
- Home
- Protocols

-

A simple and fast unidirectional strategy for chromosomal gene deletion and complementation in gram-negative bacteria
Last updated date: Jan 13, 2025 DOI: 10.21769/p2780 Views: 1204 Forks: 0
A simple and fast unidirectional strategy for chromosomal gene deletion and complementation in gram-negative bacteria
Rekha Rana1,2, Anushika Sharma1,2, Ashish Dutta1,2, Prabhu B. Patil1,2*
1Bacterial Genetics, Genomics and Evolution Laboratory, CSIR-Institute of Microbial Technology, Chandigarh, India
2The Academy of Scientific and Innovative Research, Ghaziabad, India
*Author for Correspondence: Prabhu B. Patil, Ph.D., Bacterial Genetics, Genomics and Evolution Laboratory, CSIR-Institute of Microbial Technology, Chandigarh, India. E-mail: pbpatil@imtech.res.in
Abstract
The protocols for gene deletion using the pk18mobsacB vector involve sucrose sensitivity encoding sacB as a negative selection marker and the use of multiple restriction enzyme(s) to generate sticky ends in the vector and insert for cloning. This approach is time-consuming, resource intensive and complicated. The advent of next-generation proofreading enzymes is enabling researchers to carry out long-range PCR rapidly. Hence, amplifying 5-6 kb of complete low-complex DNA cloning vectors and 2-3 kb of complex genomic regions is much easier. Here, we report a simple, accurate, rapid and unidirectional approach for chromosomal in-frame gene deletion and complementation that does not involve restriction enzymes. The method required long-range PCR using Phusion polymerase to linearize the vector (pK18mobsacB) and amplify the target gene to create a recombinant vector (pRM1) and further inverse PCR of pRM1 to create a recombinant vector (pRM4) that has a deleted version of the gene. The cloning steps involved the use of kinase for phosphorylation and ligase for ligation. The recombinant plasmid, pRM4, was finally transformed into electrocompetent cells of Xanthomonas sontii, a gram-negative phytobacterium, prepared using sucrose for final genomic integration/excision to obtain an in-frame deletion mutant (PPL1RM15). Chromosomal complementation was carried out by electroporating PPL1RM15 cells with the recombinant plasmid, pRM1, to generate PPL1RM15C. Clean gene mutation, complementation, and plasmid excision are confirmed using whole genome sequencing.
Key features
A vector digested by restriction enzymes has sticky ends that are prone to self-ligation. In this method, only the amplified gene product needs to be phosphorylated for cloning. Hence, the vector self-ligation is largely eliminated.
With the availability of high-fidelity polymerases that are much more efficient, the length of deletion will now only depend on our ability to amplify larger regions and further clone them in a suitable vector with negative selection marker.
The strategy, being PCR-based, is cost-effective as it does not require the use of multiple restriction enzymes and suitable to carry out marker-free deletion mutation in single or multiple genes in the same strain.
There is no need for a separate cloning strategy for chromosomal complementation. We can use the recombinant plasmid, which has the target gene developed in the initial steps and will meet the purpose.
Point mutations can be introduced in the desired location through site-directed mutagenesis of the cloned target gene, and mutated plasmid can be electroporated in deletion mutant for chromosomal point mutation.
Keywords
Xanthomonas sontii, pK18mobsacB, self-ligation, long-range PCR, marker-free gene deletion mutation, kinase, Inverse PCR, complementation
This protocol is used in: Applied and Environmental Microbiology (2024), DOI: 10.1128/aem.00848-24
Graphical overview
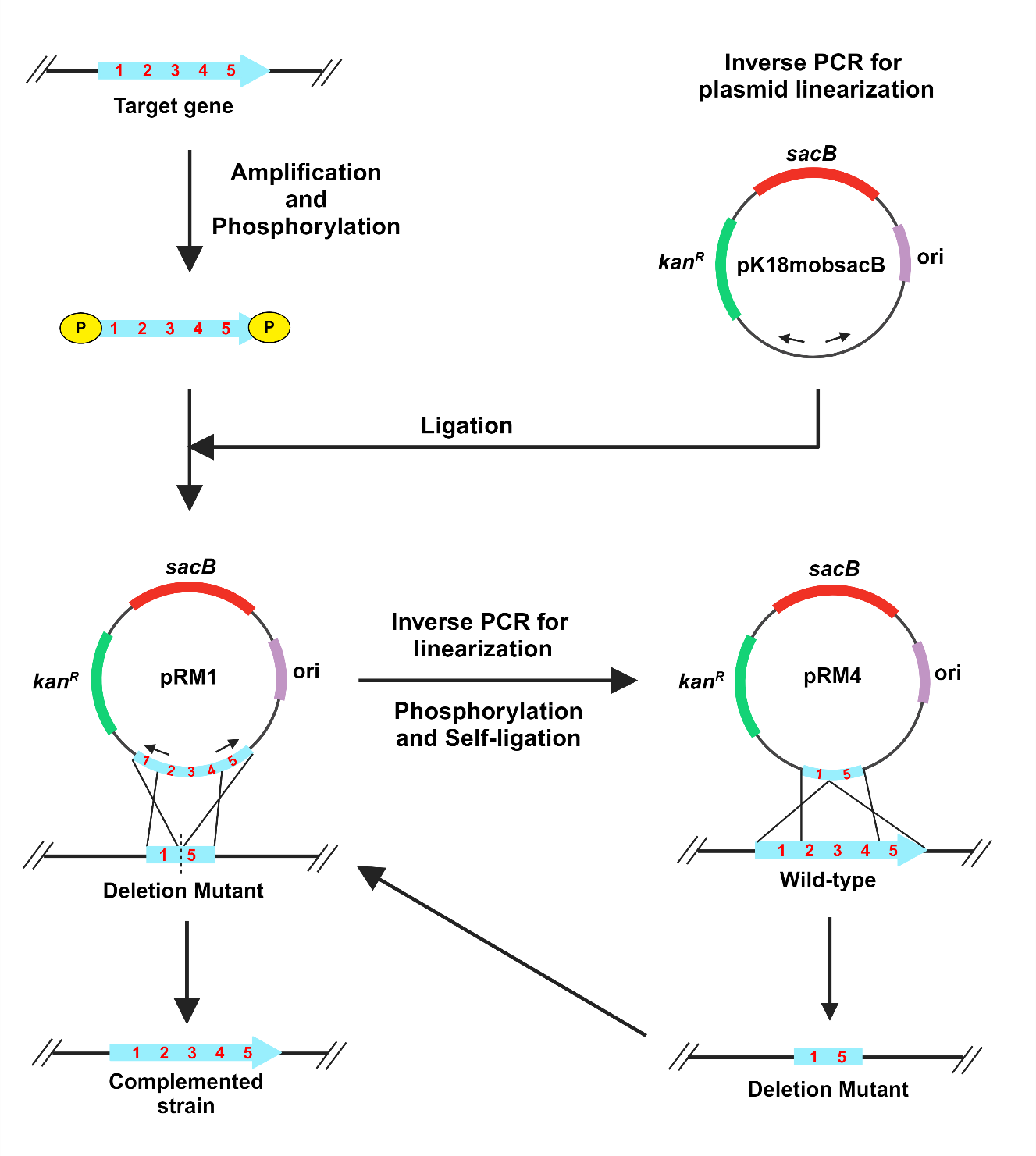
Background
The plasmid pK18mobsacB is a cloning vector that facilitates gene deletion through homologous recombination (Schäfer et al., 1994). The vector has a kanR marker, which confers kanamycin resistance and a sacB gene, which confers sucrose sensitivity through the levan synthesis in the presence of sucrose (Gay, Le Coq, Steinmetz, Berkelman, & Kado, 1985). The suicide vector pK18mobsacB is frequently used to generate gene deletions in gram-negative and gram-positive bacteria (Schulze et al., 2012; Ton‐That & Schneewind, 2003). However, restriction enzyme-mediated cloning is complicated and requires multiple time-taking steps. The first part is introducing suitable restriction sites in the target gene PCR product and vector for cloning. Second, there is a requirement to create distinct recombinant vectors for deletion and complementation (Pradhan, Ranjan, & Chatterjee, 2012). Inverse polymerase chain reaction (PCR) involves the use of oppositely directed primers to linearize the plasmid, which is ligated with PCR-amplified target gene product after phosphorylation (Imai, Matsushima, Sugimura, & Terada, 1991). With the advent of a new generation of ultra-efficient proofreading polymerases, it is becoming easier to carry out long-range PCR suitable for such purposes. Here, we describe a gene deletion and complementation method in Xanthomonas sontii, a gram-negative bacterium and rice endophyte, using a pK18mobsacB based inverse PCR strategy. The standard M13 region of pK18mobsacB is used to design inverse primers. The amplified target gene is phosphorylated with kinase and ligated with the linear plasmid. Further, to generate the in-frame deletion mutant, primers facing tail-to-tail situated at the 300-400 bp inside from the ends of the target gene are used to amplify the recombinant plasmid, excluding the gene region to be deleted. This is followed by kinase treatment and ligation. The recombinant plasmid with the deleted gene is electroporated into the Xanthomonas cells, leading to the deletion of the target gene in the chromosome through homologous recombination. In a similar way, the recombinant plasmid with the target gene is electroporated into the gene deletion mutant of Xanthomonas sontii, leading to the restoration of the full-length target gene in the chromosome through homologous recombination. The unidirectional method is fast, accurate, and can be easily used to delete any gene and also multiple genes in the chromosome and carry out chromosomal complementation independent of the reliability of restriction sites or enzymes in other gram-negative and gram-positive bacteria. Conjugation through biparental mating will further eliminate the need for electroporation in resource-poor settings.
Materials and Reagents
Biological material
Xanthomonas sontii PPL1T (Bansal et al., 2021)
Escherichia coli Top10 cells (Invitrogen)
pK18mobsacB plasmid (Schäfer et al., 1994)
Reagents
Quick-DNATM Fungal/Bacterial Miniprep Kit (Zymo Research, catalog number: D6005)
GeneJET Plasmid Miniprep Kit (Thermo Scientific, catalog number: K0502)
5X FIREPol® Master Mix (Solis BioDyne, catalog number: 04-12-00115)
Nuclease-free water (MP Biomedicals, catalog number: 112450204)
Agarose (Sigma-Aldrich, catalog number: A9539)
Ethidium bromide solution (Sigma-Aldrich, catalog number: E1510)
6X DNA loading buffer (Real-Gene Labs, catalog number: 530001)
Quick-load® 100 bp DNA ladder (New England Biolabs Inc., catalog number: N0467S)
Quick-load® 1 kb DNA ladder (New England Biolabs Inc., catalog number: N0468S)
GeneJET Gel Extraction Kit (Thermo Scientific, catalog number: K0691)
GeneJET PCR Purification Kit (Thermo Scientific, catalog number: K0701)
Phusion DNA polymerase (Thermo Scientific, catalog number: F-530S)
5X Phusion HF buffer (Thermo Scientific, catalog number: F-518)
dNTP Mix (Fermentas, catalog number: R0192)
T4 Polynucleotide kinase (New England Biolabs Inc., catalog number: M0201S)
10X T4 DNA Ligase Buffer with 10mM ATP (New England Biolabs Inc., catalog number: B0202A)
T4 DNA Ligase (New England Biolabs Inc., catalog number: M0202S)
Kanamycin Sulfate (Sigma-Aldrich, catalog number: 60615)
0.22 µm PVDF filter (WhatmanTM, catalog number: 9913-2502)
5 mL syringe (Dispo Van)
Phosphate Buffered Saline (Gibco, 10010-023)
0.025 µmole desalted primers (Sigma-Aldrich)
DifcoTM Nutrient agar (BD, catalog number: 213000)
DifcoTM Nutrient broth (BD, catalog number: 234000)
DifcoTM LB Broth, Miller (BD, catalog number: 244620)
DifcoTM LB Agar, Miller (BD, catalog number: 244520)
Sucrose (Sigma-Aldrich, catalog number: S0389)
Calcium Chloride Dihydrate (Sigma-Aldrich, catalog number: C5080-500G)
Glycerol (Qualigens, catalog number: Q15457)
Solutions
Nutrient Agar with 5% Sucrose
50X TAE (Tris-acetate-EDTA) buffer
Kanamycin Sulfate stock 50 mg/mL
300 mM Sucrose
100 mM CaCl2 + 15% glycerol
100 mM CaCl2
Recipes
Nutrient Agar with 5% Sucrose
Reagent | Quantity or volume |
Nutrient agar | 23 g |
Sucrose | 50 g |
Total volume with distilled water |
|
2. 50X TAE buffer
Reagent | Quantity or volume |
Tris Base | 242 g |
Glacial acetic acid | 57.1 mL |
0.5 mM EDTA | 18.612 g |
Total volume with distilled water | 1000 mL |
Note: 50X TAE is diluted with distilled water (1:50) to make 1X TAE for the preparation of agarose gel.
3. Kanamycin Sulfate stock 50 mg/mL
Reagent | Quantity or volume |
Kanamycin Sulfate | 250 mg |
Total volume with distilled water | 5 mL |
Note: Filter sterilize the solution with a 0.22 µm syringe filter.
4. 300 mM Sucrose
Reagent | Quantity or volume |
Sucrose | 5.134 g |
Total volume with distilled water | 50 mL |
5. 100 mM CaCl2 + 15% glycerol
Reagent | Quantity or volume |
CaCl2.2H20 | 0.735 g |
Glycerol | 7.5 mL |
Total volume with distilled water | 50 mL |
6. 100 mM CaCl2
Reagent | Quantity or volume |
CaCl2.2H20 | 1.47 g |
Total volume with distilled water | 100 mL |
Equipment
Micropipettes (Eppendorf)
Nanodrop (DeNovix)
UV Transilluminator (Wealtac)
Gel electrophoresis assembly
Mastercycler gradient (Eppendorf)
Centrifuge 5810 R (Eppendorf)
Microcentrifuge (Sigma 1-15P)
Thermomixer comfort (Eppendorf)
Electroporator (BioRad)
Incubators at 28 ºC and 37 ºC (Innova®)
Software
AmplifX v2.0.7 (Jullien)
Primer3 v0.4.0 (https://primer3.ut.ee/)
MEGA-X v10.2.6 (Kumar, Stecher, Li, Knyaz, & Tamura, 2018)
NEBiocalculator (https://nebiocalculator.neb.com/)
Procedure
A. Primer design
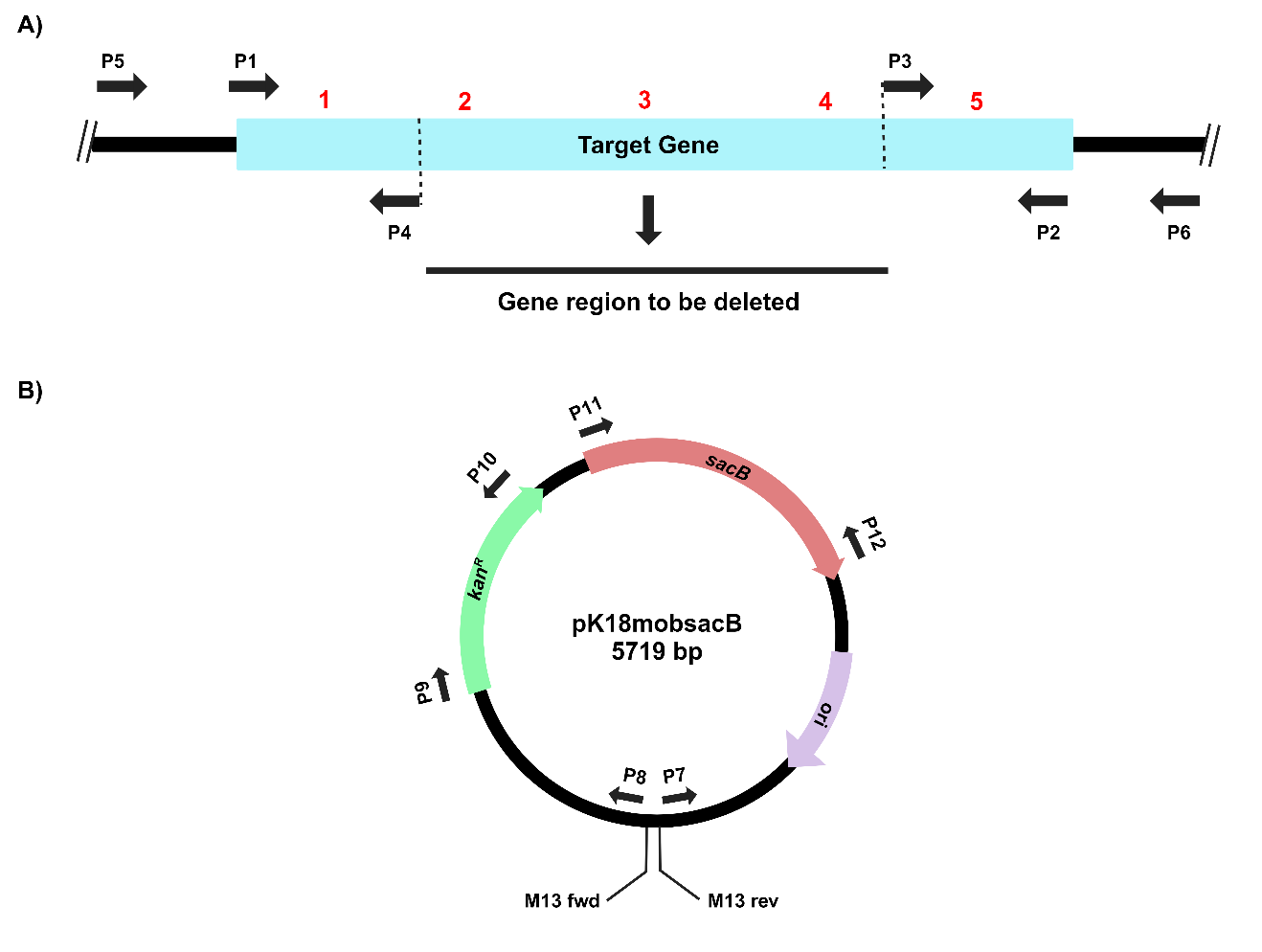
Figure 1. Primer design for PCR amplifications of A) the target gene and B) the suicide vector, pK18mobsacB, for gene cloning, deletion and complementation.
Design primers for the PCR amplification of the target gene (P1 and P2). Design tail-to-tail facing primers positioned 300-400 bp from the 3’ and 5’ ends (P3 and P4). Inverse PCR amplification using these primers will result in the deletion of a significant portion of the gene (Figure 1, Table 1). (Note: Primers can be designed in a similar way, including upstream and downstream regions of the gene for complete gene deletion)
Design primers facing tail-to-tail using the M13 forward and M13 reverse regions for inverse PCR amplification and linearization of pK18mobsacB (P7 and P8).
Design primers for PCR amplification of kanR (P9 and P10) and sacB (P11 and P12) markers of pK18mobsacB to screen integrants. Additionally, design primers for PCR amplification of the target gene, including extra 100-200 bp both upstream and downstream to screen deletion mutants (P5 and P6).
Detailed information about the set of primers used in this protocol was mentioned in an earlier publication (Rana et al., 2024).
Table 1. Oligonucleotides used in the study.
S. No. | Primer pair | Primer name | Purpose |
1 | P1 | VirD4_3_F | Target gene (virD4) amplification |
2 | P2 | VirD4_5_R | |
3 | P3 | VirD4_inv_F_2 | Linearization of pRM1 |
4 | P4 | VirD4_inv_R | |
5 | P5 | VirD4_idnt_F | Detection of gene deletion mutation in X. sontii PPL1 |
6 | P6 | VirD4_idnt_R | |
7 | P7 | Set1_M13_FP | Linearization of pK18mobsacB |
8 | P8 | Set1_M13_RP | |
9 | P9 | KanR_F | Kanamycin resistance gene amplification |
10 | P10 | KanR_R | |
11 | P11 | sacB_F | sacB gene amplification |
12 | P12 | sacB_R |
B. Target gene cloning in pK18mobsacB
The process for inverse PCR-based gene cloning using the suicide vector pK18mobsacB is depicted in Figure 2.
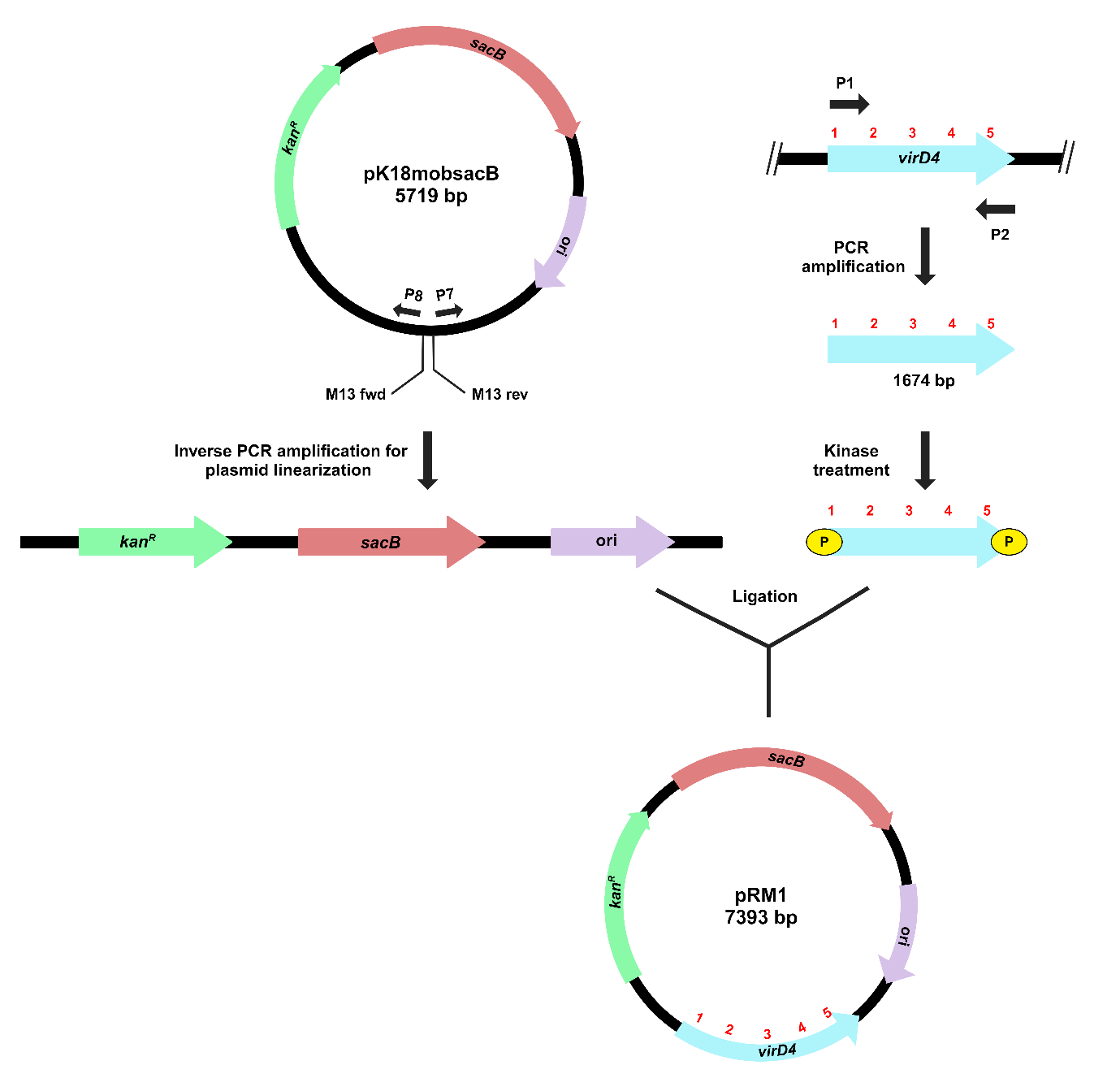
Figure 2. Schematic representation of target gene cloning in pK18mobsacB, using inverse PCR-based strategy. Outward-facing primers (P7 and P8) amplify and linearize the plasmid. Kinase treatment of the virD4 gene creates phosphorylated ends for ligation with the linearized plasmid. The recombinant plasmid, pRM1, contains a complete copy of the virD4 gene.
Streak PPL1 on NA and incubate at 28 ºC for 24-48 h. Inoculate a single colony in 10 mL NB and incubate overnight at 28 ºC, 180 rpm. Isolate genomic DNA using Quick-DNATM Fungal/Bacterial Miniprep Kit and quantify using Nanodrop.
PCR amplify the target gene (virD4) with the VirD4_3_F and VirD4_5_R primers using Phusion DNA polymerase as per the manufacturers’ instructions. Set up a 20 µL volume PCR reaction with the following conditions: (i) Initial denaturation at 98 ºC for 30 s, followed by (ii) 30 cycles of denaturation at 98 ºC for 10 s, annealing at 60 ºC for 30 s and extension at 72 ºC for 45 s, and (iii) final extension at 72 ºC for 10 min.
Confirm a 1674 bp PCR amplified gene on 1% agarose gel using the 1 kb ladder under the UV transilluminator. Purify the PCR product using GeneJET PCR purification Kit and quantify it using Nanodrop.
Streak Escherichia coli culture with pK18mobsacB on LB agar with kanamycin sulfate (50 µg/mL) and incubate at 37 ºC for 18 h. Inoculate a single colony in 10 mL LB broth with kanamycin sulfate (50 µg/mL) and incubate overnight at 37 ºC, 200 rpm. Isolate plasmid using GeneJET Plasmid Miniprep kit and quantify it using Nanodrop.
Linearize pK18mobsacB using inverse M13 region primers (Set1_M13_FP and Set1_M13_RP). Set up a 20 µL PCR amplification reaction using Phusion DNA polymerase. The PCR amplification conditions are as follows: (i) 98 ºC for 30 s, followed by 30 cycles of (ii) 98 ºC for 10 s, (iii) 60 ºC for 30 s, and (iv) 72 ºC for 3 min, and (iii) final extension at 72 ºC for 10 min.
Confirm plasmid linearization on 0.8% agarose gel with the 1 kb ladder by visualizing a 5719 bp band under the UV transilluminator. Excise the gel band and purify it using the GeneJET Gel Extraction Kit. Quantify plasmid using Nanodrop.
Set up a 50 µL kinase reaction of PCR purified target gene as given in Table 2. Incubate the reaction mixture at 37 ºC for 30 min and heat-inactivate at 65 ºC for 20 min.
Table 2. Components used for kinase treatment of the target gene.
Component | Volume/concentration |
DNA (target gene) | 19 µL (3.453 pmol of 5’ termini) |
T4 DNA ligase buffer with ATP (10x) | 5 µL |
T4 PNK | 1 µL |
Nuclease free water | 25 µL |
Total | 50 µL |
Set up a 20 µL ligation reaction with kinase-treated target gene and linearized pK18mobsacB in the ratio 1:3 as given in Table 3. Incubate the reaction mixture at 16 ºC overnight, followed by heat inactivation at 65 ºC for 10 min.
Table 3. Components used for ligation reaction.
Component | Volume/concentration |
Vector (pK18mobsacB) | 5 µL (0.055 pmol) |
Insert | 2.5 µL (0.174 pmol) |
T4 DNA ligase buffer with ATP (10x) | 2 µL |
T4 DNA ligase | 1 µL |
Nuclease free water | 9.5 µL |
Total | 20 µL |
Streak E. coli Top10 cells on LB agar plates and incubate them at 37 ºC for 18 h. Inoculate a single colony in 100 µL LB broth and prepare chemically competent cells using the previously described protocol (Chang, Chau, Landas, & Pang, 2017).
For transformation, transfer 10 µL of the ligated product in 100 µL chemical competent Top 10 cells and incubate on ice for 15 min, followed by transfer at 42 ºC for 60 s and again on ice for 2 min. Add 1 mL LB broth media to the cells and incubate at 37 ºC, 200 rpm for 1 h. Pellet down the cells at 6000 rpm for 5 min, discard the supernatant and resuspend the pellet in 100 µL LB broth. Plate the suspension on LB agar (50 µg/mL kanamycin sulfate) and incubate at 37 ºC. Transformed colonies will appear after 18-24 h.
Confirm transformed colonies using target gene (virD4) PCR amplification with the same conditions as specified in step 2.
C. Deletion of target gene region in recombinant pK18mobsacB
The method for gene deletion using inverse PCR in the recombinant pK18mobsacB is outlined in Figure 3.
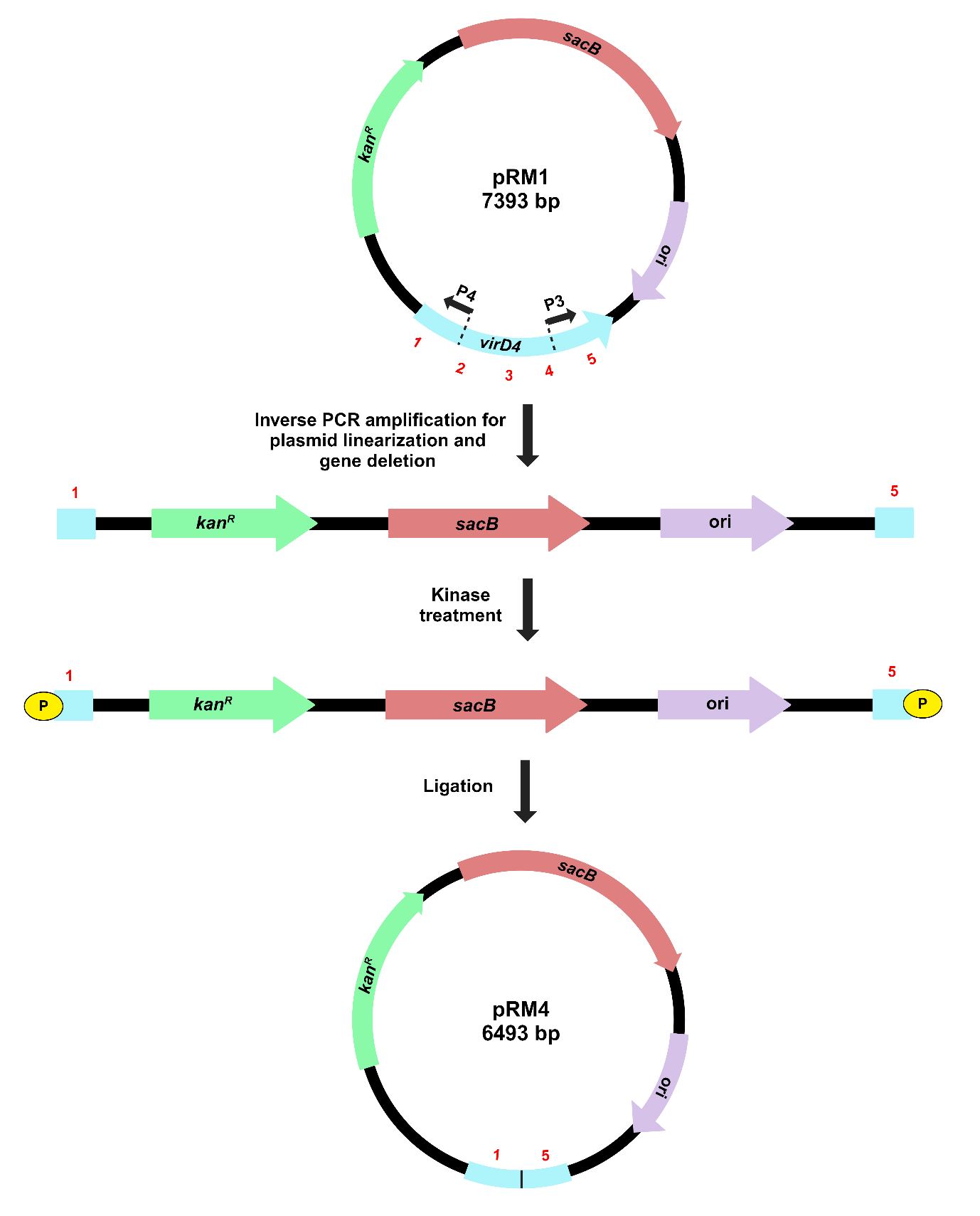
Figure 3. Illustration of target gene deletion in recombinant pK18mobsacB, using inverse PCR-based strategy. Inverse primers (P3 and P4) amplify and linearize pRM1, resulting in the deletion of a significant portion of the virD4 gene. Kinase treatment of linearized pRM1 creates phosphorylated ends for plasmid circularization through self-ligation. The recombinant plasmid, pRM4, carries a gene deletion of the virD4 gene.
Isolate recombinant plasmid (pRM1) from transformed E. coli Top10 cells using GeneJET Plasmid Miniprep Kit and quantify it using Nanodrop.
PCR amplify the recombinant plasmid using Phusion DNA polymerase and inverse primers (VirD4_inv_F2 and VirD4_inv_R) to linearize it and create target gene deletion. The PCR conditions are as follows: (i) Initial denaturation at 98 ºC for 30 s, followed by (ii) 30 cycles of denaturation at 98 ºC for 10 s, annealing at 60 ºC for 30 s and extension at 72 ºC for 3 min, and (iii) final extension at 72 ºC for 10 min.
Confirm the plasmid linearization on 0.8% agarose gel with the 1 kb ladder and by visualizing a 6439 bp band in the UV transilluminator. Cut the gel band and perform gel extraction using GeneJET Gel Extraction Kit. Quantify it using Nanodrop.
Set up a 50 µL kinase reaction of the linearized recombinant plasmid as given in Table 4. Incubate the reaction mixture at 37 ºC for 30 min, then heat-inactivate at 65 ºC for 20 min.
Table 4. Components used for kinase treatment of the target gene.
Component | Volume/concentration |
DNA | 18 µL (0.290 pmol of 5’ termini) |
T4 DNA ligase buffer with ATP (10x) | 5 µL |
T4 PNK | 1 µL |
Nuclease free water | 26 µL |
Total | 50 µL |
For self-ligation of the linearized recombinant plasmid, add 2.5 µL of T4 DNA ligase to the 50 µL kinase reaction after heat inactivation. Incubate at 16 ºC overnight, followed by heat inactivation at 65 ºC for 10 min.
Transform the chemically competent Top 10 cells by adding 10 µL of ligation mixture to 100 µL of cells following the same step as mentioned in section B (10).
Screen the colonies for the presence of gene deletion recombinant plasmid by PCR amplification using 5X FIREPol® master mix with VirD4_3_F and VirD4_5_R primers as per the following PCR conditions: (i) Initial denaturation at 95ºC for 5 min, followed by (ii) 30 cycles of denaturation at 95 ºC for 40 s, annealing at 60 ºC for 45 s and extension at 72 ºC for 1 min 30 s, and (iii) final extension at 72 ºC for 10 min.
Confirm gene deletion by running PCR product on 1.2% agarose gel with the 1 kb ladder and visualizing 774 bp DNA band under the UV transilluminator.
D. Chromosomal deletion of the target gene in X. sontii
The process for gene deletion in X. sontii PPL1 through homologous recombination is depicted in Figure 4.
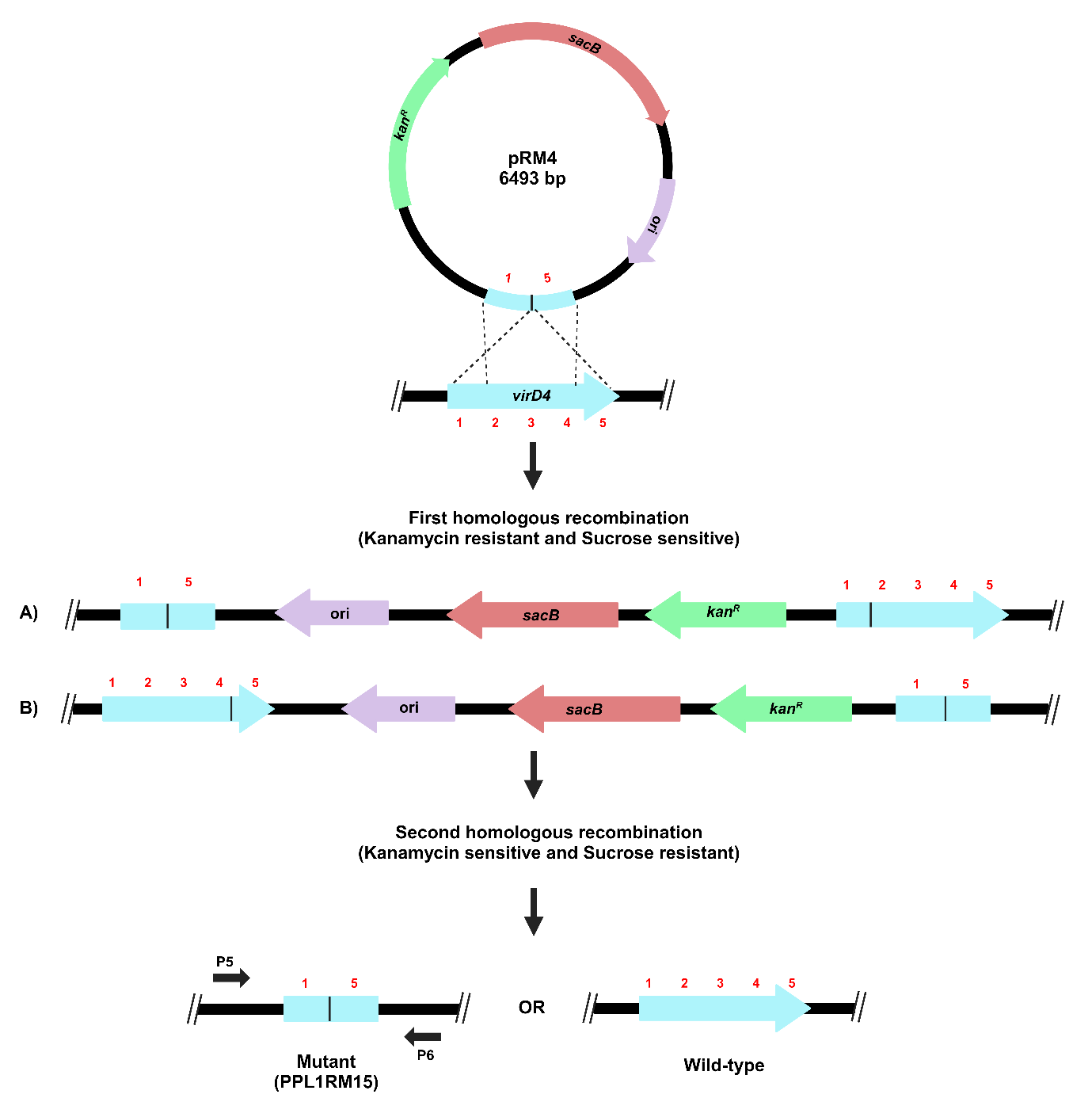
Figure 4. Schematic representation of target gene deletion in X. sontii through homologous recombination. Target gene deletion carrying plasmid, pRM4, was electroporated in X. sontii PPL1. Strains from the first homologous recombination event were screened on kanamycin, while those from the second homologous recombination event were screened on sucrose.
Extract pK18mobsacB plasmid (pRM4), carrying the deleted version of the virD4 gene, using GeneJET Plasmid Miniprep Kit and quantify it using Nanodrop.
Streak PPL1 on NA and incubate at 28 ºC for 24 to 48 h. Inoculate a single colony in 6 ml NB media and incubate at 28 ºC while shaking at 180 rpm until O.D.600nm reaches 0.7 to 0.8. Prepare electrocompetent cells of PPL1 using 300 mM sucrose, following the previously described protocol (Choi, Kumar, & Schweizer, 2006).
For electroporation, mix 1 µL (50-100 ng) plasmid in 100 µL electrocompetent cells and transfer to 2 mm electroporation cuvettes (BioRad) on ice and subject to electroporation at 2.5 kV, 25 µF, and 200 Ω using the Gene Pulser Xcell electroporation system (BioRad). Afterwards, add 1 mL NB to the cells in a 5 mL microcentrifuge tube and incubate at 28 ºC at 180 rpm for 2 h. Pellet down cells at 6000 g for 5 min, resuspend in 100 µL NB and plate on NA (50 µg/mL kanamycin sulfate). Incubate the plate at 28 ºC for 24-48 h
Screen colonies for the integration of recombinant pK18mobsacB (pRM4) into the bacterial genome (first homologous recombination) by PCR amplification of kanR (Primers, KanR_F and KanR_R) and sacB genes (Primers, SacB_F and SacB_R) using the following PCR conditions with 5X FIREPol® Master Mix: (i) Initial denaturation at 95 ºC for 5 min, followed by (ii) 30 cycles of denaturation at 95 ºC for 30 s, annealing at 66 ºC for 30 s and extension at 72 ºC for 45 s, and (iii) final extension at 72 ºC for 10 min.
Confirm PCR products by running on 1.2% agarose gel with the 100 bp ladder and visualizing ~300-400 bp DNA bands for kanR and sacB genes under the UV transilluminator.
Passage the confirmed strain from the first homologous recombination event through NB for five generations, then plate on NA + 5% sucrose. Incubate the plate at 28 ºC for 24 to 48 h.
Screen colonies for gene deletion (second homologous recombination) using PCR amplification of region ~200 bp upstream to ~180 bp downstream of virD4 gene sequence (Primers, VirD4_idnt_F and VirD4_idnt_R). The PCR amplification conditions using 5X FIREPol® Master Mix are as follows: (i) Initial denaturation at 95 ºC for 5 min, followed by (ii) 30 cycles of denaturation at 95 ºC for 30 s, annealing at 66 ºC for 45 s and extension at 72 ºC for 1 min 30 s, and (iii) final extension at 72 ºC for 10 min.
Confirm PCR product by running on 1% agarose gel with the 100 bp ladder and visualizing an 1161 bp DNA band for gene deletion mutant under the UV transilluminator.
E. Chromosomal complementation of the deletion mutant in X. sontii
The process for gene complementation in the gene deletion mutant of X. sontii through homologous recombination is described in Figure 5.
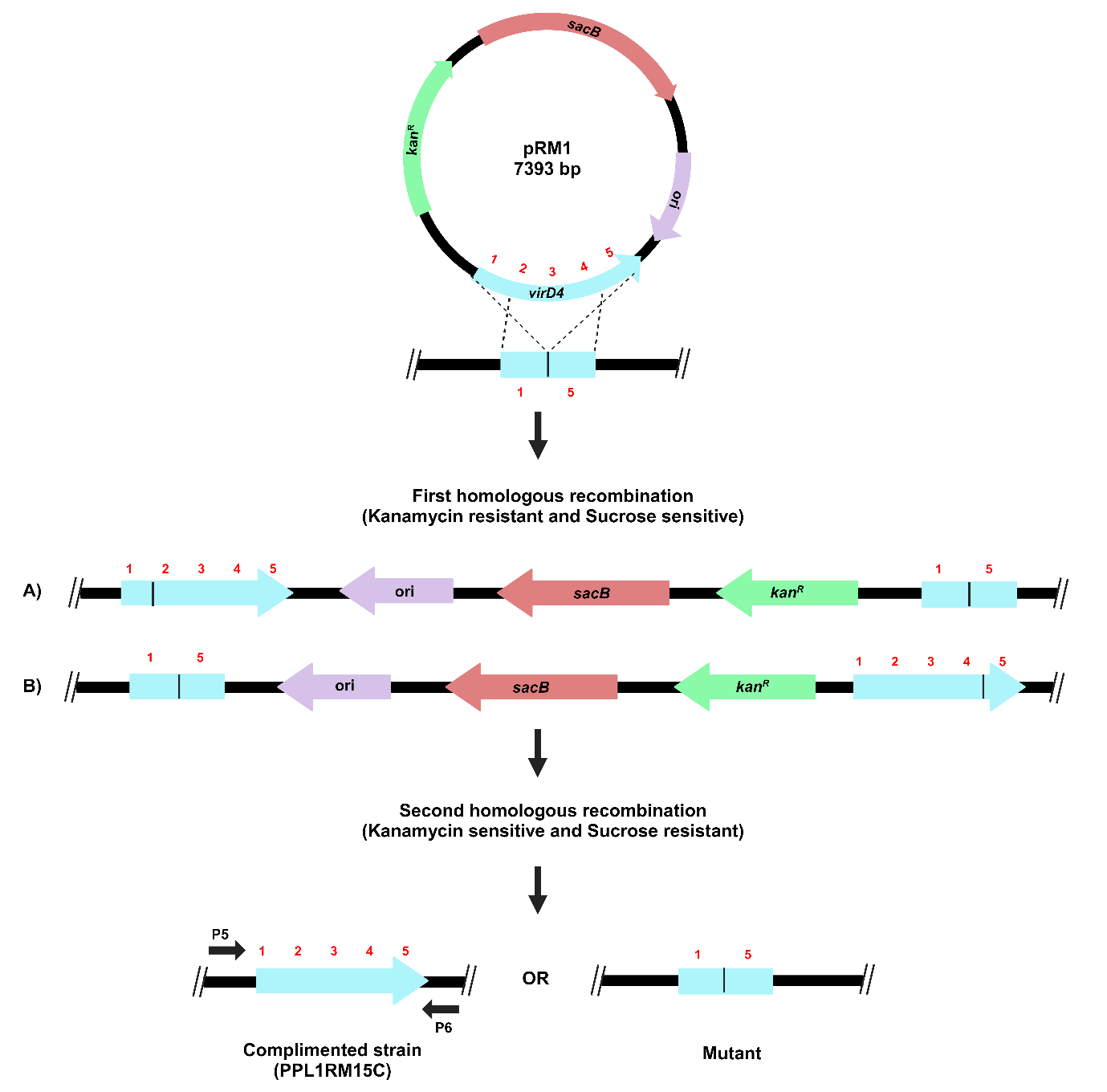
Figure 5. Schematic representation of the target gene complementation protocol through homologous recombination. The recombinant plasmid carrying a complete copy of the virD4 gene, pRM1, was electroporated in X. sontii PPL1RM15 (gene deletion mutant). Strains from the first homologous recombination event were screened on kanamycin, and from the second homologous recombination event were screened on sucrose.
For complementation, electroporate the recombinant pk18mobsacB plasmid carrying complete virD4 gene (pRM1) into electrocompetent cells of the gene deletion mutant (PPL1RM15) following steps 1 to 3, the same as mentioned in section D.
Screen colonies for the integration of pRM1 into the bacterial genome of PPL1RM15 by PCR amplification of kanR and sacB genes, following steps 4-5 (section D).
Passage the confirmed strain through NB for five generations and plate on NA + 5% sucrose. Incubate the plates at 28 ºC for 24-48 h.
Screen colonies after the second homologous recombination event by PCR amplification of region ~200 bp upstream and ~180 bp downstream of virD4 gene sequence, following step 7 from section D.
Confirm the PCR product by running on 1.2% agarose gel with the 100 bp ladder and visualizing a 2016 bp DNA band for gene complementation under the UV transilluminator.
Data analysis
Gene deletion mutant (PPL1RM15) and complemented strain of the mutant (PPL1RM15C) were further validated by whole genome sequencing, which also confirmed clean excision of pK18mobsacB. Whole genome sequencing was carried out using the NovaSeq6000 platform (Neuberg, India). Raw reads quality was assessed using FastQC v0.11.6 (https://qubeshub.org/resources/fastqc), and reads were trimmed using Trim-Galore v0.6.7 (Krueger, 2015). SPAdes v3.15.5 was used for de novo assembly (Prjibelski, Antipov, Meleshko, Lapidus, & Korobeynikov, 2020). Assembled genome quality was assessed using Quast v5.2.0 and Checkm v1.2.2 (Gurevich, Saveliev, Vyahhi, & Tesler, 2013; Parks, Imelfort, Skennerton, Hugenholtz, & Tyson, 2015). The approximate genome size of the mutant and complemented strain was 4.9 Mb assembled in 77 and 73 contigs, respectively. The nucleotide sequences of the virD4 gene and pK18mobsacB plasmid were searched in the genomes of PPL1RM15 and PPL1RM15C using NCBI local BLASTn. The pK18mobsacB sequence was not detected in any of the genomes. A clean deletion of 900 bp of the virD4 gene was present in contig 18 (genomic coordinates: 55348-55813) in PPL1RM15, and a clean, complete copy of virD4 gene was there in contig 4 (Genomic coordinates: 52202-53875) of PPL1RM15C (Figure 6). The whole genome sequences of PPL1RM15 and PPL1RM15C can be accessed on the Figshare link (https://figshare.com/s/19cfff61f91122f1b936).

Figure 6. Alignment of the virD4 gene in wild type (PPL1), mutant (PPL1RM15) and complemented strains (PPL1RM15C), respectively.
Validation of protocol
The part of the protocol related to the creation of deletion mutant has been validated in the following research article:
(Rana et al., 2024) Comparative genomics-based insights into Xanthomonas indica, a non-pathogenic species of healthy rice microbiome with bioprotection function (Figure 6C, Supplementary Figure 8).
Acknowledgements
RR planned and carried out all the experiments and drafted the manuscript. AS helped with the screening of clones and gene deletion mutants. AS and AD carried out gene complementation. PBP conceived and participated in designing of the study and finalised the manuscript. This work was funded by a grant: CRG/2022/009447 (GAP238) titled “Genetic and transcriptomics insights into anti-pathogenic activity and ecology of Xanthomonas sontii, a non-pathogenic species of rice microbiome” by the Science and Engineering Research Board, Department of Science and Technology (DST-SERB), Government of India. We also acknowledge the funding provided by the CSIR through the Microbial Type Culture Collection and Gene Bank (MTCC) project MLP-065.
Competing interests
The authors declare no competing interests.
References
Bansal, K., Kaur, A., Midha, S., Kumar, S., Korpole, S., & Patil, P. B. (2021). Xanthomonas sontii sp. nov., a non-pathogenic bacterium isolated from healthy basmati rice (Oryza sativa) seeds from India. Antonie Van Leeuwenhoek, 114, 1935-1947.
Chang, A. Y., Chau, V., Landas, J. A., & Pang, Y. (2017). Preparation of calcium competent Escherichia coli and heat-shock transformation. JEMI methods, 1(22-25).
Choi, K.-H., Kumar, A., & Schweizer, H. P. (2006). A 10-min method for preparation of highly electrocompetent Pseudomonas aeruginosa cells: application for DNA fragment transfer between chromosomes and plasmid transformation. Journal of Microbiological Methods, 64(3), 391-397.
Gay, P., Le Coq, D., Steinmetz, M., Berkelman, T., & Kado, C. (1985). Positive selection procedure for entrapment of insertion sequence elements in gram-negative bacteria. Journal of bacteriology, 164(2), 918-921.
Gurevich, A., Saveliev, V., Vyahhi, N., & Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29(8), 1072-1075.
Imai, Y., Matsushima, Y., Sugimura, T., & Terada, M. (1991). A simple and rapid method for generating a deletion by PCR. Nucleic acids research, 19(10), 2785.
Jullien, N. Retrieved from https://inp.univ-amu.fr/en/amplifx-manage-test-and-design-your-primers-for-pcr
Krueger, F. (2015). Trim Galore!: A wrapper around Cutadapt and FastQC to consistently apply adapter and quality trimming to FastQ files, with extra functionality for RRBS data. Babraham Institute.
Kumar, S., Stecher, G., Li, M., Knyaz, C., & Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35(6), 1547.
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., & Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research, 25(7), 1043-1055.
Pradhan, B. B., Ranjan, M., & Chatterjee, S. (2012). XadM, a novel adhesin of Xanthomonas oryzae pv. oryzae, exhibits similarity to Rhs family proteins and is required for optimum attachment, biofilm formation, and virulence. Molecular Plant-Microbe Interactions, 25(9), 1157-1170.
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A., & Korobeynikov, A. (2020). Using SPAdes de novo assembler. Current protocols in bioinformatics, 70(1), e102.
Rana, R., Nayak, P. K., Madhavan, V. N., Sonti, R. V., Patel, H. K., & Patil, P. B. (2024). Comparative genomics-based insights into Xanthomonas indica, a non-pathogenic species of healthy rice microbiome with bioprotection function. Applied and Environmental Microbiology, 90(9), e00848-00824.
Schäfer, A., Tauch, A., Jäger, W., Kalinowski, J., Thierbach, G., & Pühler, A. (1994). Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene, 145(1), 69-73.
Schulze, S., Kay, S., Büttner, D., Egler, M., Eschen‐Lippold, L., Hause, G., . . . Scheel, D. (2012). Analysis of new type III effectors from Xanthomonas uncovers XopB and XopS as suppressors of plant immunity. New Phytologist, 195(4), 894-911.
Ton‐That, H., & Schneewind, O. (2003). Assembly of pili on the surface of Corynebacterium diphtheriae. Molecular microbiology, 50(4), 1429-1438.
- Rana, R, Sharma, A, Dutta, A and Patil, P B(2025). A simple and fast unidirectional strategy for chromosomal gene deletion and complementation in gram-negative bacteria. Bio-protocol Preprint. 10.21769/p2780.
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.