solution:
Confirm effective competition conditions (e.g., sufficient spermidine concentration). Validate DHPS knockout efficiency by Western blot prior to proteomics. Increase replicate number to improve statistical robustness. Re-evaluate fold change and significance thresholds if overly stringent.
Notes
All reagents were purchased from commercial suppliers and used without further purification.
- The 500 mM TCEP and 500 mM IAA stock solution should be aliquoted, stored at −20 °C, and freshly diluted prior to use.
- NEM is used to block free cysteines, thereby minimizing non-specific probe labeling. NEM blocks free cysteines by undergoing a Michael addition at near-neutral pH, forming an irreversible covalent thioether bond with the sulfhydryl group (-SH) to permanently mask it from subsequent labeling or oxidation.
- For mass spectrometry analysis, proceed only up to step 10 in section C and continue with the next section without performing the elution step.
- Before the section E, prepare a fresh 8 M urea (in 100 mM Tris-HCl, pH 8.5) solution.
- For each step in section E involving solvent replacement or sample loading, the remaining liquid from the previous step should be centrifuged until the liquid level aligns with the top of the C18 packing material.
- For each treatment group, analyze the four biological replicates consecutively by LC-MS/MS, with a wash run (involving sequential high-organic and aqueous phases) inserted prior to the next treatment group to eliminate potential column carryover.
- The in-house packed C18 analytical column (150 mm × 100 μm i.d., 1.9 μm resin) can be substituted with a commercially available column of comparable dimensions and particle size.
In section G, protein quantitative abundances were first transformed to log2 scale prior to imputation using the normal distribution parameters described in step 2-c.
Acknowledgments
This project was supported (to Junying Yuan) by the National Natural Science Foundation of China (82188101), Brain Science and Brain-like Intelligence Technology-National Science and Technology Major Project (2025ZD0214800), the Shanghai Basic Research Pioneer Project, the Shanghai Municipal Science and Technology Major Project and the Shanghai Key Laboratory of Aging Studies (19DZ2260400), and from the National Natural Science Foundation of China (32370796 to Bing Shan).
Author contributions
J.Y., B.S. and T.Z. conceived of this project. T.Z. and J.L. performed the majority of the experiments. Y.Z. and B.S. directed J.L. to perform the mass spectrometry experiments. Z.W. directed X.H. in the probe synthesis. J.Y., B.S., T.Z., J.L. and X.H. wrote the paper.
Declaration of interests
The authors declare no competing interests.
References
1. Zhang, T., Fu, W., Zhang, H., Li, J., Xing, B., Cai, Y., Zhang, M., Liu, X., Qi, C., Qian, L., et al. (2024). Spermidine mediates acetylhypusination of RIPK1 to suppress diabetes onset and progression. Nat Cell Biol. 10.1038/s41556-024-01540-6.
2. Madeo, F., Eisenberg, T., Pietrocola, F., and Kroemer, G. (2018). Spermidine in health and disease. Science 359. 10.1126/science.aan2788.
3. Park, M.H., Cooper, H.L., and Folk, J.E. (1981). Identification of hypusine, an unusual amino acid, in a protein from human lymphocytes and of spermidine as its biosynthetic precursor. Proc Natl Acad Sci U S A 78, 2869-2873. 10.1073/pnas.78.5.2869.
4. Park, M.H., Cooper, H.L., and Folk, J.E. (1982). The biosynthesis of protein-bound hypusine (N epsilon -(4-amino-2-hydroxybutyl)lysine). Lysine as the amino acid precursor and the intermediate role of deoxyhypusine (N epsilon -(4-aminobutyl)lysine). J Biol Chem 257, 7217-7222.
5. Cooper, H.L., Park, M.H., Folk, J.E., Safer, B., and Braverman, R. (1983). Identification of the hypusine-containing protein hy+ as translation initiation factor eIF-4D. Proc Natl Acad Sci U S A 80, 1854-1857. 10.1073/pnas.80.7.1854.
6. Pegg, A.E. (2013). Toxicity of polyamines and their metabolic products. Chem Res Toxicol 26, 1782-1800. 10.1021/tx400316s.
7. Holbert, C.E., Dunworth, M., Foley, J.R., Dunston, T.T., Stewart, T.M., and Casero, R.A., Jr. (2020). Autophagy induction by exogenous polyamines is an artifact of bovine serum amine oxidase activity in culture serum. J Biol Chem 295, 9061-9068. 10.1074/jbc.RA120.013867.
8. Pledgie, A., Huang, Y., Hacker, A., Zhang, Z., Woster, P.M., Davidson, N.E., and Casero, R.A., Jr. (2005). Spermine oxidase SMO(PAOh1), Not N1-acetylpolyamine oxidase PAO, is the primary source of cytotoxic H2O2 in polyamine analogue-treated human breast cancer cell lines. J Biol Chem 280, 39843-39851. 10.1074/jbc.M508177200.
9. Tyanova, S., Temu, T., and Cox, J. (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature protocols 11, 2301-2319.
10. Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M.Y., Geiger, T., Mann, M., and Cox, J. (2016). The Perseus computational platform for comprehensive analysis of (prote) omics data. Nature methods 13, 731-740.
11. Kirkeby, S., Moe, D., Bog-Hansen, T.C., and van Noorden, C.J. (1993). Biotin carboxylases in mitochondria and the cytosol from skeletal and cardiac muscle as detected by avidin binding. Histochemistry 100, 415-421. 10.1007/BF00267821.
12. Chandler, C.S., and Ballard, F.J. (1986). Multiple biotin-containing proteins in 3T3-L1 cells. Biochem J 237, 123-130. 10.1042/bj2370123.
- Zhang, T, Li, J, Hu, X, Wang, Z, Zhang, Y, Yuan, J and Shan, B(2026). Proteomic profiling of hypusinated protein candidates using a clickable spermidine probe. Bio-protocol Preprint. bio-protocol.org/prep2940.
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.