
- Home
- Protocols

-

m6A-RIP-qPCR
Last updated date: Sep 24, 2023 Views: 872 Forks: 0
MeRIP-qPCR
Yang lab protocol
⚫ Material and Reagents:
DEPC treated H2O: add 1 ml DEPC (Amresco) in 1,000 ml miliQ H2O, Mix and incubate at RT for 4 h, autoclave.
1M Tris-HCl (pH 7.4) (100ml): weigh 12.11g Tris powder, dissolve in 90 ml DEPC H2O, adjust pH to 7.4 with HCl, then add DEPC H2O to make the volume 100 ml.
5M NaCl (100ml): dissolve 29.22 g NaCl in 80 ml DEPC H2O, then add DEPC H2O to make the final volume 100 ml.
10% NP-40 (50ml): dilute 5 ml NP-40 solution in 45 ml DEPC H2O, mix throughly on the shaker at RT.
RNasin (Promega, N2515, 10,000U)

m6A antibody (Synaptic systems, 202003, 1μg/μl)
Dynabeads Protein A for Immunoprecipitation (Thermo, 10001D)
Nuclease Free water (Ambion, AM9937)
Dynabeads® mRNA Purification Kit (Ambion, 61006)
DNase I (NEB, M0303, 2 U/μl)
RNA Fragmentation Reagents (Ambion, AM8740)
N6-methyladenosine (Berry & Associates, P3732)
Acid Phenol: Chloroform (pH 4.3-4.7) (Ambion, AM9720)
Glycogen (20mg/ml) (Thermo, #R0551)
3M Sodium Acetate, pH5.5 (Ambion, AM9740)
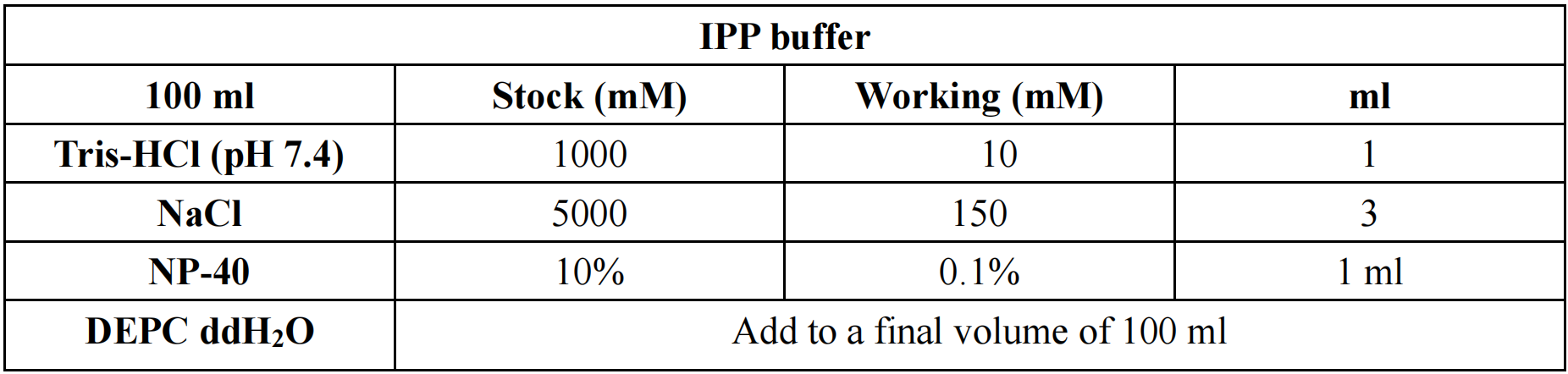
IPP buffer:
⚫ Procedure:
Day1.
1. mRNA preparation with the Ambion Dynabeads mRNA purification kit
1.1 Prepare RNA
- Adjust the volume of your 75 μg total RNA to 100 μl with distilled DEPC-treated water, or with 10 mM Tris-HCl, pH 7.5. Omit this step if only a small adjustment is needed (see also step 3 under "Prepare Dynabeads®").
- Heat to 65°C for 2 min to disrupt secondary structures. Place on ice.
1.2 Prepare Dynabeads®
- Transfer 200 μl (1 mg) of well resuspended Dynabeads® to a microcentrifuge tube. Place the tube on the magnet for 30 s, or until all Dynabeads® have migrated to the tube wall.
- Discard the supernatant, remove the tube from the magnet, and add 100 μl Binding Buffer to calibrate the beads. Put the tube back on the magnet and remove the supernatant. Remove the tube from the magnet.
- Add 100 μl Binding Buffer to the Dynabeads®. Optimal hybridization conditions are obtained in Binding Buffer added in a 1:1 ratio relative to sample volume. If your total RNA is more dilute than 75 μg/100 μl, then simply add an equal volume of Binding Buffer to the Dynabeads®.
1.3 Isolate mRNA
- Add the total RNA to the Dynabeads®/Binding Buffer suspension. Mix thoroughly, and rotate on a roller or mixer for 10 min at room temperature to allow mRNA to anneal to the oligo (dT) on the beads.
- Place the tube on the magnet until solution is clear. Remove the supernatant.
- Remove the tube from the magnet and wash the mRNA-bead complex twice with 200 μL Washing Buffer B. Remove all the supernatant between each washing step with the help of the magnet (this is important because washing can remove the ribosome RNA, so when you washing you need to pipette the sample with the tipe several times, at least 15 times). (这步非常重要,因为在洗的过程中用枪头多吹打几次可以洗去核糖体 RNA,所以至少每次洗 都应吹打 15 次左右)
- Add 100 μl 10 mM Tris-HCl, pH 7.5 to elute. Heat to 70 °C for 2 min and place the tube immediately on the magnet.
- Transfer the eluted mRNA to a new RNase-free tube.
1.4 Rebound RNA
- Rewash the beads with 100ul binding buffer and 100 μl 10 mM Tris-HCl, pH 7.5 (1:1), pipette several times. Place the tube on the magnet for 30 sec, or until all Dynabeads® have migrated to the tube wall. Discard the supernatant.
- Add 100 μl Binding Buffer and your sample (1:1) to the beads. Mix thoroughly, and rotate on a roller for 10 min at room temperature to allow mRNA to anneal to the beads.
- Remove the tube from the magnet and wash the mRNA-bead complex twice with 200 μl Washing Buffer B. Remove all the supernatant between each washing step with the help of the magnet (this is important because washing can remove the ribosome RNA, so when you washing you need to pipette the sample with the tipe several times, at least 15 times). (这步非常重要,因为在洗的过程中用枪头多吹打几次可以洗去核糖体 RNA,所以至少每次洗都应吹打 15 次左右)
- If elution is required, add the desired amount (10–20 μl, or down to 5 μl) of 10 mM Tris-HCl, pH 7.5. Heat to 70°C for 2 min and place the tube immediately on the magnet.
- Transfer the eluted mRNA to a new RNase-free tube.
(The procedures marked in RED are very important to in case the rRNA contamination.)
Note: The following are the components of individual buffer of the mRNA purification kit.
Binding Buffer 4 ml
20 mM Tris-HCl (pH 7.5)
1.0 M LiCl
2 mM EDTA
Washing Buffer B 4 ml
10 mM Tris-HCl (pH 7.5)
0.15 M LiCl
1 mM EDTA
10 mM Tris-HCl (pH 7.5) 4 ml
1.5 DNase I digestion
- Add 5 μl 1x DNase I buffer and 1-3ul DNase I (NEB, M0303) (the volume and amount of DNase I can be ajusted according to the volume of the RNA) to the purified mRNA with the final volume of 50ul, and incubate at 37℃ for 10 min in order to digest the potential DNA contamination.
- Add 1 μl glycogen, 5 μl 3 M NaOAc (pH5.5) (to 10 fold dilution), and 190 μl pure ethanol, and incubate overnight at -80℃.
Day2.
2.RNA fragmentation and purification
- mRNA fragments in pure ethanol need to be precipitated by centrifuging at 4℃,14,800 rpm for 40min.
- Wash RNA pellet once with 1 ml 70% ethanol.
- Spin at 4℃ 14,800 rpm for 10min.
- Discard the supernatant.
- Spin at 14,800 rpm for 1min to remove the rest ethanol, taking care not to disrupt RNA pellet, which is easily visible because of the presence of glycogen. (Usually, 60%-80% mRNA could be recovered after centrifugation.)
- Let the pellet air-dry for 10 min on ice.
- Resolve the RNA pellet with 20 μl nuclease free water.
- Dilute 0.5 μl fragmented RNA with 0.5 l nuclease free water to measure the RNA concentration with NanoDrop spectrophotometer.
- Leave 100~200 ng mRNA (about ~100nt) as Input.
(Note: In our study, we use normal RNA-seq data (double-end, 200~300 bp) as Input to call m6A peaks).
10. For 100 nt size, use Ambion kit AM8740 to react at 90℃ for 1 min, 10 μl/tube (no more than 2 μg/tube).
11. Pool the tubes together for 50 μl.
(Note: For m6A-IP, at least 1 μg of mRNA was used.)
12. Add 1 μl glycogen, 5 μl 3 M NaOAc (pH5.2) (to 10 fold dilution), and 190 μl pure ethanol, and incubate overnight at -80℃.
Day3.
3.m6A-mRNA pull down
- mRNA fragments in pure ethanol need to be precipitated by centrifuging at 4℃,14,800 rpm for 40min.
- Wash RNA pellet once or twice with 1 ml 70% ethanol.
- Spin at 4℃ 14,800 rpm for 10min.
- Discard the supernatant.
- Spin at 14,800 rpm for 1 min to remove the rest ethanol, taking care not to disrupt RNA pellet, which is easily visible because of the presence of glycogen. (Usually, 60% - 80% mRNA could be recovered after centrifugation.)
- Let the pellet air-dry for 10min on ice.
- Resolve the RNA pellet with 20 μl nuclease free water.
- Dilute 0.5 μl fragmented RNA with 0.5 μl nuclease free water to measure the RNA concentration with NanoDrop spectrophotometer.
4. Preparation of Dynabeads Protein A for Immunoprecipitation.
- Use large-orifice tips or cut off the end of regular micropipette tips for transferring 40 μl beads (5 μg mRNA == 10 μg m6A-Ab == 40 μl beads)
(Note: The bindign capacity of dynabeads protein A is8 μg human IgG/mg beads. Dynabeads protein A contains 30 mg Dynabeads/ml; 8μg antibody equals 33 μl beads).
2. Wash beads three times in 1 ml 1x IPP buffer for 3 min with gentle rotation at R.T.
3. Discard the supernatant.
4. Resuspend the beads in 500 μl 1x IPP buffer with 0.5 μl RNasin. Incubate 10 μg anti-m6A polyclonal antibody with protein A beads for 1 h with gentle rotation at R.T.
5. Immunoprecipitation
- Fragmented RNA in nuclease free water was denatured by heating at 75°C for 5 min, and then cooled on ice for 2-3 min.
- 5 μg fragmented RNA was added into the beads-antibody mixture, and incubated for 4 h with gentle rotation at 4ºC.
6. Elution of m6A-containing mRNA
- Spin down and collect the beads, transfer the supernatant to a new 1.5 ml tube. If the RIP-qPCR is needed, then do step 2. Otherwise, go to step 3 directly.
- (For qPCR) Take 150 ul supernatant to extract unbinding RNA by Trizol and chloroform for binding efficiency verification. The rest supernatant is stored in 80℃ freezer. (In my experiment I add 1ml Trizol per 150 μl supernatant, vortex. Incubate on ice for 5 min. Add 200 μl chloroform to each sample, vortex. Put on ice and incubate for 5 min. Centrifuge at 14,800 rpm for 5 min at 4°C. Transfer the supernatant to a new 1.5 ml tube. Add 3 volumes of ethanol. Incubate the mixture at -80°C for more than 1 h. Then extract RNA.)
- Wash beads three times with 1 ml 1x IPP buffer for 2 min with gentle rotation at RT.
- The beads were eluted with 300 μl 0.5 mg/ml N 6 -Methyladenosine in IPP buffer (with RNasin) with gentle rotation at RT for 1 h.
- Repeat step 4 once more.
- Combine the two eluates, add equal volume (600 μl) of Acid Phenol (pH 4.3-4.7) into the eluates, and vortex thoroughly, incubate on ice for 5min.
- Centrifuge at 14,800 rpm for 15 min at 4°C, transfer the supernatant to two new 1.5 ml tubes (300 μl/tube).
- (It is strongly recommended to) Add 30 μl 3 M NaOAc (pH5.5), vortex to mix thoroughly.
- Add 20 μg of glycogen in total and vortex to mix throughly.
(In my experiments, 1 ul for each tube, and totally 2 ul for each sample).
10. Add 3 volumes (990 μl) of pure ethanol to each tube, incubate the mixture at -80°C for more than 1 h. (The longer the better, usually overnight).
Day4.
- Centrifuge the mixture for 30 min at 14,800 rpm. Discard the supernatant.
- Wash the pellet with 1 ml cold 75% ethanol. Then the samples can be used for direct library construction or put into 95% ethanol for longer preservation.
7. RNA quantification and qRT-PCR
- Dissolve the flow-through (FL) RNA and IP-RNA in 11 μl of DEPC-treated Water, take 0.5 μl RNA samples and dilute it with 0.5 μl nuclease-free water for RNA quantification.
- Take equal amount of Input-RNA, FL-RNA, and IP-RNA from control and treated samples to do RT, and take equal volume of cDNA to do PCR.
- Design primers for specific genes around the m6A peaks identified from MeRIP-seq.
- The relative enrichment of IP compared to Input is calculated according to the 2^(delta (delta Ct)) method.
- Ye, L, Yang, Y and Yu, S(2023). m6A-RIP-qPCR. Bio-protocol Preprint. bio-protocol.org/prep2427.
- Yao, Y., Yang, Y., Guo, W., Xu, L., You, M., Zhang, Y., Sun, Z., Cui, X., Yu, G., Qi, Z., Liu, J., Wang, F., Liu, J., Zhao, T., Ye, L., Yang, Y. and Yu, S.(2021). METTL3-dependent m6A modification programs T follicular helper cell differentiation. Nature Communications 0(0). DOI: 10.1038/s41467-021-21594-6
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.