
- Home
- Protocols

-

The method for in vitro screening of unknown interacting proteins using GST Pull-down MS and the method for validating protein interactions using pGEX4T-1 and pColdTF vectors based on the principle of GST Pull-down
Last updated date: Nov 10, 2024 DOI: 10.21769/p2750 Views: 255 Forks: 0
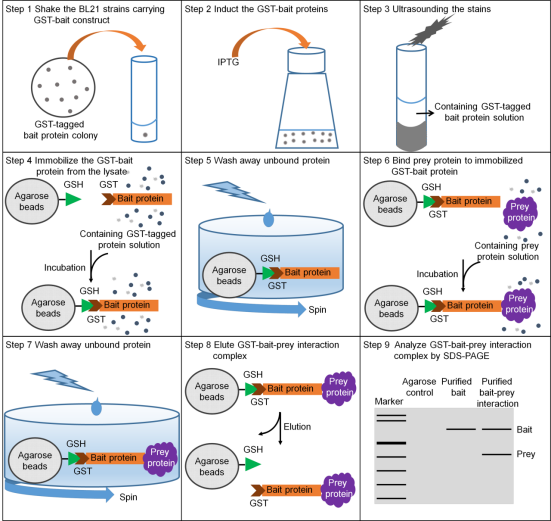
In vitro screening of unknown interacting proteins using GST Pull-down MS method
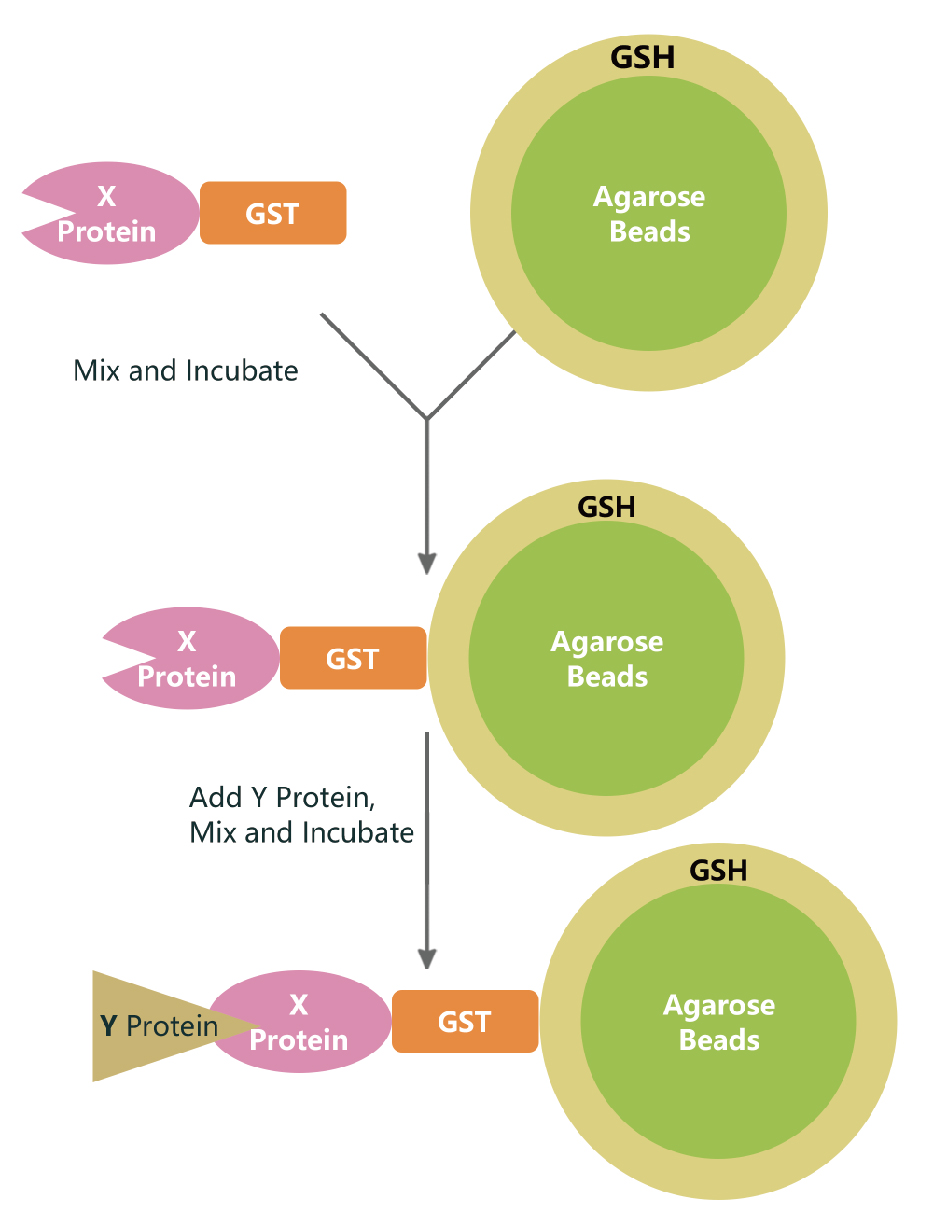
Validation of protein interactions using pGEX4T-1 and pColdTF vectors based on the principle of GST Pull-down
Experimental principle
Pull-down technology (also known as in vitro protein binding assay) is an effective method for validating the yeast two-hybrid system in vitro experiments (Wissmueller et al., 2011). As early as 1988, Smith and Johnson (1988) used glutathione-S-transferase (GST fusion tags) to purify GST fusion proteins in one step from bacteria. Since then, GST fusion proteins have been greatly promoted in the field of protein interaction research. The basic principle of using GST fusion proteins for pull-down is to immobilize the target protein-GST fusion protein on glutathione beads (as a support for affinity with the target protein, acting as a bait protein). The target protein solution is incubated with it, and the interacting proteins (target proteins) can be captured from it. After washing and eluting the bound proteins, the interaction between the two proteins can be confirmed or the corresponding target proteins can be screened through SDS-PAGE (Sodium Dodecyl-Sulfate-Polyacrylamide Gel Electrophoresis) electrophoresis analysis. Bait proteins and captured proteins can be obtained from cell lysates, purified proteins, expression systems, and in vitro transcription-translation systems. In pull-down experiments, designing appropriate control experiments is very important. Controls can be lysates from cells expressing GST (non-bait fusion proteins), or GST can be added to lysates from non-transformed cells. The GST fusion protein pull-down method can be used to identify unknown proteins that interact with known fusion proteins, as well as to determine whether two known proteins interact with each other. The method is simple, easy to perform, and convenient. However, the results of pull-down do not necessarily reflect the actual interaction between proteins, that is, they do not mean that they can bind under physiological conditions, because they may not meet in subcellular space within living organisms.
Instruments:
PCR Machine
Temperature-Controlled Water Bath
Vortex Mixer
Centrifuge
Gel Electrophoresis Apparatus
UV-Visible Spectrophotometer
Microplate Reader
Sonicator
Magnetic Rack
Incubator
Shaking Incubator
Gradient Gel Caster
Gel Documentation System
Western Blot Transfer Apparatus
Film Developer
Scanner
Mass Spectrometer
Reagents:
pGEX4T-1 Vector
pColdTF Vector
IPTG (Isopropyl β-D-1-thiogalactopyranoside)
PBS Buffer (Phosphate-Buffered Saline)
NETN Buffer
SDS (Sodium Dodecyl Sulfate)
Bromophenol Blue
β-mercaptoethanol
TBST (Tris-Buffered Saline with Tween 20)
Tris Base
Sodium Chloride (NaCl)
Tween 20
Laemmli Buffer
Protease Inhibitor Cocktail
Triton X-100
PMSF (Phenylmethylsulfonyl fluoride)
Glutathione Sepharose 4B
Imidazole
Nickel-Agarose Gel
BCA Reagent Kit
Bovine Serum Albumin (BSA) Standard
KOD FX DNA Polymerase
dNTPs (Deoxyribonucleotide Triphosphates)
EcoR I-HF Restriction Enzyme
rCutSmart Buffer
Agarose
Ethidium Bromide
DNA Gel Extraction Kit
ClonExpress® Ultra One Step Cloning Kit
Solution formula
1× PBS buffer (pH7.4):
NaCl: 137 mM
KCl: 2.7 mM
Na2HPO4: 10 mM
KH2PO4: 2 mM
NETN buffer:
Tris-HCl (pH8.0): 20 mM
NaCl: 100 mM
EDTA: 0.5 mM
Nonidet P-40 (NP-40): 0.5% (v/v)(optional)
3× SDS sample buffer:
Tris-HCl (pH6.8): 167 mM
Glycerin: 33% (v/v)
Sodium dodecyl sulfate: 6.6% (w/v)
Bromophenol blue: 0.01% (w/v)
β-mercaptaethanol: 7.5% (v)
TBST:
Tris-Buffered Saline (TBS):
6.05 grams Tris base
85.0 grams Sodium Chloride (NaCl)
Adjust pH to 7.4-7.6 with HCl
Add water to make up to 1000 ml
Tween 20:
1 ml (final concentration of 0.1%)
Preparation Steps:
Prepare TBS solution: Dissolve 6.05 grams of Tris base and 85.0 grams of Sodium Chloride in a certain volume of water, adjust the pH to 7.4-7.6 with HCl, then add water to reach a final volume of 1000 ml.
Add Tween 20: To the 1000 ml of TBS solution, add 1 ml of Tween 20.
4x Laemmli Buffer (for SDS-PAGE):
250 mM Tris-HCl (pH 6.8) 20 ml
8% SDS 6.4 g
40% Glycerol 32 ml
4% β-Mercaptoethanol 3.2 ml
0.01% Bromophenol Blue 0.008 g
Note: Bromophenol Blue does not need to be weighed precisely; add while observing the color change, use just enough to achieve the desired color, as too much can cause the color to turn red.
Total Protein Extraction Buffer:
50 mM Tris-HCl (pH 7.5) 500 μl
150 mM NaCl 300 μl
0.5% Triton X-100 250 μl
Protease Inhibitor (Roche) 200 μl
Note: If the extracted total protein is to be used directly for SDS-PAGE detection, SDS can be used in place of Triton X-100. Due to its stronger denaturing ability compared to Triton X-100, SDS can better extract proteins from the nucleus; however, if the extracted total protein is for the separation and purification of proteins or complexes, SDS should not be used.
Experimental Group Settings:
Experimental Group 1: GST-X fusion protein with His-Y fusion protein GST pull-down.
Experimental Group 2: GST protein with His-Y fusion protein GST pull-down.
Control Group Settings:
Control Group 1: Western blot of GST-X fusion protein with His-Y fusion protein.
Control Group 2: Western blot of GST protein with His-Y fusion protein.
Experimental steps:
1.Construct pGEX4T-1-X Vector
1.1 PCR Amplification of X Linker Sequence X Sequence Synthesis Rules:
X-F: ccgcgtggatccccggaa + first 20 bp of X CDS
X-R: ctcgagtcgacccgggaa + last 20 bp of X CDS (reverse complement)
1.2 Configuration of PCR Amplification System with Linker Gene X (Using KOD FX, TOYOBO KFX-101)
Before preparing the reaction mixture, please thoroughly mix all reagents except for KOD FX (enzyme solution). Frozen reagents should be completely thawed on ice before use.
2x PCR buffer 25 μl
2mM dNTPs 10 μl
X-F Primer 1.5 μl
X-R Primer 1.5 μl
Plant cDNA 0.2 μg
KOD FX (1.0U/μl) 1 μl
ddH2O up to 50 μl Please add KOD FX (enzyme solution) last, and mix the reaction mixture thoroughly with a Vortex or similar device, then spin down before proceeding with PCR.
1.3 PCR Amplification of Gene X with Linkers
Subsequently, use a temperature-controlled PCR machine with the following program:
Predenature 94℃, 2 min.
Denature 98℃, 10 sec
Annealing (Tm-5)℃, 30 sec
Extension 68℃, 1kb/min Set Denature to Extension for 33 cycles.
Final extension 68℃, 7 min. After the reaction is complete, transfer to a 4℃ refrigerator for storage.
1.4 Take the pGEX4T-1 plasmid out of the -20℃ freezer, place 10x rCutSmart buffer on ice, and after complete dissolution, configure the following system:
10x rCutSmart buffer 5 μl
EcoR I-HF 1 μl
pGEX4T-1 Plasmid DNA 1 μg
ddH2O up to 50 μl Subsequently, use a temperature-controlled PCR machine with the following program:
37℃ 45 min
65℃ 45 sec After the reaction is complete, transfer to a 4℃ refrigerator for storage.
1.5 Agarose Gel Electrophoresis and Gel Recovery (EasyPure Quick Gel Extraction Kit, TransGen Biotech, EG101)
(The method can refer to Wang, S, Huang, Z, Liu, Y, Shao, S, Li, L and Ma, M(2024). Application of the Nicotiana Allergic Necrosis Assay for the Validation of Protein-Protein Interactions between Fungal Effectors and Plant Receptor Kinases. Bio-protocol Preprint. bio-protocol.org/prep2729.)
1.6 Construct pGEX4T-1-X Vector by Homologous Recombination (ClonExpress® Ultra One Step Cloning Kit, Vazyme, C115)
Calculate the dosage of linearized vector and insert fragment:
Optimal cloning vector dosage = [0.02 × cloning vector base pairs] ng (0.03 pmol)
Optimal insert fragment dosage = [0.04 × insert fragment base pairs] ng (0.06 pmol) Note: Calculate the required DNA amount for the recombination reaction according to the formula. To ensure the accuracy of pipetting, dilute the linearized vector and insert fragment appropriately before preparing the recombination reaction system, with each component's volume not less than 1μl. Prepare the following reaction system on ice:
Linearized vector pGEX4T-1 X μl
Insert fragment X μl
2 × ClonExpress Mix 5 μl
ddH2O to 10 μl Gently pipette to mix (do not vortex), briefly centrifuge to collect the reaction liquid at the bottom of the tube. Subsequently, use a temperature-controlled PCR machine with the following program:
50℃ 30 min After the reaction is complete, transfer to a 4℃ refrigerator for storage.
2.Construct pColdTF-Y Vector
2.1 PCR Amplification of X Linker Sequence X Sequence Synthesis Rules:
Y-F: accctcgagggatccgaa + first 20 bp of X CDS
Y-R: caggtcgacaagcttgaa + last 20 bp of X CDS (reverse complement)
2.2 Configuration of PCR Amplification System with Linker Gene X (Using KOD FX, TOYOBO KFX-101)
Before preparing the reaction mixture, please thoroughly mix all reagents except for KOD FX (enzyme solution). Frozen reagents should be completely thawed on ice before use.
2x PCR buffer 25 μl
2mM dNTPs 10 μl
Y-F Primer 1.5 μl
Y-R Primer 1.5 μl
Plant cDNA 0.2 μg
KOD FX (1.0U/μl) 1 μl
ddH2O up to 50 μl Please add KOD FX (enzyme solution) last, and mix the reaction mixture thoroughly with a Vortex or similar device, then spin down before proceeding with PCR.
2.3 PCR Amplification of Gene Y with Linkers
Subsequently, use a temperature-controlled PCR machine with the following program:
Predenature 94℃, 2 min.
Denature 98℃, 10 sec
Annealing (Tm-5)℃, 30 sec
Extension 68℃, 1kb/min Set Denature to Extension for 33 cycles.
Final extension 68℃, 7 min. After the reaction is complete, transfer to a 4℃ refrigerator for storage.
2.4 Take the pColdTF plasmid out of the -20℃ freezer, place 10x rCutSmart buffer on ice, and after complete dissolution, configure the following system:
10x rCutSmart buffer 5 μl
EcoR I-HF 1 μl
pColdTF Plasmid DNA 1 μg
ddH2O up to 50 μl Subsequently, use a temperature-controlled PCR machine with the following program:
37℃ 45 min
65℃ 45 sec After the reaction is complete, transfer to a 4℃ refrigerator for storage.
2.5 Agarose Gel Electrophoresis and Gel Recovery (EasyPure Quick Gel Extraction Kit, TransGen Biotech, EG101)
(The method can refer to Wang, S, Huang, Z, Liu, Y, Shao, S, Li, L and Ma, M(2024). Application of the Nicotiana Allergic Necrosis Assay for the Validation of Protein-Protein Interactions between Fungal Effectors and Plant Receptor Kinases. Bio-protocol Preprint. bio-protocol.org/prep2729.)
2.6 Construct pColdTF-X Vector by Homologous Recombination (ClonExpress® Ultra One Step Cloning Kit, Vazyme, C115)
Calculate the dosage of linearized vector and insert fragment:
Optimal cloning vector dosage = [0.02 × cloning vector base pairs] ng (0.03 pmol)
Optimal insert fragment dosage = [0.04 × insert fragment base pairs] ng (0.06 pmol) Note: Calculate the required DNA amount for the recombination reaction according to the formula. To ensure the accuracy of pipetting, dilute the linearized vector and insert fragment appropriately before preparing the recombination reaction system, with each component's volume not less than 1μl. Prepare the following reaction system on ice:
Linearized vector pColdTF X μl
Insert fragment X μl
2 × ClonExpress Mix 5 μl
ddH2O to 10 μl Gently pipette to mix (do not vortex), briefly centrifuge to collect the reaction liquid at the bottom of the tube. Subsequently, use a temperature-controlled PCR machine with the following program:
50℃ 30 min After the reaction is complete, transfer to a 4℃ refrigerator for storage.
3.Prokaryotic induction expression of GST-X fusion protein and GST protein.
3.1 Transfer the plasmids expressing pGEX4T-1-X (experimental group) and pGEX4T-1 (control group) into the BL21 strain separately.
3.2 Pick positive single colonies from each and inoculate into 3 mL of LB liquid medium containing antibiotics, culture overnight at 37°C with 200 rpm.
3.3 Transfer the overnight culture into 300 mL (1:100) of LB liquid medium containing antibiotics, shake at 37°C with 200 rpm for 2.5–3 hours until the OD600 reaches around 0.8.
3.4 Take 0.5 mL of the bacterial culture and store at 4°C (as pre-induction bacterial culture). At the same time, add IPTG to a final concentration of 1 mmol·L–1 to the remaining culture, induce at 16–27°C with 100–200 rpm for 8–16 hours. After induction, take 0.5 mL of the bacterial culture and store at 4°C (as post-induction bacterial culture).
3.5 Centrifuge at 4°C, 4500 ×g for 15 minutes, collect the induced bacteria, resuspend the pellet in 20 mL of PBS solution, and transfer to a 50 mL centrifuge tube.
3.6 Centrifuge at 4°C, 4500 ×g for 10 minutes, discard the supernatant, quickly freeze in liquid nitrogen, and store at –80°C (for purification).
3.7 Centrifuge at 4°C, 10,000 ×g for 2 minutes, collect the pre-induction and post-induction bacterial cultures, resuspend the pellets in 80 µL of 1×SDS loading buffer.
3.8 Preparation of 50% GST Sepharose 4B slurry: Homogenize the original 75% Glutathione Sepharose 4B slurry. Take 677 µL of the original slurry per tube, centrifuge at 1,000 ×g for 5 minutes, discard the supernatant. Add 500 µL of PBS, mix by inversion, centrifuge at 1,000 ×g for 5 minutes, discard the supernatant, repeat this process five times. Add 500 µL of PBS, mix by inversion, and prepare 50% Glutathione Sepharose 4B, keep on standby.
4.Prokaryotic induction expression of His-Y fusion protein.
4.1 Transfer the plasmid expressing pColdTF-Y into the BL21 strain.
4.2 Pick positive single colonies and inoculate into 3 mL of LB liquid medium containing antibiotics, culture overnight at 37°C with 200 rpm.
4.3 Transfer the overnight culture into 300 mL (1:100) of LB liquid medium containing antibiotics, shake at 37°C with 200 rpm for 2.5–3 hours until the OD600 reaches around 0.8.
4.4 Take 0.5 mL of the bacterial culture and store at 4°C (as pre-induction bacterial culture). At the same time, add IPTG to a final concentration of 1 mmol·L–1 to the remaining culture, induce at 16–27°C with 100–200 rpm for 8–16 hours. After induction, take 0.5 mL of the bacterial culture and store at 4°C (as post-induction bacterial culture).
4.5 Centrifuge at 4°C, 4500 ×g for 15 minutes, collect the induced bacteria, resuspend the pellet in 20 mL of PBS solution, and transfer to a 50 mL centrifuge tube.
4.6 Centrifuge at 4°C, 4500 ×g for 10 minutes, discard the supernatant, quickly freeze in liquid nitrogen, and store at –80°C (for purification).
4.7 Centrifuge at 4°C, 10,000 ×g for 2 minutes, collect the pre-induction and post-induction bacterial cultures, resuspend the pellets in 80 µL of 1×SDS loading buffer.
5. Purification of GST Fusion Protein
5.1 Thawing and Resuspension: Remove the frozen samples from the -80°C refrigerator. After thawing, add 4 mL of PBS, 40 μL of PMSF, and 40 μL of Protease Inhibitor Cocktail to the pGEX4T-1-X (experimental group) and pGEX4T-1 bacterial bodies to fully resuspend.
5.2 Sonication and Centrifugation: Sonicate the bacterial bodies with a frequency of 100–200 J·S–1; sonicate for 40 seconds, then pause for 20 seconds, repeat this cycle 5 times (until the bacteria become clear from turbid). Centrifuge at 4°C, 15,000 ×g for 40 minutes.
5.3 Transfer and Incubation: Transfer the supernatant of the cell lysate to a clean 10 mL centrifuge tube on ice. Add 50 μL of 50% Glutathione Sepharose 4B to the supernatant and gently rotate incubate at 4°C for 30–60 minutes on a silent mixer.
5.4 Centrifugation and Washing: Centrifuge at 1,500 ×g for 5 minutes, discard the supernatant. The GST fusion protein or GST is now bound to the Sepharose. Add 500 μL of pre-chilled PBS to the tube (add along the wall gently to avoid breaking the connection between the beads and the protein), gently resuspend the beads, and wash the Sepharose once. Centrifuge at 1,500 ×g for 3 minutes, and discard the supernatant.
5.5 Repeated Washing and Transfer: Wash the Sepharose with PBS three times. Finally, use a small pipette tip to remove the water film on the surface of the beads to obtain the Sepharose bound with GST-X fusion protein or GST. Transfer the Sepharose to a 1.5 mL conical bottom centrifuge tube using a large-bore pipette tip.
5.6 Preparation for Detection: If used for detection, add 15–20 μL of 1×SDS protein loading buffer to the Sepharose, boil in water for 3 minutes, centrifuge at 15,000 ×g for 1 minute, and collect the supernatant for SDS-PAGE electrophoresis.
6. Purification of His-fusion Protein
6.1 Thawing and Resuspension: Remove the frozen samples from the -80°C refrigerator. After thawing, add 4 mL of PBS, 40 μL of PMSF, and 40 μL of Protease Inhibitor Cocktail to the pColdTF-Y bacterial bodies to fully resuspend.
6.2 Sonication and Centrifugation: Sonicate the bacterial bodies with a frequency of 100–200 J·S–1; sonicate for 40 seconds, then pause for 20 seconds, repeat this cycle 5 times (until the bacteria become clear from turbid). Centrifuge at 4°C, 15,000 ×g for 40 minutes.
6.3 Transfer and Imidazole Addition: Transfer the supernatant of the cell lysate to a clean 10 mL centrifuge tube on ice. Add 2M imidazole solution to the supernatant to achieve a final concentration of 20mM, with a sample volume of 10 mL.
6.4 Filtration and Buffer Preparation: It is best to filter the sample through a 0.45μm filter membrane before column chromatography to avoid clogging. Equilibration buffer: 50mM phosphate buffer pH 7.4 with 0.5M NaCl, containing 20mM imidazole. Elution buffer: 50mM phosphate buffer pH 7.4 with 0.5M NaCl, containing 500mM imidazole.
6.5 Column Loading: Take a 1 mL nickel-agarose gel FF or nickel-NTA-agarose gel FF pre-packed column, equilibrate with 10 mL of equilibration buffer, then load 10 mL of the lysate supernatant at a flow rate of 0.5 mL/min, and collect fractions in 2 mL tubes.
6.6 Washing Unbound Samples: Wash away unbound samples with 15 mL of equilibration buffer at a flow rate of 1-2 mL/min, collecting fractions in 2 mL tubes.
6.7 Elution of Bound Samples: Elute bound samples with 5 mL of elution buffer at a flow rate of 1-2 mL/min, collecting fractions in 2 mL tubes.
6.8 Column Re-equilibration and Storage: Re-equilibrate the column with 5 mL of equilibration buffer, then fill with 20% ethanol, seal, and store for future use. If the result is not satisfactory, use a gradient elution with 50 mM, 100 mM, 300 mM, and 500 mM imidazole, eluting for about 5-10 column volumes, at a flow rate of 1-2 mL/min, collecting fractions in 2 mL tubes.
6.9 SDS-PAGE Analysis: Take samples from the fractions eluted at various imidazole concentrations and perform SDS-PAGE.
7. Pre-Pull Down Western Blot (WB) Detection
7.1 Gradient Gel Preparation: Prepare a 4%-20% gradient pre-cast gel and select an appropriate concentration based on the molecular weight of the target proteins X and Y.
7.2 Sample Loading and Electrophoresis: Record the order of samples to be loaded, the amount of sample, the name of the primary antibody, the size of the target protein, the dilution ratio of the primary antibody, and the species of the secondary antibody in the lab notebook.
Use a 10 μL/50 μL pipette to load the appropriate amount of boiled samples into the wells, and run the gel at 100V for 60-70 minutes until the purple bromophenol blue band migrates to within 5-10 mm of the bottom of the gel.
7.3 Transfer Membrane: Mark the PVDF membrane in advance, soak it in methanol for 30-60 seconds, discard the methanol, rinse the PVDF membrane with distilled water, and finally soak it in transfer buffer while shaking. Transfer conditions (adjustable as needed) are as follows: for proteins with a molecular weight of 1~15 kD, transfer for about 3 minutes; 15~30 kD for about 5 minutes; 30~150 kD for about 8 minutes; 150~300 kD for about 13 minutes; and above 300 kD for about 22 minutes.
After transferring, soak the PVDF membrane in 1x TBS for 5 minutes, then wash it with distilled water for 10 minutes.
7.4 Blocking: Prepare a blocking solution with 5% skim milk or 1% casein. Remove the PVDF membrane from the transfer apparatus and soak it in TBS solution, then place it in an incubation box with the blocking solution (submerge the PVDF membrane), and shake at room temperature for 1-2 hours.
7.5 Primary Antibody Incubation: Discard the blocking solution, dilute the primary antibody with antibody dilution buffer (initially follow the manufacturer's recommended dilution ratio, adjust as needed based on results), and add it to the corresponding incubation box. Place the box on a shaker at 4°C overnight.
7.6 Washing: Discard or recover the primary antibody dilution buffer, and wash the PVDF membrane with TBST solution (submerge the membrane). Wash 3-4 times, about 10 minutes each, and shake on a shaker.
7.7 Secondary Antibody Incubation: Choose the secondary antibody based on the source of the primary antibody, usually mouse or rabbit. Dilute it with antibody dilution buffer, typically around 1:10000. Discard the incubation box's wash liquid, add the diluted secondary antibody to the corresponding incubation box, and then place the box on a shaker at room temperature for about 1-1.5 hours.
7.8 Washing: Discard or recover the secondary antibody dilution buffer, and wash the PVDF membrane with TBST solution (submerge the membrane). Wash 3-4 times, about 10 minutes each, and shake on a shaker.
7.9 Development: Soak the washed PVDF membrane in TBS solution, lay plastic wrap, place the PVDF membrane on filter paper, and prepare the luminescent solution. The luminescent solution consists of A and B liquids, mixed in a 1:1 ratio, and let it stand in the dark for about 3 minutes. Place the PVDF membrane on filter paper, dry the TBS solution, place the PVDF membrane neatly on plastic wrap, add 1 mL of luminescent solution per membrane. Re-cover, place the PVDF membrane on the re-covered film, add another layer of plastic wrap, trim, and fix the trimmed PVDF membrane with adhesive in a dark box for development.
Enter the darkroom and expose the film for 2 seconds, 10 seconds, 30 seconds, 2 minutes, and 10 minutes (adjustable as needed). Place the film in the developer for about 30 seconds, then in the fixer for about 30 seconds. Wash the film, dry it, draw markers, and label necessary information such as the name of the primary antibody, the size of the target protein, the dilution ratio of the primary antibody, project number, development time, and date.
7.10 Scanning: Scan the film using a scanner, save it in TIFF format. Name the image with the format of date + project number + development time, and finally save it in the corresponding folder. Note: The pull-down experiment should only be conducted after the target band is detected in the pre-pull down WB detection.
8.GST In Vitro Protein Binding Assay (GST Pull-down)
8.1 emove Glutathione Sepharose 4B from the 4°C refrigerator and mix thoroughly by inverting several times to ensure the magnetic beads and solution are evenly mixed.
8.2 Transfer 50 μL of the magnetic bead solution into two clean 1.5 mL EP tubes, labeled as the control group and the experimental group.
8.3 Add 0.5 mL of pre-chilled IP wash buffer to resuspend the magnetic beads, place on a magnetic rack for 1 minute to separate the beads from the solution, and carefully aspirate the supernatant using a pipette.
8.4 Repeat step 3 two more times, for a total of three washes.
8.5 Add 300-1000 μg of bait protein GST-X to the experimental group, and 300-1000μg of tagged GST protein to the control tube, supplement the volume to 1 mL with IP dilution buffer, and mix quietly at room temperature for 2 hours.
8.6Place both tubes on a magnetic rack for 1 minute to separate the beads from the solution, and carefully aspirate the supernatant or collect it (store the collected sample at -20°C).
8.7 Add 0.5 mL of pre-chilled IP wash buffer, place on a magnetic rack for 1 minute to separate the beads from the solution, and carefully aspirate the supernatant.
8.8 Repeat step 3 two more times, for a total of three washes.
8.9 Add 300-1000 μg of His-Y or 500-2000 μg of total protein to both tubes, supplement the volume to 1 mL with IP dilution buffer, and mix quietly at 4°C overnight (approximately 16 hours).
8.10 Place both tubes on a magnetic rack for 1 minute to separate the beads from the solution, and carefully aspirate the supernatant or collect it (store the collected sample at -20°C).
8.11 Add 0.5 mL of pre-chilled IP wash buffer, place on a magnetic rack for 1 minute to separate the beads from the solution, and carefully aspirate the supernatant.
Repeat step 3 two more times, for a total of three washes.
8.12 Add 100 μL of elution buffer to both tubes, incubate at 95°C for 5 minutes, centrifuge at 12,000 rpm for 5 minutes, collect the supernatant, add 20 μL of 6× Loading buffer, and incubate in boiling water for 8-10 minutes, labeled as the control group and the experimental group.
8.13 Take two clean EP tubes, label them as Input Control and Input Experimental, add 50 μL of GST-tagged protein and target protein to the Input Control, and add 50 μL of bait protein and target protein to the Input Experimental (if the target protein is unknown, take only 100 μL of the Input Experimental plus 100 μL of total protein). Add 20 μL of 6× Loading buffer, and incubate in boiling water for 8-10 minutes.
8.14 Store the control group, experimental group, and Input at -20°C for later use.
Preparation for Mass Spectrometry Samples (if needed),Before the final wash before elution, take 1/4 of the mixed magnetic bead eluate, wash with PBS three times, discard the wash buffer, and store the magnetic beads at -20°C.
SDS-PAGE and Western Blotting Detection
9.Post-Pull Down Western Blot (WB) Detection
Note:Protein detection is required before and after the pull-down experiment)
9.1 If both the bait and prey proteins are known, perform GST pull-down followed by Western Blot (GST pull-down-WB).
9.2 If the bait protein is known and the prey protein is unknown, perform GST pull-down followed by Mass Spectrometry (GST pull-down-MS).
BCA Method for Total Protein Concentration Measurement
Dilution of BSA Standard: Dilute the BSA standard with the same diluent used for the protein samples to be measured (1x PBS solution or 0.9% physiological saline).
Reagents: The BCA reagent is divided into A, B, and C solutions. For each set of samples to be measured, the required amounts of the three solutions are 4 μl of C solution, 100 μl of B solution, and 104 μl of A solution.
Total Reagent Volume: For n samples of different concentrations, prepare reagents for n+8 sets.
Operation (using one set as an example): Take 198 μl of 1x PBS into an EP tube labeled with the sample name; add 2 μl of the sample lysate; then add 200 μl of the prepared BCA mixed reagent (which is a mixture of 4 μl of C solution, 100 μl of B solution, and 104 μl of A solution), making a total of 400 μl of solution, mix well; load the microplate, add 300 μl of the mixed solution into the microplate wells, record the order of the samples in the wells, cover the wells with a large label, and incubate in a 60°C oven for 1 hour.
Measurement with a Microplate Reader: Adjust the wavelength to 562 nm, select quick measurement, and record the data.
Data Input: Enter the recorded measurement data into an EXCEL spreadsheet.
Calculation of Sample Concentration: Subtract the reference G value from the measured data of the samples, input the adjusted data into the formula derived from step 6, divide by 1000, and multiply by 100 to obtain the concentration of the samples in mg/ml.
Precautions:
The detection range of the BCA method for protein concentration is 20-2000 μg/ml. The absorbance value will continue to deepen over time. Therefore, all sample measurements must be completed within 3-5 minutes to ensure the accuracy of protein quantification.
It is recommended to use a standard 96-well microplate for measurement, with 300 μl/well being optimal.
The absorbance values of the standard (7 reference groups) and the samples must be subtracted by the absorbance value of the blank control (reference group G) before plotting the standard curve.
If the readings of the standard deviate significantly from the linear curve due to operational errors, they should be discarded.
The unknown sample concentration can be calculated from the standard curve equation, and the actual concentration needs to be multiplied by the dilution factor of the sample.
If the obtained protein concentration is out of the detection range, please re-dilute the sample and measure again.
If we are extracting unknown interacting proteins of protein X from plant total proteins, we will need the GST pull-down MS technology mentioned earlier. The procedures for purification and separation are the same as for GST pull-down, with the only additional steps being mass spectrometry analysis and total protein extraction. Here are the supplementary steps:
10.Plant Total Protein Extraction
Plant Tissue Collection and Grinding: Take approximately 0.3g of cleaned plant tissue and finely chop it using a disinfected pair of scissors in a sterile, pre-chilled mortar. Grind the tissue into a powder with liquid nitrogen and collect it into a 1.5ml EP tube.
10.2 Addition of Extraction Buffer: Add 1ml of Total Protein Extraction Buffer to the powdered tissue.
10.3 Ice Bath and Vortexing: Let the mixture stand on ice for 20 minutes, vortexing every 5 minutes for 10 seconds each time.
10.4 Sonication: Sonicate the mixture for 8 minutes using an ultrasonic cell disruptor at 20% power, with a 3-second working interval and a 3-second rest interval, all while keeping the sample on ice.
10.5 Centrifugation: Centrifuge the sample at 4°C, 12000 rpm for 10 minutes using a desktop high-speed refrigerated centrifuge. Collect the supernatant, which is the total protein extract.
Notes: The entire protein extraction process should be conducted on ice to minimize protein degradation caused by high temperatures; avoid bubble formation during sonication to reduce protein degradation. Store the total protein at -20°C to prevent repeated freeze-thaw cycles; a portion can be kept at 4°C for immediate use.
11.MS analyse
11.1 Data Preprocessing: Peak Detection, Identification, and Extraction:
Utilize software such as XCMS for data preprocessing to identify chromatographic peaks from raw data. For instance, the findChromPeaks() function can recognize chromatographic peaks and perform data extraction.
Set parameters such as ppm (allowed mass-to-charge ratio deviation), peakwidth (the width range of chromatographic peaks), etc., to optimize the peak detection process.
11.2 Signal Denoising: Perform signal denoising through software tools to remove noise and impurity signals, thereby enhancing data quality. This can be achieved by setting an intensity threshold value (noise parameter), with signals below this value considered instrumental noise.
11.3 Feature Extraction: Extracting Mass-to-Charge Ratios (m/z) and Retention Times:
Extract meaningful features from complex mass spectrometry data, such as mass-to-charge ratios and retention times. This can be achieved through feature detection algorithms, for example, using the centWave algorithm for chromatographic peak extraction.
11.4 Data Alignment: Using Alignment Algorithms: Due to differences between samples, mass spectrometry data may experience drift and shifts. Utilize alignment algorithms such as the edit distance algorithm (Levenshtein distance) for data alignment.
These algorithms match data between different samples by calculating the minimum number of operations (insertions, deletions, substitutions) to ensure that data can be compared and analyzed.
11.5 Feature Quantification:Determining Feature Abundance: Determine the relative or absolute abundance of each feature using standard curve methods, internal standard methods, or statistical model-based approaches.
This can be achieved by comparing the response of known concentration standards with the sample response.
11.6 Data Interpretation and Statistical Analysis: Interpreting and Analyzing Mass Spectrometry Data: Interpret and analyze mass spectrometry data to derive the final analytical results. This may include the use of multivariate statistical analysis methods, such as Principal Component Analysis (PCA) or Partial Least Squares Discriminant Analysis (PLS-DA).
11.7 Analysis: Confirming Molecular Ion Peaks:Confirm molecular ion peaks through mass spectrometry spectra and derive relative molecular masses and chemical formulas from them.
Calculate the degree of unsaturation, identify major ion peaks, and record their mass-to-charge ratios (m/z values) and relative intensities.
11.8 MS-MS Analysis: Use MS-MS technology to identify parent and daughter ions, or use metastable scanning techniques to identify metastable ions.
11.9 Complementing with Other Analytical Techniques: Complement with elemental analysis, UV, IR, NMR, and the physicochemical properties of the sample to propose the structure of the sample.
Check whether the proposed structure conforms to the fragmentation patterns of the corresponding compounds. If there are no contradictions, it is possible to determine the likely structure.
1.Vector Name: pCold-TF
Vector Tag: N-His, N-Trigger-factor, N-HRV 3C, N-Thrombin, N-FactorXa
Vector Resistance: Ampicillin
Vector size:5769bp
Note: Temperature-inducible, cold shock protein expression vector.
pCold-TF-F: 5'-GCGAAAGTGACTGAAAAAG-3
pCold-TF-R: 5'-GGCAGGGATCTTAGATTCTG-3'
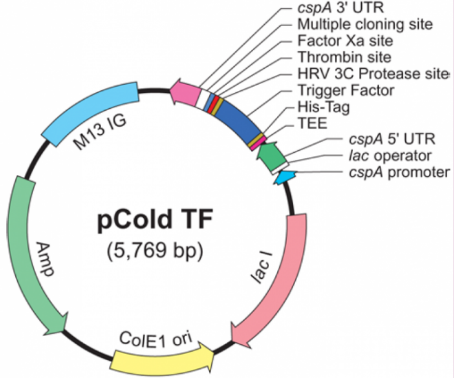
>pCold-TF
AAGGAATGGTGTGGCCGATTAATCATAAATATGAAAAATAATTGTTGCATCACCCGCCAATGCGTGGCTTAATGCACATCAAATTGTGAGCGGATAACAATTTGATGTGCTAGCGCATATCCAGTGTAGTAAGGCAAGTCCCTTCAAGAGTTATCGTTGATACCCCTCGTAGTGCACATTCCTTTAACGCTTCAAAATCTGTAAAGCACGCCATATCGCCGAAAGGCACACTTAATTATTAAGAGGTAATACACCATGAATCACAAAGTGCATCATCATCATCATCACATGCAAGTTTCAGTTGAAACCACTCAAGGCCTTGGCCGCCGTGTAACGATTACTATCGCTGCTGACAGCATCGAGACCGCTGTTAAAAGCGAGCTGGTCAACGTTGCGAAAAAAGTACGTATTGACGGCTTCCGCAAGGGCAAAGTGCCAATGAATATCGTTGCTCAGCGTTATGGCGCGTCTGTACGCCAGGACGTTCTGGGTGACCTGATGAGCCGTAACTTCATTGACGCCATCATTAAAGAAAAAATCAATCCGGCTGGCGCACCGACTTATGTTCCGGGCGAATACAAGCTGGGTGAAGACTTCACTTACTCTGTAGAGTTTGAAGTTTATCCGGAAGTTGAACTGCAAGGTCTGGAAGCGATCGAAGTTGAAAAACCGATCGTTGAAGTGACCGACGCTGACGTTGACGGCATGCTGGATACTCTGCGTAAACAGCAGGCGACCTGGAAAGAAAAAGACGGCGCTGTTGAAGCAGAAGACCGCGTGACCATCGACTTCACCGGTTCTGTAGACGGCGAAGAGTTCGAAGGCGGTAAAGCGTCTGATTTCGTACTGGCGATGGGCCAGGGTCGTATGATCCCGGGCTTTGAAGACGGTATCAAAGGCCACAAAGCTGGCGAAGAGTTCACCATCGACGTGACCTTCCCGGAAGAATACCACGCAGAAAACCTGAAAGGTAAAGCAGCGAAATTCGCTATCAACCTGAAGAAAGTTGAAGAGCGTGAACTGCCGGAACTGACCGCAGAGTTCATCAAACGTTTCGGCGTTGAAGATGGTTCCGTAGAAGGTCTGCGCGCTGAAGTGCGTAAAAACATGGAGCGCGAGCTGAAGAGCGCCATCCGTAACCGCGTTAAGTCTCAGGCGATCGAAGGTCTGGTAAAAGCTAACGACATCGACGTACCGGCTGCGCTGATCGACAGCGAAATCGACGTTCTGCGTCGCCAGGCTGCACAGCGTTTCGGTGGCAACGAAAAACAAGCTCTGGAACTGCCGCGCGAACTGTTCGAAGAACAGGCTAAACGCCGCGTAGTTGTTGGCCTGCTGCTGGGCGAAGTTATCCGCACCAACGAGCTGAAAGCTGACGAAGAGCGCGTGAAAGGCCTGATCGAAGAGATGGCTTCTGCGTACGAAGATCCGAAAGAAGTTATCGAGTTCTACAGCAAAAACAAAGAACTGATGGACAACATGCGCAATGTTGCTCTGGAAGAACAGGCTGTTGAAGCTGTACTGGCGAAAGCGAAAGTGACTGAAAAAGAAACCACTTTCAACGAGCTGATGAACCAGCAGGCGTCCGCGGGTCTGGAAGTTCTGTTCCAGGGGCCCTCCGCGGGTCTGGTGCCACGCGGTAGTGGTGGTATCGAAGGTAGGCATATGGAGCTCGGTACCCTCGAGGGATCCGAATTCAAGCTTGTCGACCTGCAGTCTAGATAGGTAATCTCTGCTTAAAAGCACAGAATCTAAGATCCCTGCCATTTGGCGGGGATTTTTTTATTTGTTTTCAGGAAATAAATAATCGATCGCGTAATAAAATCTATTATTATTTTTGTGAAGAATAAATTTGGGTGCAATGAGAATGCGCAGGCCCTTTCGTCTCGCGCGTTTCGGTGATGACGGTGAAAACCTCTGACACATGCAGCTCCCGGAGACGGTCACAGCTTGTCTGTAAGCGGATGCCGGGAGCAGACAAGCCCGTCAGGGCGCGTCAGCGGGTGTTGGCGGGTGTCGGGGCTGGCTTAACTATGCGGCATCAGAGCAGATTGTACTGAGAGTGCACCATAAAATTGTAAACGTTAATATTTTGTTAAAATTCGCGTTAAATTTTTGTTAAATCAGCTCATTTTTTAACCAATAGGCCGAAATCGGCAAAATCCCTTATAAATCAAAAGAATAGCCCGAGATAGGGTTGAGTGTTGTTCCAGTTTGGAACAAGAGTCCACTATTAAAGAACGTGGACTCCAACGTCAAAGGGCGAAAAACCGTCTATCAGGGCGATGGCCCACTACGTGAACCATCACCCAAATCAAGTTTTTTGGGGTCGAGGTGCCGTAAAGCACTAAATCGGAACCCTAAAGGGAGCCCCCGATTTAGAGCTTGACGGGGAAAGCCGGCGAACGTGGCGAGAAAGGAAGGGAAGAAAGCGAAAGGAGCGGGCGCTAGGGCGCTGGCAAGTGTAGCGGTCACGCTGCGCGTAACCACCACACCCGCCGCGCTTAATGCGCCGCTACAGGGCGCGTACTATGGTTGCTTTGACGTATGCGGTGTGAAATACCGCACAGATGCGTAAGGAGAAAATACCGCATCAGGCGTCAGGTGGCACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTATGAGTATTCAACATTTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAACGCTGGTGAAAGTAAAAGATGCTGAAGATCAGTTGGGTGCACGAGTGGGTTACATCGAACTGGATCTCAACAGCGGTAAGATCCTTGAGAGTTTTCGCCCCGAAGAACGTTTTCCAATGATGAGCACTTTTAAAGTTCTGCTATGTGGCGCGGTATTATCCCGTATTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATACACTATTCTCAGAATGACTTGGTTGAGTACTCACCAGTCACAGAAAAGCATCTTACGGATGGCATGACAGTAAGAGAATTATGCAGTGCTGCCATAACCATGAGTGATAACACTGCGGCCAACTTACTTCTGACAACGATCGGAGGACCGAAGGAGCTAACCGCTTTTTTGCACAACATGGGGGATCATGTAACTCGCCTTGATCGTTGGGAACCGGAGCTGAATGAAGCCATACCAAACGACGAGCGTGACACCACGATGCCTGTAGCAATGGCAACAACGTTGCGCAAACTATTAACTGGCGAACTACTTACTCTAGCTTCCCGGCAACAATTAATAGACTGGATGGAGGCGGATAAAGTTGCAGGACCACTTCTGCGCTCGGCCCTTCCGGCTGGCTGGTTTATTGCTGATAAATCTGGAGCCGGTGAGCGTGGGTCTCGCGGTATCATTGCAGCACTGGGGCCAGATGGTAAGCCCTCCCGTATCGTAGTTATCTACACGACGGGGAGTCAGGCAACTATGGATGAACGAAATAGACAGATCGCTGAGATAGGTGCCTCACTGATTAAGCATTGGTAACTGTCAGACCAAGTTTACTCATATATACTTTAGATTGATTTAAAACTTCATTTTTAATTTAAAAGGATCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTCCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCTGCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGCCGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATACCAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCACCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAAGTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCGGGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAACTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGGCGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTCCAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAGCGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACGCGGCCTTTTTACGGTTCCTGGCCTTTTGCTGGCCTTTTGCTCACATAGTCATGCCCCGCGCCCACCGGAAGGAGCTGACTGGGTTGAAGGCTCTCAAGGGCATCGGTCGAGATCCCGGTGCCTAATGAGTGAGCTAACTTACATTAATTGCGTTGCGCTCACTGCCCGCTTTCCAGTCGGGAAACCTGTCGTGCCAGCTGCATTAATGAATCGGCCAACGCGCGGGGAGAGGCGGTTTGCGTATTGGGCGCCAGGGTGGTTTTTCTTTTCACCAGTGAGACGGGCAACAGCTGATTGCCCTTCACCGCCTGGCCCTGAGAGAGTTGCAGCAAGCGGTCCACGCTGGTTTGCCCCAGCAGGCGAAAATCCTGTTTGATGGTGGTTAACGGCGGGATATAACATGAGCTGTCTTCGGTATCGTCGTATCCCACTACCGAGATATCCGCACCAACGCGCAGCCCGGACTCGGTAATGGCGCGCATTGCGCCCAGCGCCATCTGATCGTTGGCAACCAGCATCGCAGTGGGAACGATGCCCTCATTCAGCATTTGCATGGTTTGTTGAAAACCGGACATGGCACTCCAGTCGCCTTCCCGTTCCGCTATCGGCTGAATTTGATTGCGAGTGAGATATTTATGCCAGCCAGCCAGACGCAGACGCGCCGAGACAGAACTTAATGGGCCCGCTAACAGCGCGATTTGCTGGTGACCCAATGCGACCAGATGCTCCACGCCCAGTCGCGTACCGTCTTCATGGGAGAAAATAATACTGTTGATGGGTGTCTGGTCAGAGACATCAAGAAATAACGCCGGAACATTAGTGCAGGCAGCTTCCACAGCAATGGCATCCTGGTCATCCAGCGGATAGTTAATGATCAGCCCACTGACGCGTTGCGCGAGAAGATTGTGCACCGCCGCTTTACAGGCTTCGACGCCGCTTCGTTCTACCATCGACACCACCACGCTGGCACCCAGTTGATCGGCGCGAGATTTAATCGCCGCGACAATTTGCGACGGCGCGTGCAGGGCCAGACTGGAGGTGGCAACGCCAATCAGCAACGACTGTTTGCCCGCCAGTTGTTGTGCCACGCGGTTGGGAATGTAATTCAGCTCCGCCATCGCCGCTTCCACTTTTTCCCGCGTTTTCGCAGAAACGTGGCTGGCCTGGTTCACCACGCGGGAAACGGTCTGATAAGAGACACCGGCATACTCTGCGACATCGTATAACGTTACTGGTTTCACATTCACCACCCTGAATTGACTCTCTTCCGGGCGCTATCATGCCATACCGCGAAAGGTTTTGCGCCATTCGATGGTGTCCGGGATCTCGACGCTCTCCCTTATGCGACTCCTGCATTAGGAAGCAGCCCAGTAGTAGGTTGAGGCCGTTGAGCACCGCCGCCGC
2.Vector Name: pGEX-4T-1
Vector Promoter: Tac
Vector Size: 4969 bp
Primer for 5' Sequencing : pGEX5': GGGCTGGCAAGCCACGTTTGGTG
Primer for 3' Sequencing: pGEX3': CCGGGAGCTGCATGTGTCAGAGG
Vector Tag: N-GST
Vector Resistance: Ampicillin
Note: The replication origin is pMB1.
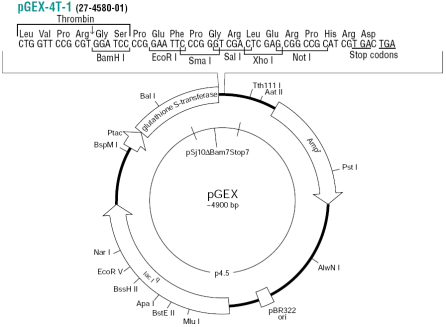
>pGEX4T-1
ACGTTATCGACTGCACGGTGCACCAATGCTTCTGGCGTCAGGCAGCCATCGGAAGCTGTGGTATGGCTGTGCAGGTCGTAAATCACTGCATAATTCGTGTCGCTCAAGGCGCACTCCCGTTCTGGATAATGTTTTTTGCGCCGACATCATAACGGTTCTGGCAAATATTCTGAAATGAGCTGTTGACAATTAATCATCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGTATTCATGTCCCCTATACTAGGTTATTGGAAAATTAAGGGCCTTGTGCAACCCACTCGACTTCTTTTGGAATATCTTGAAGAAAAATATGAAGAGCATTTGTATGAGCGCGATGAAGGTGATAAATGGCGAAACAAAAAGTTTGAATTGGGTTTGGAGTTTCCCAATCTTCCTTATTATATTGATGGTGATGTTAAATTAACACAGTCTATGGCCATCATACGTTATATAGCTGACAAGCACAACATGTTGGGTGGTTGTCCAAAAGAGCGTGCAGAGATTTCAATGCTTGAAGGAGCGGTTTTGGATATTAGATACGGTGTTTCGAGAATTGCATATAGTAAAGACTTTGAAACTCTCAAAGTTGATTTTCTTAGCAAGCTACCTGAAATGCTGAAAATGTTCGAAGATCGTTTATGTCATAAAACATATTTAAATGGTGATCATGTAACCCATCCTGACTTCATGTTGTATGACGCTCTTGATGTTGTTTTATACATGGACCCAATGTGCCTGGATGCGTTCCCAAAATTAGTTTGTTTTAAAAAACGTATTGAAGCTATCCCACAAATTGATAAGTACTTGAAATCCAGCAAGTATATAGCATGGCCTTTGCAGGGCTGGCAAGCCACGTTTGGTGGTGGCGACCATCCTCCAAAATCGGATCTGGTTCCGCGTGGATCCCCGGAATTCCCGGGTCGACTCGAGCGGCCGCATCGTGACTGACTGACGATCTGCCTCGCGCGTTTCGGTGATGACGGTGAAAACCTCTGACACATGCAGCTCCCGGAGACGGTCACAGCTTGTCTGTAAGCGGATGCCGGGAGCAGACAAGCCCGTCAGGGCGCGTCAGCGGGTGTTGGCGGGTGTCGGGGCGCAGCCATGACCCAGTCACGTAGCGATAGCGGAGTGTATAATTCTTGAAGACGAAAGGGCCTCGTGATACGCCTATTTTTATAGGTTAATGTCATGATAATAATGGTTTCTTAGACGTCAGGTGGCACTTTTCGGGGAAATGTGCGCGGAACCCCTATTTGTTTATTTTTCTAAATACATTCAAATATGTATCCGCTCATGAGACAATAACCCTGATAAATGCTTCAATAATATTGAAAAAGGAAGAGTATGAGTATTCAACATTTCCGTGTCGCCCTTATTCCCTTTTTTGCGGCATTTTGCCTTCCTGTTTTTGCTCACCCAGAAACGCTGGTGAAAGTAAAAGATGCTGAAGATCAGTTGGGTGCACGAGTGGGTTACATCGAACTGGATCTCAACAGCGGTAAGATCCTTGAGAGTTTTCGCCCCGAAGAACGTTTTCCAATGATGAGCACTTTTAAAGTTCTGCTATGTGGCGCGGTATTATCCCGTGTTGACGCCGGGCAAGAGCAACTCGGTCGCCGCATACACTATTCTCAGAATGACTTGGTTGAGTACTCACCAGTCACAGAAAAGCATCTTACGGATGGCATGACAGTAAGAGAATTATGCAGTGCTGCCATAACCATGAGTGATAACACTGCGGCCAACTTACTTCTGACAACGATCGGAGGACCGAAGGAGCTAACCGCTTTTTTGCACAACATGGGGGATCATGTAACTCGCCTTGATCGTTGGGAACCGGAGCTGAATGAAGCCATACCAAACGACGAGCGTGACACCACGATGCCTGCAGCAATGGCAACAACGTTGCGCAAACTATTAACTGGCGAACTACTTACTCTAGCTTCCCGGCAACAATTAATAGACTGGATGGAGGCGGATAAAGTTGCAGGACCACTTCTGCGCTCGGCCCTTCCGGCTGGCTGGTTTATTGCTGATAAATCTGGAGCCGGTGAGCGTGGGTCTCGCGGTATCATTGCAGCACTGGGGCCAGATGGTAAGCCCTCCCGTATCGTAGTTATCTACACGACGGGGAGTCAGGCAACTATGGATGAACGAAATAGACAGATCGCTGAGATAGGTGCCTCACTGATTAAGCATTGGTAACTGTCAGACCAAGTTTACTCATATATACTTTAGATTGATTTAAAACTTCATTTTTAATTTAAAAGGATCTAGGTGAAGATCCTTTTTGATAATCTCATGACCAAAATCCCTTAACGTGAGTTTTCGTTCCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTTTTTTCTGCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGTTTGTTTGCCGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAGAGCGCAGATACCAAATACTGTCCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAAGAACTCTGTAGCACCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGCTGCCAGTGGCGATAAGTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGATAAGGCGCAGCGGTCGGGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCGAACGACCTACACCGAACTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGCTTCCCGAAGGGAGAAAGGCGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGGAGAGCGCACGAGGGAGCTTCCAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCGGGTTTCGCCACCTCTGACTTGAGCGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGAGCCTATGGAAAAACGCCAGCAACGCGGCCTTTTTACGGTTCCTGGCCTTTTGCTGGCCTTTTGCTCACATGTTCTTTCCTGCGTTATCCCCTGATTCTGTGGATAACCGTATTACCGCCTTTGAGTGAGCTGATACCGCTCGCCGCAGCCGAACGACCGAGCGCAGCGAGTCAGTGAGCGAGGAAGCGGAAGAGCGCCTGATGCGGTATTTTCTCCTTACGCATCTGTGCGGTATTTCACACCGCATAAATTCCGACACCATCGAATGGTGCAAAACCTTTCGCGGTATGGCATGATAGCGCCCGGAAGAGAGTCAATTCAGGGTGGTGAATGTGAAACCAGTAACGTTATACGATGTCGCAGAGTATGCCGGTGTCTCTTATCAGACCGTTTCCCGCGTGGTGAACCAGGCCAGCCACGTTTCTGCGAAAACGCGGGAAAAAGTGGAAGCGGCGATGGCGGAGCTGAATTACATTCCCAACCGCGTGGCACAACAACTGGCGGGCAAACAGTCGTTGCTGATTGGCGTTGCCACCTCCAGTCTGGCCCTGCACGCGCCGTCGCAAATTGTCGCGGCGATTAAATCTCGCGCCGATCAACTGGGTGCCAGCGTGGTGGTGTCGATGGTAGAACGAAGCGGCGTCGAAGCCTGTAAAGCGGCGGTGCACAATCTTCTCGCGCAACGCGTCAGTGGGCTGATCATTAACTATCCGCTGGATGACCAGGATGCCATTGCTGTGGAAGCTGCCTGCACTAATGTTCCGGCGTTATTTCTTGATGTCTCTGACCAGACACCCATCAACAGTATTATTTTCTCCCATGAAGACGGTACGCGACTGGGCGTGGAGCATCTGGTCGCATTGGGTCACCAGCAAATCGCGCTGTTAGCGGGCCCATTAAGTTCTGTCTCGGCGCGTCTGCGTCTGGCTGGCTGGCATAAATATCTCACTCGCAATCAAATTCAGCCGATAGCGGAACGGGAAGGCGACTGGAGTGCCATGTCCGGTTTTCAACAAACCATGCAAATGCTGAATGAGGGCATCGTTCCCACTGCGATGCTGGTTGCCAACGATCAGATGGCGCTGGGCGCAATGCGCGCCATTACCGAGTCCGGGCTGCGCGTTGGTGCGGATATCTCGGTAGTGGGATACGACGATACCGAAGACAGCTCATGTTATATCCCGCCGTTAACCACCATCAAACAGGATTTTCGCCTGCTGGGGCAAACCAGCGTGGACCGCTTGCTGCAACTCTCTCAGGGCCAGGCGGTGAAGGGCAATCAGCTGTTGCCCGTCTCACTGGTGAAAAGAAAAACCACCCTGGCGCCCAATACGCAAACCGCCTCTCCCCGCGCGTTGGCCGATTCATTAATGCAGCTGGCACGACAGGTTTCCCGACTGGAAAGCGGGCAGTGAGCGCAACGCAATTAATGTGAGTTAGCTCACTCATTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCATGATTACGGATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGTAATAGCGAAGAGGCCCGCACCGATCGCCCTTCCCAACAGTTGCGCAGCCTGAATGGCGAATGGCGCTTTGCCTGGTTTCCGGCACCAGAAGCGGTGCCGGAAAGCTGGCTGGAGTGCGATCTTCCTGAGGCCGATACTGTCGTCGTCCCCTCAAACTGGCAGATGCACGGTTACGATGCGCCCATCTACACCAACGTAACCTATCCCATTACGGTCAATCCGCCGTTTGTTCCCACGGAGAATCCGACGGGTTGTTACTCGCTCACATTTAATGTTGATGAAAGCTGGCTACAGGAAGGCCAGACGCGAATTATTTTTGATGGCGTTGGAATT
Reference
Smith, D. B., & Johnson, K. S. (1988). Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene, 67(1), 31-40. DOI: 10.1016/0378-1119(88)90203-3
Prabu Gnanasekaran, Hanu R Pappu. (2023). Detecting Protein-Protein Interactions Using Glutathione S-Transferase (GST) Pull-Down Assay. Methods in Molecular Biology, DOI: 10.1007/978-1-0716-3327-4_10
Melnikov, A. D., Tsentalovich, Y. P., & Yanshole, V. V. (2019). Deep learning enables precise peak detection in high-resolution LC-MS data. Analytical Chemistry, 91(24), 15899-15906. DOI: 10.1021/acs.analchem.9b04811
Reiz, B., Kertesz-Farkas, A., Pongor, S., & Myers, M. P. (2012). Data preprocessing and filtering in mass spectrometry-based proteomics. Current Bioinformatics, 7(2), 96-108. DOI: 10.2174/157489312800604363
Dai, H., Xu, A., & Sun, W. (2016). Signal Denoising Method Based on Improved Singular Spectrum Analysis. Beijing Institute of Technology. DOI:10.15918/j.tbit1001-0645.2016.07.013
Wissmueller, B., Boeddrich, A., & Erdmann, H. S. (2011). The yeast two-hybrid system and beyond: exploring and exploiting protein-protein interactions. ChemMedChem, 6(2), 290-306. DOI: 10.1002/cmdc.201000329
Smith, D. B., & Johnson, K. S. (1988). Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene, 67(1), 31-40. DOI: 10.1016/0378-1119(88)90203-3
Wang, S., Huang, Z., Liu, Y., Shao, S., Li, L., & Ma, M. (2024). Application of the Nicotiana Allergic Necrosis Assay for the Validation of Protein-Protein Interactions between Fungal Effectors and Plant Receptor Kinases. Bio-protocol Preprint. bio-protocol.org/prep2729. DOI: 10.21769/BioProtoc.2729
XU CY (2020).Pull-down and Co-immunoprecipitation assays of interacting proteins in plants.Chin Bul Bot 55,62-68.
Results interpretation:
1.
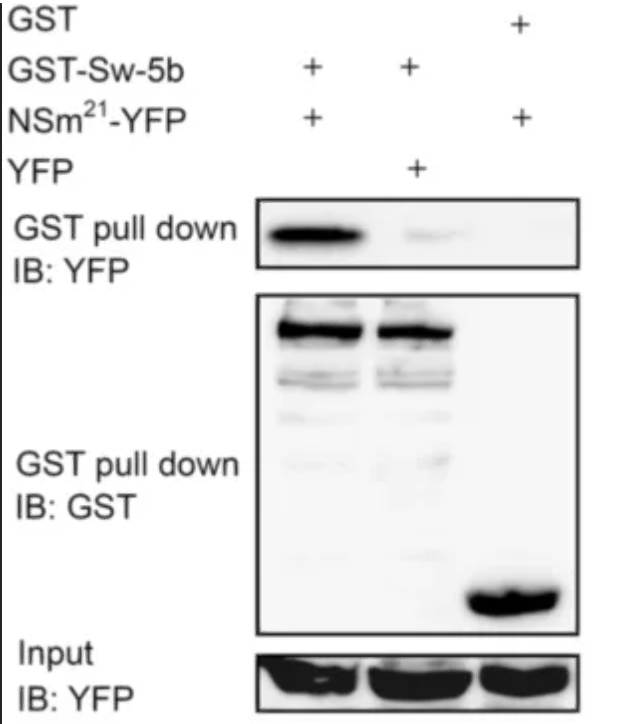
GST Pull-Down with YFP Immunoblot (IB: YFP):
A band is present in the GST-Sw-5b lane, indicating that the GST-Sw-5b fusion protein has pulled down YFP.
No band is present in the GST lane, suggesting that GST alone does not interact with YFP or the interaction is too weak to be detected by this assay.
A band is present in the NSm^21^-YFP lane, indicating that the NSm^21^-YFP fusion protein is present and can be detected by the YFP antibody.
GST Pull-Down with GST Immunoblot (IB: GST):
Bands are present in both the GST-Sw-5b and NSm^21^-YFP lanes, confirming that the GST fusion proteins are present in the pull-down complexes.
The presence of GST in both lanes serves as a positive control for the pull-down assay.
Input Control with YFP Immunoblot (IB: YFP):
Bands are present in all lanes, indicating that the YFP fusion proteins were present in the input samples before the pull-down assay.
This control confirms that the YFP fusion proteins were expressed and available for the pull-down.
Conclusion:
The GST-Sw-5b fusion protein interacts with YFP, as evidenced by the presence of YFP in the GST pull-down assay.
GST alone does not appear to interact with YFP, as no band is detected in the corresponding lane.
The NSm^21^-YFP fusion protein is present in the sample but does not show a significant interaction with GST in this assay, as no band is detected in the GST pull-down lane for YFP.
2.
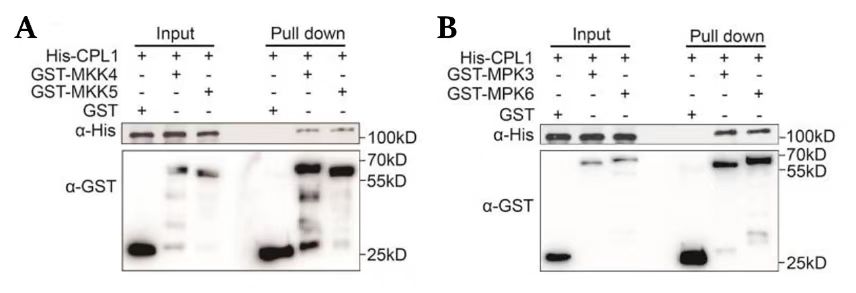
Panel A:
Experimental Design: The assay includes His-CPL1 as a control and two experimental conditions with GST-MKK4 and GST-MKK5.
Input: The presence of His-CPL1, GST-MKK4, and GST-MKK5 is confirmed in the input lanes, indicating that the proteins were expressed and present before the pull-down.
Pull-Down:
His-CPL1 is pulled down by GST-MKK4 but not by GST-MKK5, as indicated by the presence of the His-CPL1 band in the GST-MKK4 pull-down lane and its absence in the GST-MKK5 pull-down lane.
The GST-MKK4 and GST-MKK5 bands are present in their respective pull-down lanes, confirming the successful immobilization of the GST fusion proteins on the beads.
Panel B:
Experimental Design: The assay includes His-CPL1 as a control and two experimental conditions with GST-MPK3 and GST-MPK6.
Input: The presence of His-CPL1, GST-MPK3, and GST-MPK6 is confirmed in the input lanes.
Pull-Down:
His-CPL1 is pulled down by both GST-MPK3 and GST-MPK6, as indicated by the presence of the His-CPL1 band in both pull-down lanes.
The GST-MPK3 and GST-MPK6 bands are present in their respective pull-down lanes, confirming the successful immobilization of the GST fusion proteins.
Conclusion:
In Panel A, His-CPL1 interacts with GST-MKK4 but not with GST-MKK5.
In Panel B, His-CPL1 interacts with both GST-MPK3 and GST-MPK6.
The GST control in Panel A shows no interaction with His-CPL1, which is expected as it is only a tag without the protein of interest.
3.
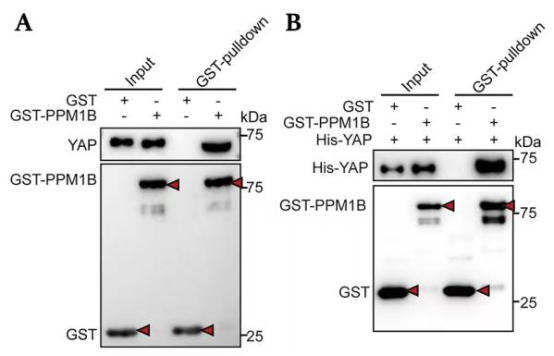
Panel A:
Experimental Design: The assay includes GST alone and GST-PPM1B as bait proteins, with YAP as the prey protein.
Input: The presence of GST, GST-PPM1B, and YAP is confirmed in the input lanes, indicating that the proteins were expressed and present before the pull-down.
Pull-Down:
YAP is pulled down by GST-PPM1B but not by GST alone, as indicated by the presence of the YAP band in the GST-PPM1B pull-down lane and its absence in the GST pull-down lane.
The GST-PPM1B band is present in its pull-down lane, confirming the successful immobilization of the GST fusion protein on the beads.
Panel B:
Experimental Design: The assay includes GST alone and GST-PPM1B as bait proteins, with His-YAP as the prey protein.
Input: The presence of GST, GST-PPM1B, and His-YAP is confirmed in the input lanes.
Pull-Down:
His-YAP is pulled down by GST-PPM1B but not by GST alone, as indicated by the presence of the His-YAP band in the GST-PPM1B pull-down lane and its absence in the GST pull-down lane.
The GST-PPM1B band is present in its pull-down lane, confirming the successful immobilization of the GST fusion protein.
Conclusion:
In both panels A and B, YAP (or His-YAP) interacts with GST-PPM1B but not with GST alone.
The results suggest a specific interaction between YAP and GST-PPM1B, which can be further investigated for their biological significance.
4.
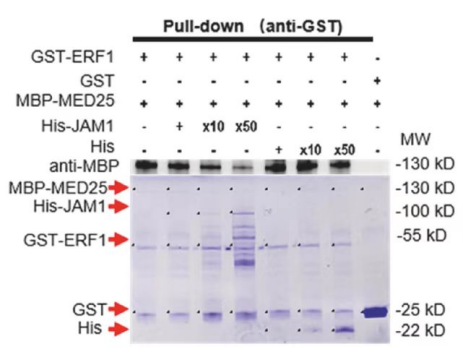
GST-ERF1:
A band is present in the GST-ERF1 lane, indicating that GST-ERF1 is successfully pulled down.
The band is not present in the GST lane, suggesting that GST alone does not interact with the prey proteins or the interaction is too weak to be detected.
MBP-MED25:
A band is present in the MBP-MED25 lane, indicating that MBP-MED25 is successfully pulled down.
The band is not present in the GST lane, similar to the GST-ERF1 result.
His-JAM1:
Bands are present in the His-JAM1 lanes at different dilutions (x10 and x50), indicating that His-JAM1 is pulled down and its presence is detected by the anti-His antibody.
The presence of His-JAM1 in the pull-down suggests an interaction with the GST fusion proteins, although the specific interacting partner is not clear from this assay alone.
Conclusion:
GST-ERF1 and MBP-MED25 are both pulled down, indicating they may interact with one or more of the prey proteins in the assay.
His-JAM1 is also pulled down, suggesting it interacts with the GST fusion proteins, but further analysis is needed to determine the specific interacting partner(s).
5.
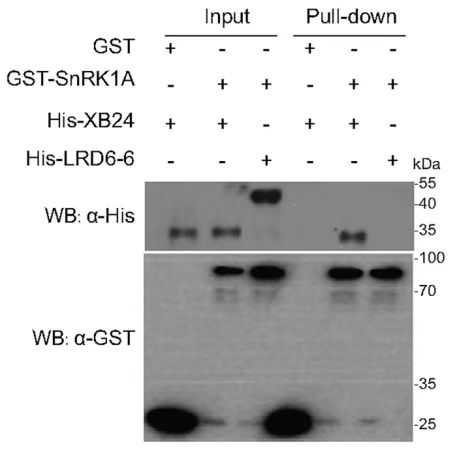
Input Lanes:
All proteins (GST, GST-SnRK1A, His-XB24, His-LRD6-6) are present in the input lanes, indicating successful expression and loading of the proteins.
Pull-Down Lanes:
GST-SnRK1A: A band is present in the pull-down lane when GST-SnRK1A is used as bait, indicating that GST-SnRK1A interacts with one or more of the prey proteins (His-XB24 or His-LRD6-6).
His-XB24: A band is present in the pull-down lane when His-XB24 is used as bait, suggesting an interaction with GST or GST-SnRK1A.
His-LRD6-6: A band is present in the pull-down lane when His-LRD6-6 is used as bait, indicating an interaction with GST or GST-SnRK1A.
Western Blot (WB) Analysis:
α-His: The Western blot using an anti-His antibody shows bands corresponding to His-XB24 and His-LRD6-6 in their respective pull-down lanes, confirming the successful pull-down of these proteins.
α-GST: The Western blot using an anti-GST antibody shows bands corresponding to GST and GST-SnRK1A in their respective pull-down lanes, confirming the successful pull-down of these proteins.
Conclusion:
GST-SnRK1A interacts with both His-XB24 and His-LRD6-6, as indicated by the presence of bands in the pull-down lanes.
His-XB24 and His-LRD6-6 also show interactions with GST or GST-SnRK1A, as indicated by the bands in their respective pull-down lanes.
- Wang, S, Dong, J, Wang, S, Shi, X, Wang, S and Li, L(2024). The method for in vitro screening of unknown interacting proteins using GST Pull-down MS and the method for validating protein interactions using pGEX4T-1 and pColdTF vectors based on the principle of GST Pull-down. Bio-protocol Preprint. 10.21769/p2750.
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.