
- Home
- Protocols

-

Transcriptomic and metabolomic co-extraction protocol for the characterization of biofilms


Last updated date: Dec 12, 2024 Views: 120 Forks: 0
Transcriptomic and metabolomic co-extraction protocol for the characterization of biofilms
Anaïs Séguéla1,2, Oriane Della-Negra3, Roselyne Gautier1,2, Jérôme Hamelin3, Kim Milferstedt3, Rémi Servien3, Marie-Ange Teste4,5, and Cécile Canlet1,2, *
1Toxalim (Research Centre in Food Toxicology), Toulouse University, INRAE, ENVT, INP-Purpan, Toulouse, France
2MetaboHUB-MetaToul, National Infrastructure of Metabolomics and Fluxomics, Toulouse, France
3INRAE, Univ. Montpellier, LBE, Narbonne, France
4TBI, Université de Toulouse, CNRS, INRAE, INSA, Toulouse, France
5IBISBA-GeT-Biopuces, Toulouse, France
*For correspondence: cecile.canlet@inrae.fr
Abstract
Capturing metabolite exchanges (“metabolomics”) and microbial activities (“transcriptomics”) require the co-extraction of RNA and metabolites. Multi-omics approaches, and notably the combination of metabolomics and transcriptomic analyses, are required for understanding the functional changes and adaptation of microorganisms when they experience different physico-chemical and environmental conditions. A protocol was developed to extract total RNA and metabolites from less than 6 mg of a kind of phototrophic biofilm: oxygenic photogranules. These granules are aggregates of a size of several hundreds of micrometres up to several millimetres. They harbour heterotrophic bacteria and phototrophs. After a common step for cell disruption by bead-beating, half of the volume was recovered for RNA extraction and the other half was used for the methanol- and dichloromethane-based extraction of metabolites. The solvents enabled the separation of two phases (aqueous and lipid) containing hydrophilic and lipophilic metabolites respectively. 1H NMR analysis of these extracts showed spectra with a signal-to-noise ratios superior of ten for more than a hundred signals and led to the identification of dozens of metabolites per sample. Total RNA was purified using a commercially available kit, yielding sufficient concentration and quality for metatranscriptomic analysis.
This new co-extraction method for RNA and metabolites on a complex biofilm matrix provides metabolomic and metatranscriptomic profiles from a small biofilm sample or one individual photogranule, thereby limiting the bias associated with sampling variability. This protocol was applied to more than 200 samples. It has proven its robustness and can be applied to high-throughput analysis for the integration of multi-omics approaches to objects with spatial structure.
Key features
- Co-extraction of metabolites and total RNA from 6 mg of dry biomass of phototrophic biofilms, notably oxygenic photogranules.
Biphasic metabolome extraction for the characterization of hydrophilic and lipophilic metabolites using 1H NMR.
Total RNA extraction with sufficient quality and quantity for analysis of (meta)transcriptome.
Keywords: Biofilm, NMR, RNA, metabolomics, (meta)transcriptomic, oxygenic photogranules, multi-omic
Graphical overview
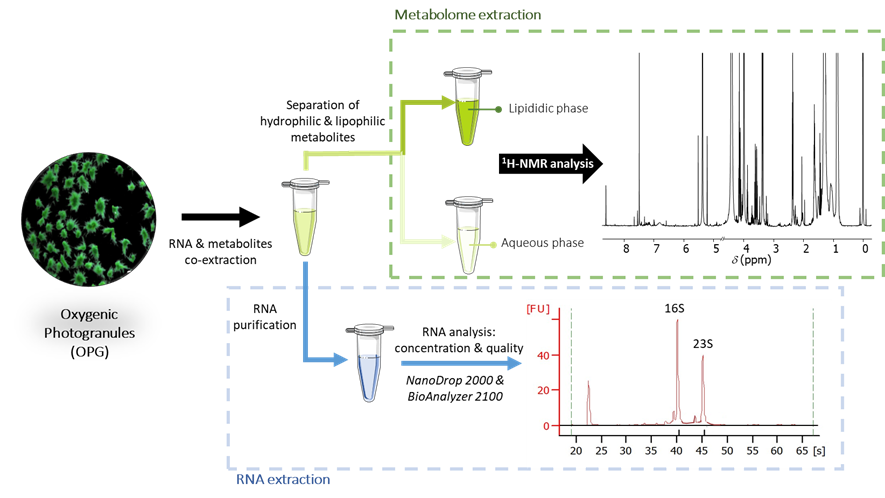
Metabolomic and RNA co-extraction from oxygenic photogranules.
Background
In contrast to suspended individual cells, biofilms are spatialized microbial structures composed of diverse interacting microbial populations. Gradients created by energy and mass transfer as well as the production and consumption of metabolites in the biofilm matrix generate a wide range of physiological states of the resident microbial community. The biofilm environment implies genetic and phenotypic heterogeneity, with distinct metabolic pathways and varied responses to stress and environmental conditions along the gradients. These sources of heterogeneity affect functional and metabolic responses in space and time[1, 2].
Elucidating the physiologies of biofilm-associated micro-organisms, by capturing metabolite exchange and gene expression, is the key to decipher how bacteria develop and interact in heterogeneous systems.
Various methods are currently available to study the dynamic of biofilms, including metabolomic[3–5] and transcriptomic[6, 7] approaches, but to our knowledge no multi-omics approach, combining metatranscriptomics and metabolomics has been developed for the application for biofilms.
Metabolomics focuses on the detection of metabolites (for example produced during biofilm formation) and characterises the state of the metabolome at a given time. In this way, metabolomics provides a view of changes in metabolism by covering all the major metabolic pathways. Metabolite profiles are functional signatures that can indicate an actual phenotype[5]. Nuclear Magnetic Resonance (NMR) spectroscopy is increasingly used in metabolomics and biofilm characterisation due to the simplicity of measurements compared with other methods (such as mass spectrometry). The NMR method includes a simplified sample pre-treatment and allows immediate (semi) quantification of metabolites and robust metabolite assignment[8]. However, this method is less sensitive than mass spectrometry[9], only focuses on the major changes in metabolic pathways and is not sufficient to profile all the metabolites and identify all the functional changes.
Metatranscriptomic analysis enables the study of the total mRNA transcripts and reveals the global gene expression profiles and changes. It can give insights into the functional components activated or inhibited during biofilm growth or decay. It can also be used to monitor biofilm dynamics in response to changes in physico-chemical or environmental conditions[7].
Currently, most omics studies are conducted separately, resulting in a partial picture of the state of the biological system. Combining metabolomics and metatranscriptomic approaches can help to identify new correlations between metabolites and transcripts providing insight into the complex interactions that drive biofilm formation and associate underlying biological processes[7, 10].
Here, oxygenic photogranules (OPGs) were used as model biofilm system for the development of a co-extraction protocol of metabolites and RNA for respective NMR-based metabolomics and metatranscriptomic analyses. OPGs are three-dimensional aggregates containing a syntrophic community composed of heterotrophic and phototrophic microorganisms[11]. The simultaneous extraction overcomes analysis bias due to sampling variability and enables the integration of multi-omics analysis for OPGs and more globally for biofilm characterisation.
Materials and reagents
Biological materials
Metabolite and RNA were co-extracted from OPGs (Figure 1). The photogranules were produced using the previously described protocols[11, 12]. Briefly, OPGs were grown in an open sequencing batch reactor operated at an average temperature of 25°C with overhead stirring of 110 rpm. LED panels provided white light at 4000 K yielding 96 µmol·m-2·s-1 photosynthetically active radiation at the outside of the vertical surfaces of the reactor. The reactor had a working volume of 4 liters. A sequencing batch cycle had a length of three hours. Two liters of bulk phase were replaced in every cycle with a synthetic medium at a carbon concentration of 33 mg.L-1, as chemical oxygen demand, resulting in a hydraulic retention time of six hours.
For the co-extraction experiment, OPGs were sieved to obtain a biomass with a narrower size distribution ranging from 0.6 to 1 mm. Altogether, 200 samples were prepared. Each sample corresponded to 50 photogranules (about 6 mg dry weight in total) that were manually placed in 500 mL of tap water contained in 2 mL screw cap vials.
Note: The minimum quantity of biomass required for the validity of the method may vary. It is therefore recommended to carry out preliminary tests to ensure that the method works properly.
The tubes containing the OPGs were exposed for 16 hours to a light of 70 µmol·m-2·s-1 photosynthetically active radiation in a growth chamber to assimilate all the residual available carbon. After this time, the light was turned off for 3 hours to stop photosynthetic activity. The end of the dark period marked the beginning to the experiment. Over the four hours of the experiment the biomass was exposed to light and dark phases in the presence or absence of carbon substrates to stimulate photosynthetic and/or heterotrophic activities. At defined intervals, tubes were sacrificed and prepared for the co-extraction.
To stop all metabolic activities, the tubes after sampling were immediately immersed in liquid nitrogen and stored at -80°C until the co-extraction of metabolites and RNA. Eight replicated vials were collected for each of the tested conditions. The lysing matrix E was added later while samples were maintained on a bed of dry ice.
Note: Here RNA and metabolites were extracted from oxygenic photogranules and supernatant (biofilm in suspension), however, this protocol could be adapted to solid pellets (raw biofilm).

Figure 1. Stereomicroscopic image of photogranules obtained for the co-extraction.
Reagents
Ethanol absolute (Fisher, catalog number: 10048291)
Methanol (Fisher, catalog number: 10499560)
Dichloromethane (Fisher, catalog number: 10626642)
Disodium hydrogen phosphate anhydrous (Na2HPO4), (SigmaAldrich, catalog number : 71640)
Potassium phosphate monobasic (KH2PO4), (SigmaAldrich, catalog number : P5655)
3-(Trimethylsilyl)propionic acid-d4 sodium salt (TSP), (SigmaAldrich, catalog number : 269913)
Deuterium oxide (D2O), (SigmaAldrich, catalog number : 151882)
Methanol-d4 (CD3OD) (SigmaAldrich, catalog number: 151947)
Chloroform-d + 0.03% trimethylsilane (CDCl3) (SigmaAldrich, catalog number: 343803)
Solutions
1. 70% ethanol solution (See Recipes)
2. Buffer phosphate solution (See Recipes)
Recipes
1. 70% ethanol solution
Reagent | Final concentration | Quantity or Volume |
Ethanol absolute | 70% | 700 mL |
MilliQ water (H2O) | n/a | 300 mL |
Total | n/a | 1000 mL |
2. Buffer phosphate solution
To prepare the buffer phosphate solution, weigh the components mentioned in the table below and dissolve in 100 mL of deuterated water. Measure the pH and adjust the pH to 7.0 with a chlorhydric acid solution 5 M.
Reagent | Final concentration | Quantity or Volume |
Na2HPO4 | 200 mM | 2.885 g |
KH2PO4 | 40 mM | 595 mg |
TSP | 0.2 mM | 3.45 mg |
D2O | n/a | 100 mL |
Total | n/a | 100 mL |
Laboratory supplies
Sampling
Lysing Matrix E (MP Biomedicals, catalog number : 1169140-CF)
Sieves of 0.6 and 1.0 mm (Retsch 200 mm DIA × 50 mm ISO 3310-1 0,6 mm; catalog number : 60131000600; catalog number : 23208274, Fisher Scientific 200 mm DIA × 50 mm ISO 3310-1 1 mm; catalog number : 10536222; catalog number : 7156588)
Liquid nitrogen
Dry ice
Co-extractions
QIAshredder (Qiagen, catalog number : 79654)
RNeasy Mini Kit (Qiagen, catalog number : 74104)
1.5 mL microtubes (e.g., Eppendorf, catalog number : 0030125150)
Filter tips 1250 µL, 200 µL, 20µl (e.g., Clearline, catalog number : 713119;713117, 713115)
2, 10, 20, 200, and 1,000 µL pipettes (e.g., Clearline or Gilson)
NMR analysis
1.75 mL Eppendorf tubes (Dutscher, Greiners, catalog number: 2710757)
Pipet tips 1250 µL, 200 µL (Dutscher, Clearline, catalog number : 713113; 713111)
Pasteur pipet (Dutscher, catalog number: 2517260)
NMR tubes 3 mm including caps (Bruker, catalog number: Z112272)
Cap sealing balls (Bruker, catalog number: Z147554)
RNA quality control
RNaseZap (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9780)
RNA 6000 Nano kit (Agilent, catalog number: 5067-11511)
Qubit 1X dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q33230)
DNA-free™ DNA Removal Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM1906)
Equipment
Laboratory fume hood (e.g.,Waldner, MC6 1500 mm)
FastPrep-24™ 5G bead beating grinder and lysis system (MP Biomedicals, catalog number : 116005500)
Microcentrifuge (e.g., Eppendorf, model: 5425)
Refrigerated centrifuge (e.g., Sartorius, model Centrisart A-14C; Mikkro 220R, Dutscher, catalog number : 472328)
Multivortex Multireax (Heidolph, Dutscher, catalog number : 498446)
Bioanalyzer 2100 (Agilent, catalog number : G2939BA)
Nanodrop ND2000 (Thermo Scientific, catalog number : ND-2000)
Qubit 4 fluorometer (Thermo Scientific, catalog number : Q33238)
-20°C Freezer
-80°C Freezer
pH-meter HI 3221 (Hanna Instruments, Dutscher, catalog number : 054585)
SpeedVac SPD 300 DDA (Dutscher, Fischer, catalog number : 22880, 228324, 228731)
Sample preparation robot (Tecan, Fluent 780)
Software and datasets
TopSpin (Bruker Biospin, Germany, V3.6.4, February 2023)
R software (https://cran.r-project.org/, V4.2.2, November 2022)
ASICS R package (V2.20.1)
2100 expert software (Agilent, V B.02.10.764, July 2018)
Nanodrop 2000 software (V1.6.198, August 2014)
Procedure
A. Sampling
24h before the experiment, store the tubes containing the lysing matrix E at -80°C.
Note: The lysing matrix E, used for OPG grinding, contained 1.4 mm ceramic spheres, 0.1 mm silica spheres and one 4 mm glass bead.
Sort the oxygenic photogranules by size using sieves of 0.6 and 1.0 mm.
Prepare batches of 50 small OPGs (approximatively 6 mg of dry biomass) in screw-cap tubes.
Note: As mentioned in Biological Materials, OPGs were exposed to different environmental conditions: phases of light and dark, in the presence or absence of carbon substrates in order to stimulate the photosynthetic and/or heterotrophic activities.
Immediately after sampling, freeze the biomass in liquid nitrogen.
Note: It is important to make sure that the biomass is at the bottom of the vials so that the buffer can be added easily for further treatment of the vials.
Critical: Very-low temperature freezing is mandatory to instantaneously stop metabolic and transcriptomic activity, maintain RNA and metabolome integrity and avoid loss of information and avoid the production of erroneous results. Here we recommend the use of liquid nitrogen that meet the temperature requirements and ensures efficient temperature transfer via an optimum contact surface.
Addition of the beads to the samples:
Remove the lysing matrix from the -80°C freezer and store the tubes on dry ice.
Remove the samples from the -80°C freezer and put them into a cryobox containing a bed of dry ice.
Open the sample tubes and discard the caps.
Transfer the lysing matrix E contained in the cold tubes to the samples.
Using the caps of the lysing matrix tubes, close the tubes now containing photogranules and matrix E. This step simplifies the handling of the tubes.
Note: Switching the caps simplified the workflow. Sometimes, the O-ring of the initial caps would fall out of the cap when the cap was unscrewed. Putting it back in place is a hassle that can be avoided by using the new caps.
Put back the samples at -80°C until the co-extractions.
Note: We recommend to store the samples at -80°C for no longer than 12 months to avoid any degradation of the metabolome or RNA
B. Common steps of the co-extraction
Put the samples in ice and add 600 µL of buffer RLT (cf: RNeasy mini kit) to the tubes (see General Note 1).
Critical: in order to avoid breaking the cold chain, it is important to perform this step on ice bed.
Grind the samples with the FastPrep for 20 seconds at 6 m/s.
Place the samples in the ice bed for 1 minute.
Repeat the grinding step.
Note: Depending on the matrix, the grinding step must be optimized. If the sample is not enough ground, add another cycle. Between each cycle, put the sample on ice bed for one minute to avoid overheating.
Centrifuge for 2 min at 16000 g at room temperature to allow the large particles to settle.
At room temperature, collect the solution and transfer it to QIAshredder spin Column.
Note: Low temperature can alter the column, so it is recommended to work at room temperature.
Centrifuge for 2 min at 16000 g at room temperature to remove microbeads and particles
Split the eluate in two:
300 µl for RNA extraction step (see D), without resuspending the pellet.
450 µL of the remaining volume for metabolite extraction (see C).
Note: The volume may vary depending on the sample but is limited by QIAshredder spin Column capacity (< 750 mL).
C. Metabolite extraction
Note: For the following steps, it is advisable to place samples on an ice bed to avoid any degradation during the extraction and to improve stability.
Add 400 µL of methanol in the tube with the remaining volume and pellet from step B.
Caution: perform this step in a fume hood because of the toxicity of methanol.
Vortex for 2 min with a multivortex.
Add 200 µL dichloromethane and vortex for 2 min.
Caution: perform this step in a fume hood because of the toxicity of dichloromethane.
Vortex for 2 min with a multivortex.
Repeat steps 3 and 4.
Place the sample on ice for 15 minutes at 4 °C to allow the two phases to separate.
Centrifuge for 15 minutes at 2200 g at 4 °C in a refrigerated centrifuge.
Separate the two phases obtained. The aqueous phase is above the organic phase:
Aqueous phase: collect 700 µL of this phase for each sample.
Organic phase: collect 200 µL of this phase for each sample
Note: There are approximatively 400 µL of organic phase but in order to be sure to not take the aqueous phase or the pellet, only 200 µL of organic phase are taken.
Note: Take the same volume between each sample for repeatability.
Caution: perform this steps in a fume hood.
At this point, samples can be stored at -80°C until the evaporation step.
Note: Samples should not be stored for longer than 3 months to avoid any degradation.
Evaporation of the different phases:
Aqueous phase:
Remove the sample from the freezer and thaw it.
Evaporate the aqueous phase using a Speedvac: no temperature, manual program, ramp 3.
Note: To avoid contaminations, the Eppendorf tube should not be more than half full. If it there is too much volume available for one run split the volumes. In our case, we proceeded as follows:
Take 400 µL and put them in another Eppendorf tube and store them at -80°C.
Transfer the remaining 300 µL in the initial tube which has been evaporate.
Evaporate using the same parameters than ii.
Store the samples at -80°C.
Note: Samples should be stored for no longer than 3 months to avoid any degradation.
b. Organic phase
Evaporate the organic phase under nitrogen flow at room temperature over a minimum of 2 hours.
Caution: perform this step in a fume hood.
Note: Once dry, organic samples may have stability problems and can no longer be stored at -80°C. We therefore recommend transferring them into NMR tubes for immediate analysis.
D. NMR tubes preparation
Aqueous phase:
Prepare the deuterated buffer phosphate solution (see Recipes 2).
Using the Tecan Robot, add 270 µL of deuterated buffer to each dry samples and mix with the needles.
Note: in the absence of Tecan Robot, manually add 270 µL of buffer and vortex the samples using a multivortex.
Centrifuge for 15 min at 1200 g at 4°C with a refrigerated centrifuge.
With the Tecan Robot, take 25 µL of each sample and pool the aliquots in a 15 ml centrifuge tube to perform a quality control.
Transfer 200 µL of the samples in NMR tubes of 3 mm of diameter.
Note: Perform steps iv. and v. manually with the same volumes if you do not have a Tecan Robot.
Fill the openings of the NMR tubes with beads.
Note: Close the NMR tubes with a suitable cap if you do not have a Tecan Robot.
b. Organic phase :
Caution: perform the following steps in a fume hood.
Prepare a solution of deuterated chloroform with 0.03% TMS and deuterated methanol (2:1 in volume).
Add 270 µL of this solution to each sample using a Pasteur pipette.
Take 25 µL of each sample and put them into a glass tube for the quality control.
Transfer 200 µL of the samples in NMR tubes of 3 mm of diameter.
Fill the openings of the NMR tubes with cap sealing balls [see above].
Note: Close the NMR tubes with a suitable cap.
E. RNA extraction
Take the 300 µl of RNA extraction eluate (see B.8 a) and add 300 µl of 70% ethanol (see Recipes 1) - Mix by pipetting.
Transfer the approximatively 600 µl into the RNeasy Spin column.
Note: Keep always the spin columns at room temperature
Centrifuge for 1 minute at maximum speed (approximately 16000 g) - Discard the eluate.
Add 700 µl of RW1 buffer.
Centrifuge for 1 minute at maximum speed (approximately 16000 g) - Discard the eluate.
Add 700 µl of RPE buffer.
Centrifuge for 1 minute at maximum speed (approximately 16000 g) - Discard the eluate.
Add 500 µl of RPE buffer.
Centrifuge for 1 minute at maximum speed (approximately 16000 g) - Discard the eluate.
Centrifuge for 2 minutes at maximum speed (approximately 16000 g) to eliminate all the RPE buffer.
Place the column in a new Eppendorf tube (final tube).
Add 35 µl of water in the centre of the column.
Centrifuge for 2 minutes at maximum speed (approximately 16000 g).
Take the eluate and place it again on the column.
Centrifuge for 2 minutes at maximum speed (approximately 16000 g).
Keep an aliquot (5µl) for quantification and quality control of your sample (e.g., Nanodrop or Qubit 4 fluorometer & BioAnalyzer) to avoid freeze-thaw cycles of your RNA sample and store the rest at -20°C. (see Data Analysis-B)
Data analysis
NMR analysis
1H-NMR spectra were obtained with a 300 K Bruker Avance III HD 600 MHz NMR spectrometer (Bruker Biospin, Rheinstetten, Germany), operating at 600.13 MHz for 1H resonance frequency, equipped with a 5 mm 1H-13C-15N-31P cryoprobe attached to a Cryoplatform (the preamplifier unit). Probe tuning and matching, locking, shims tuning, pulse duration (90°) and gain computation were automatically performed for each sample. For aqueous samples, 1H-NMR spectra were acquired using the noesy-presat 1H experiment (noesypr1d: sequence used for suppression of the water signal) whereas for organic phase, 1H-NMR spectra were acquired using a standard 1H experiment (sequence : zg). A total of 256 transients were collected into 64k data points using a spectral width of 20 ppm, a relaxation delay of 5 s and an acquisition time of 2.72 s. Prior to Fourier transformation, an exponential line broadening function of 0.3 Hz was applied to the free induction decays (FID). All NMR spectra were phase- and baseline-corrected and referenced to the chemical shift of TSP or TMS (0 ppm) using the Topspin software. Data analysis was carried out using R version V 4.2.2[13], using the ASICS package version V2.20.1[14]. This package is based on a statistical linear model to identify and quantify metabolites in complex NMR spectra using a library containing pure metabolite spectra. The processing method includes automatic baseline correction, normalisation, alignment, and exclusion of undesired areas (D2O 4.5-5.1 ppm, CD3OD 3.33-3.38 ppm, CDCl3 7.2-7.3 ppm, dichloromethane 4.88-5.31 ppm). 1H-NMR spectra were cleaned by setting a noise threshold of 0.1 for NMR spectra from the aqueous phase and 0.02 for those from the organic phase.
B. RNA quality control
Measurement of RNAs concentration
Total RNA concentration was quantified using Nanodrop 2000. In order to achieve the ribodepletion for the metatranscriptomics experiments, the concentration of extracted RNA must be high enough (in our case around 100 ng/µl). As the Nanodrop measures total nucleic acids (DNA and RNA) it is essential to know the real concentration of total RNAs. We therefore measured DNA contamination with the Qubit 4 fluorometer using 1µL of extracted RNAs in 199 µL of Qubit 1X dsDNA HS Assay Kit buffer.
Note: If the DNA contamination is more than 10% it will be necessary to treat the total RNAs with DNase (see Figure 2).
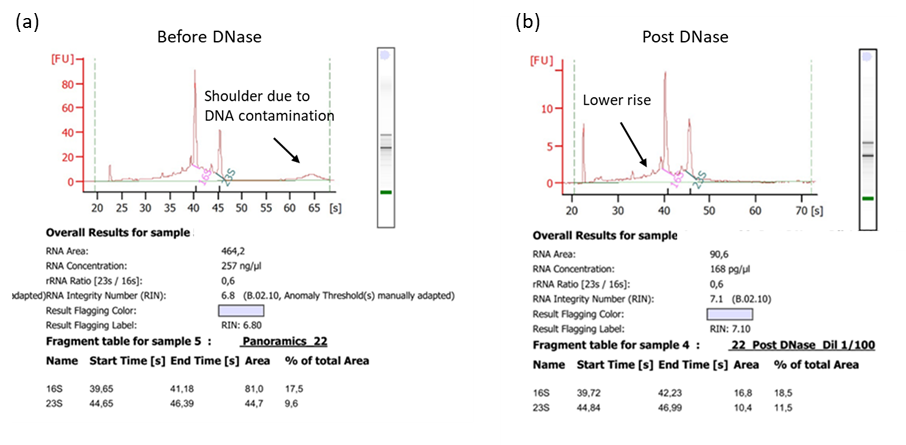
Figure 2. RNA profile (from Qubit 4 fluorometer) (a) before DNase treatment and (b) after DNase treatment.
2. Treatment of RNA eluate with DNase
Add 2 µL of DNase Buffer and 1 µL of DNase to 20 μL of eluted mRNA (2-2.5 µg of total RNAs)
Incubate 20 minutes at 37°C
Add 2.3 µL of inactivation reagent, incubate 2 min at room temperature with occasional mixing
Centrifuge and transfer the supernatant in a clean tube
Note: Perform reactions in small tubes (0.2 mL or 0.5 mL) to simplify removal of supernatant after treatment with the DNase inactivation reagent.
After DNase treatment measure again the RNA concentration with the Qubit 4 fluorometer as describe above. The measured value will be used for the rest of the experiments.
3. Measurement of quality and integrity of the extracted RNA
The quality and integrity of the extracted RNA were checked using the Bioanalyzer 2100 and Bioanalyzer RNA 6000 Nano kit. The RNAs were denatured 2 minutes at 70°C and put on ice; 1 µL was used according the manufacturer recommendations.
Validation of protocol
This protocol was tested on a set of 200 samples, demonstrating its robustness and reproducibility, obtaining sufficient concentrations of metabolites and RNA for subsequent metabolomic and metatranscriptomic analyses, while complying with the quality criteria for NMR spectra and RNA.
NMR Analysis
In NMR, the standardized requirement for the limit of detection (LOD) and quantification (LOQ) are signal-to-noise ratio ≥ 3 and 10, respectively[15].
In our case, most of the NMR spectra that were obtained had enough signals with a signal-to-noise ratio greater than 10.
In the aqueous phase, on average 120 peaks were detected per sample and 12 ± 4 metabolites were identified per sample using the ASICS package (55 different metabolites were identified in aqueous phase depending on the condition). The citrate signal at 2.67 ppm, coming from the RLT buffer and displayed in Figure 3, masked some of the information and is certainly responsible for the relative small number of metabolites identified here (see General Note 1).
In the organic phase, around 150 peaks were detected per sample and 17 ± 4 metabolites were identified per sample (altogether 88 different metabolites were identified in the organic phase depending on the condition).
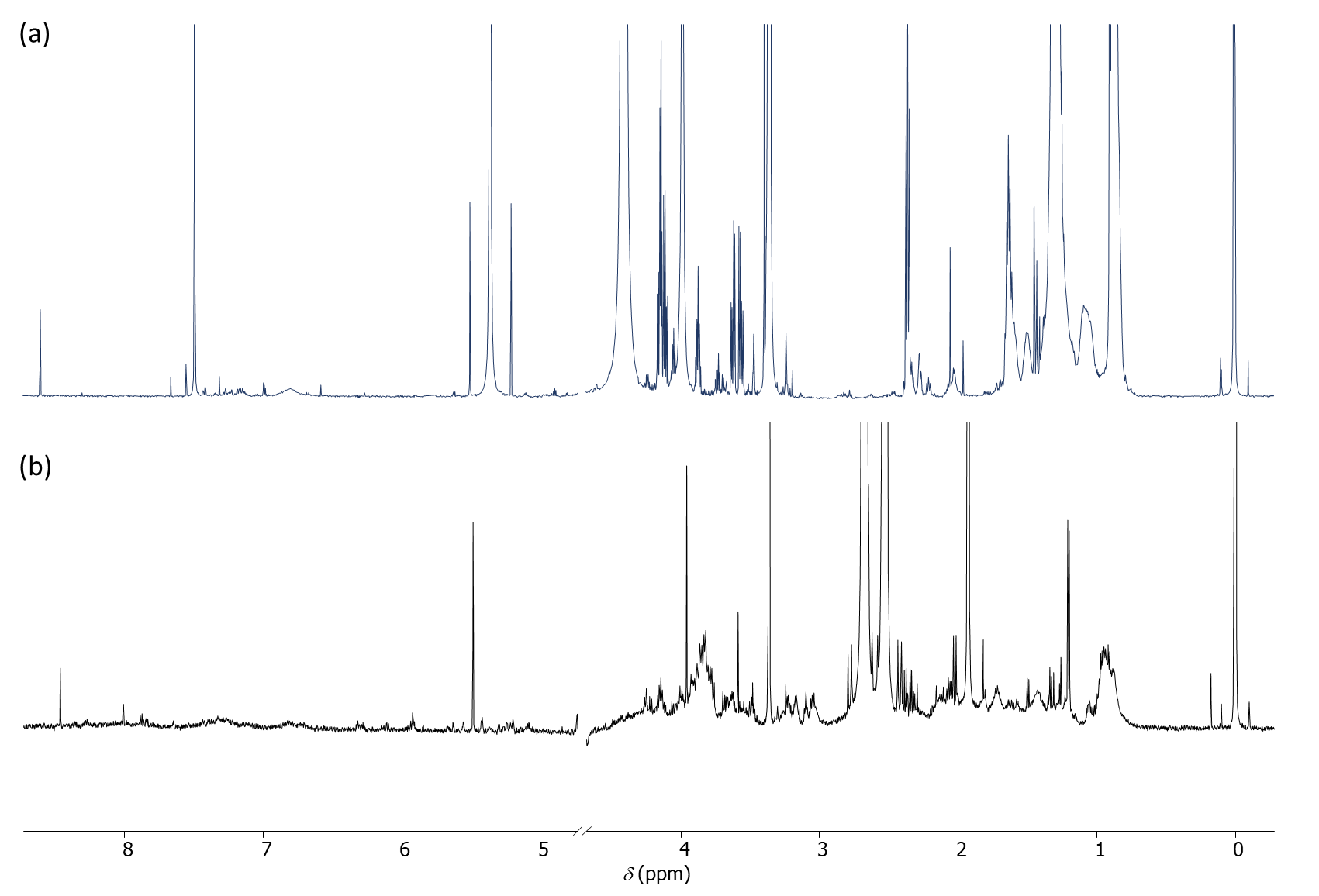
Figure 3. (a) 1H-NMR spectra of the lipidic phase in CDCl3/CD3OD (2/1). (b) 1H-NMR spectra of the aqueous phase in D2O.
The quality of the analysis was controlled at multiple points for both phases to assure that there was no drift or other introduction of error in the results. For this, dedicated controls samples (pool of all samples) were analysed repeatedly after a series of ten unknown samples. Identical spectra were expected for the quality controls in the aqueous and lipid phases. Principal component analysis is one way of demonstrating that, as expected, controls for both phases clustered together, indicating the absence of analytical derivation (Figure 4) and thus validating the analytical method.
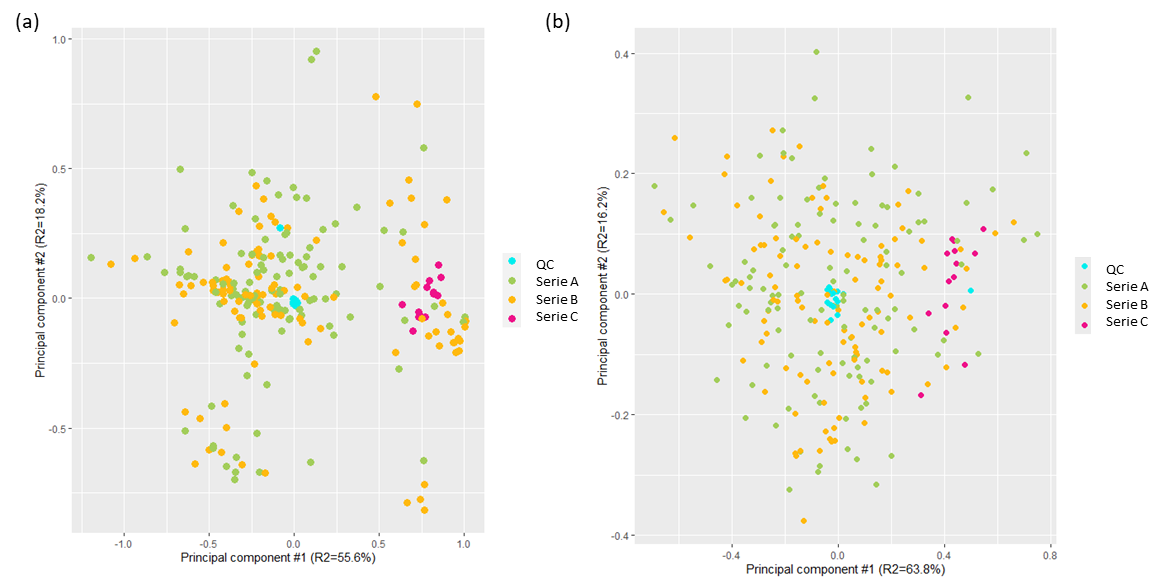
Figure 4. Principal Component Analysis obtained from NMR spectra of (a) the lipidic samples and (b) the aqueous samples. Quality controls are depicted in blue (QC).
To estimate the recovery efficiency and reproducibility of the extraction method, two conditions were tested with OPGs: (i) a condition containing OPGs in water (ii) and another containing OPGs in water and acetate as carbon source. As shown in Figure 3, the extraction method coupled to the identification method with ASICS enabled the identification and the recovery of acetate in NMR spectra. Furthermore, as shown in Figure 5, a relative quantification carried out on 70 samples with and without acetate clearly showed a significant difference (p-value < 0.05) between the two conditions, demonstrating the reproducibility and the recovery efficiency of the extraction method.
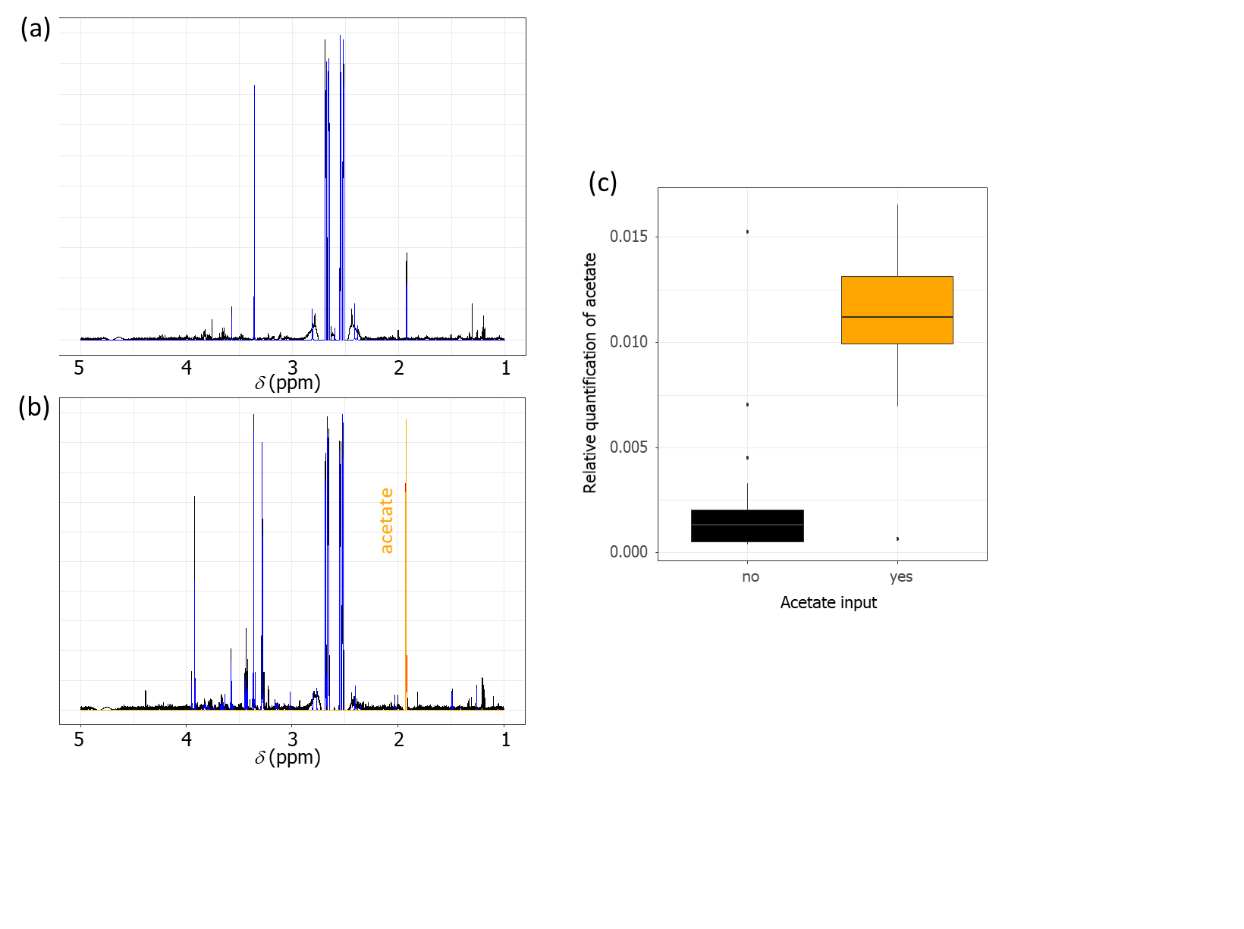
Figure 5. (a) 1H-NMR spectrum (D2O) of an aqueous phase extracted from OPG samples without acetate input. (b) 1H-NMR spectrum (D2O) of an aqueous phase extracted from OPG sample with acetate input. (c) Boxplot showing the relative quantification of acetate found in samples with or without acetate (35 replicates for each condition).
B. RNA quality control
When measuring the nanodrop concentration the absorbance ratios at wavelengths 260/230 nm and 260/280 nm measured must be greater than 1.8 to consider sufficient RNA purity.
After the DNase treatment the profile of all the sample must be similar. Furthermore, the higher the RIN, the better the RNA integrity. In our case we consider only the samples with RIN>7.
C. Mass range for method validity (minimum quantity of biomaterial)
Preliminary tests aiming at optimising the method were carried out on samples containing 20 OPGs (approximately 3 mg of dry biomass) and 50 OPGs (approximately 6 mg of dry biomass).
With 20 OPGs, the resulting NMR spectra were of poorer quality, insufficient for metabolomic analysis. For example, only 6 metabolites were detected in the aqueous phase extracted from 20 OPGs versus an average of 12 for 50 OPGs. Similarly, in the organic phase, 8 metabolites were identified with 20 OPGs versus an average of 17 for 50 photogranules.
Furthermore, as mentioned above, 50 OPGs were sufficient to obtain RNA of good quality to later perform metatranscriptomic analysis (with 20 OPGs the quantity was not sufficient).
General notes and troubleshooting
General notes
1. The buffer RLT was analysed by 1H NMR during the optimisation of the protocol and showed the presence of citrate at 2.67 ppm.
Troubleshooting
| Problem or issue | Likely cause(s) | Possible solution(s) |
No metabolites were extracted / they underwent degradation | OPG metabolome was not frozen correctly. | Freeze OPG metabolome in liquid nitrogen. |
The cold chain was interrupted. | Keep the samples on dry ice during the transfer between rooms or labs and directly put them into a -80°C freezer. | |
The protocol was performed by one person only, resulting in too long lag times. | The metabolite and RNA extractions are best carried out by two people to avoid lag times after the steps common to both extractions. | |
Extraction times between the first and last samples in a series were not identical. | In the case of high-throughput extraction, be careful to produce small series (8 samples per series). | |
No RNA was extracted /it was degraded | The cold chain was broken when adding the RLT buffer and/or during the grinding steps. | Use an ice bed during this steps to maintained the OPGs at a low temperature. |
RNA extraction was performed at a wrong temperature. | RNA extraction must be carried out at room temperature to avoid damaging the RNA kit columns after the addition of 70% ethanol (procedures D 8-a). | |
RNA was contaminated by DNA | No optimal DNase treatment | Perform a new DNase digestion on the RNA eluate. Alternatively, use the RNeasy Plus mini kit (Qiagen, reference: 74134) which contains a supplemental step with gDNA Eliminator Spin Columns |
Acknowledgments
This work was supported by ANR Project Panoramics (grant number ANR-21-CE45-0036-0). This work benefited from the Environmental Biotechnology and Biorefinery Facility (Bio2E) of INRAE-LBE (doi.org/10.15454/1.557234103446854E12). We thank the Metabohub-Metatoul platform for NMR analyses that were performed on instruments funded by the French National Infrastructure for Metabolomics and Fluxomics MetaboHUB (ANR-11-INBS-010). We thank the GeT-Biopuces platform (IBSIBA.fr https://doi.org/10.15454/08BX-VJ91, GenoToul, Toulouse, France) for performing the RNA quality control and subsequent metatranscriptomics experiments.
Competing interests
The authors declare that they have no competing interests.
Ethical considerations
No human or animal subjects were included in this study.
References
1. Flemming, H.-C., van Hullebusch, E. D., Neu, T. R., Nielsen, P. H., Seviour, T., Stoodley, P., et al. (2023). The biofilm matrix: multitasking in a shared space. Nat Rev Microbiol. 21: 70–86. https://doi.org/10.1038/s41579-022-00791-0
2. Stewart, P. S. and Franklin, M. J. (2008). Physiological heterogeneity in biofilms. Nat Rev Microbiol. 6: 199–210. https://doi.org/10.1038/nrmicro1838
3. Cook, J. M., Edwards, A., Bulling, M., Mur, L. A. J., Cook, S., Gokul, J. K., et al. (2016). Metabolome-mediated biocryomorphic evolution promotes carbon fixation in Greenlandic cryoconite holes. Environ Microbiol. 18: 4674–4686. https://doi.org/10.1111/1462-2920.13349
4. Doose, C. and Hubas, C. (2024). The metabolites of light: Untargeted metabolomic approaches bring new clues to understand light-driven acclimation of intertidal mudflat biofilm. Sci Total Environ. 912: 168692. https://doi.org/10.1016/j.scitotenv.2023.168692
5. Zhang, B. and Powers, R. (2012). Analysis of bacterial biofilms using NMR-based metabolomics. Future Med Chem. 4: 1273–1306. https://doi.org/10.4155/fmc.12.59
6. Shi, Y., Xu, C., Ji, B., Li, A., Zhang, X. and Liu, Y. (2024). Microalgal-bacterial granular sludge can remove complex organics from municipal wastewater with algae-bacteria interactions. Commun Earth Environ, Nature Publishing Group. 5: 1–10. https://doi.org/10.1038/s43247-024-01499-0
7. Vohra, M., Kour, A., Kalia, N. P., Kumar, M., Sharma, S., Jaglan, S., et al. (2024). A comprehensive review of genomics, transcriptomics, proteomics, and metabolomic insights into the differentiation of Pseudomonas aeruginosa from the planktonic to biofilm state: A multi-omics approach. Int J Biol Macromol. 257: 128563. https://doi.org/10.1016/j.ijbiomac.2023.128563
8. Powers, R., Andersson, E. R., Bayless, A. L., Brua, R. B., Chang, M. C., Cheng, L. L., et al. (2024). Best practices in NMR metabolomics: Current state. TrAC Trends in Analytical Chemistry. 171: 117478. https://doi.org/10.1016/j.trac.2023.117478
9. Emwas, A.-H. M. (2015). The Strengths and Weaknesses of NMR Spectroscopy and Mass Spectrometry with Particular Focus on Metabolomics Research. In Metabonomics: Methods and Protocols (Bjerrum, J. T., ed.), pp 161–193, Springer, New York, NY https://doi.org/10.1007/978-1-4939-2377-9_13
10. Borgan, E., Sitter, B., Lingjærde, O. C., Johnsen, H., Lundgren, S., Bathen, T. F., et al. (2010). Merging transcriptomics and metabolomics--advances in breast cancer profiling. BMC Cancer. 10: 628. https://doi.org/10.1186/1471-2407-10-628
11. Milferstedt, K., Kuo-Dahab, W. C., Butler, C. S., Hamelin, J., Abouhend, A. S., Stauch-White, K., et al. (2017). The importance of filamentous cyanobacteria in the development of oxygenic photogranules. Sci Rep. 7: 17944. https://doi.org/10.1038/s41598-017-16614-9
12. Galea-Outón, S., Milferstedt, K. and Hamelin, J. (2024). High methane potential of oxygenic photogranules decreases after starvation. Bioresource Technology. 406: 130986. https://doi.org/10.1016/j.biortech.2024.130986
13. R: A Language and Environment for Statistical Computing | BibSonomy https://www.bibsonomy.org/bibtex/7469ffee3b07f9167cf47e7555041ee7
14. Lefort, G., Liaubet, L., Canlet, C., Tardivel, P., Père, M.-C., Quesnel, H., et al. (2019). ASICS: an R package for a whole analysis workflow of 1D 1H NMR spectra. Bioinformatics.35: 4356–4363. https://doi.org/10.1093/bioinformatics/btz248
15. Lucas-Torres, C. and Wong, A. (2019). Current Developments in µMAS NMR Analysis for Metabolomics. Metabolites. 9: 29. https://doi.org/10.3390/metabo9020029
- Séguéla, A, Della-Negra, O, Gautier, R, Hamelin, J, Milferstedt, K, Servien, R, Teste, M and Canlet, C(2024). Transcriptomic and metabolomic co-extraction protocol for the characterization of biofilms. Bio-protocol Preprint. bio-protocol.org/prep2767.
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.