
- Home
- Protocols

-

In-situ hybridization for mouse brain sections

Last updated date: Nov 3, 2021 Views: 1282 Forks: 0
In-situ hybridization for mouse brain sections
Yue Ying1, Jinyun Wu1, *, Miao He1, *
Institutes of Brain Sciences, Fudan University, Shanghai 200032, China
*For correspondence:19211520004@fudan.edu.cnhem@fudan.edu.cn
Abstract
In-situ hybridization (ISH) is a method for qualitative analysis of RNA expression. During ISH, labelled nucleic acid probe binds to the specific RNA through complementary base pairing, forming a hybrid molecule. The molecule label on the probe can then be recognized by its antibody, and through a chromogenic reaction, the expression of the target RNA can be directly observed on the tissue section. This protocol will introduce how to perform ISH on mouse brain sections using Digoxin (DIG)-labelled nucleic acid probes.
Key words: In-situ-hybridization, complementary base pairing, antigen antibody combination, chromogenic reaction
Materials and Reagents:
- Deionized water (Milipore)
- Diethylpyrocarbonate (DEPC)-treated deionized water
- 4% paraformaldehyde (PFA)
- DEPC treated phosphate buffer saline (PBS) (pH 7.4-7.5)
- 20%, 30% sucrose (Invitrogen, catalog number:15503-022) solution (prepared with DEPC-treated 1×PBS)
- OCT embedding medium (SAKURA, catalog number: 4583)
- Hydroxymethyl aminomethane (Tris) (BBI, catalog number: A600194-0500)
- Hydrochloric acid (HCl) (Sangon, catalog number: 10011018)
- Triethylamine alcohol (TEA) (1M) (VETEC, catalog number: V900257)
- Proteinase K (ThermoFisher,catalog number: 25530-049)
- Proteinase K buffer (see Recipes)
- Anti-DIG alkaline phosphatase (Anti-DIG-AP) (Roche, catalog number: 11093275910)
- RNA probe
- 100% Methanol (Hushi, catalog number: 10014118)
- Mounting Medium (Beyotime, catalog number: P0126)
- Saline Sodium Citrate (SSC) buffer (pH7.0)
- Hybridization solution (see Recipes)
- Buffer B1 (see Recipes)
- Buffer B2 (see Recipes)
- Buffer B3 (see Recipes)
- Buffer B4 (store in the dark) (see Recipes)
Equipment
- Cryostat microtome (Leica, catalog number: CM1950) (Figure 1A)
- Anti-Roll Plate (Leica, 14047742497)
- Measuring cylinder (Shenbo)
- Hybridization oven (UVP, catalog number: HL-2000) (Figure 1B)
- Microcentrifuge (Thermo Fisher, catalog number:75002420) (Figure 1C)
- Dry bath incubator (Allsheng, catalog number: MK2000-2E) (Figure 1D)
- Orbital shaker (QILINBEIER, catalog number: TS-1) (Figure 1E)
- Magnetic hotplate stirrer (SCILOGEX, catalog number: MS7-H550-Pro)
- Water bath (Jinghong, DK-S24 )
- Microscope cover glass (Fisherbrand, catalog number: 12-544-E) (Figure 2A)
(Or Electron Microscopy Sciences, catalog number: 70329-60; Or LifterSlip, catalog number: 100499-626) - Microscope slides (Fisherbrand, catalog number: 12-550-15) (Figure 2B)
- Hydrophobic barrier PAP pen (ImmEdge, catalog number: H-4000) (Figure 2C)
- Hybridization box (LOCK&LOCK) (with sponge inside) (Figure 2D)
- Deionized water box (LOCK&LOCK) (with sponge inside) (Figure 2E)
- Plastic staining box (Sangon, E678012) (multiple slides), or 50 ml centrifuge tube (Thermo Fisher,339652) (no more than two slides) (Figure 2F, G)
- Incubator (Jinghong, catalog number: SHP-250)


Procedure
Note: During step 1 and 2, all reagents should be RNA-free. See Recipes. Masks, lab coats and gloves should be put on to avoid RNase contamination.
A. Preparations for ISH (tissue section)
- Before ISH, tissue should be fixed in advance . Embryonic tissue or small tissue should be immersed in 4% PFA at 4℃ overnight. Whereas for larger grown-up mouse tissue, such as brain tissue, it should be perfused by 1×PBS and 4% PFA, and then fixed by 4% PFA at 4℃ overnight.
- After fixation, the tissue sample should be transferred to a centrifuge tube with 1×PBS in it. Then it should be rinsed for 3 times, in order to wash off PFA.
- Dehydrate the tissue with 20% sucrose solution (for about one day). When the tissue sample sinks, it should be transferred to 30% sucrose. Wait for another one to two days until the sample sinks again.
- Dry the sucrose-sunk tissue sample and place it in a size-suitable mold. Slowly add OCT into the mold to embed the sample. Make sure the tissue is completely immersed in OCT.
- Quickly store the sample at -80℃ until tissue sectioning, or directly perform tissue sectioning.
- Before tissue sectioning, freeze the tissue by keeping it inside the cryostat for at least 30 mins. The temperature for tissue sectioning is normally -20℃.
- Perform tissue sectioning in the cryostat microtome. The recommended thickness is 12-22um. The sections should be smoothed flat and transferred to the slide.
- Before ISH, the slide should be rewarmed and dried at room temperature for 2 hours, and be dried at 50℃ for 15 mins. The above procedures prevent the sections from detaching the slide. As for slides not for use immediately, 1-hour drying at room temperature is required, and these slides should be stored at -80℃. Before ISH, such slides should be dried for 1 hour at room temperature and 15mins at 50℃.
B. The first day of ISH
Note: the plastic staining boxes (or centrifuge tubes) need to be washed by deionized water, dried in an incubator under 60℃, and washed for 3 times by deionized water before being used. Measuring cylinder and microscope cover glasses should be put into the 180℃ incubator in advance.
- Prepare 3×(200ml 1×PBS) ①②③, PK buffer, 1×TAE, 4%PFA in the staining box. (If centrifuge tubes are used instead, 25ml each would be sufficient.)
- Dry the slides for 15-30 mins at 50℃.
- At room temperature, fix the slides by immersing them in 4% PFA for 20 mins.
- At room temperature, rinse the slides by immersing them in 1×PBS①, then in 1×PBS②, each for 5 mins.
- At room temperature, immerse the slides in PK buffer, which contains proteinase K, for 10 mins.
- At room temperature, rinse the slides by immersing them in 1×PBS②.
- At room temperature, fix the slides by immersing them in 4% PFA for 10 mins.
- At room temperature, rinse the slides with 1×PBS①, then with 1×PBS②, each for 5 mins.
- At room temperature, immerse the slides in 0.1M TEA for 10 mins.
Note: a magnetic stirrer can be used at proper rotate speed. Orbital shaker can also be applied. - At room temperature, rinse the slides with 1×PBS②, then with 1×PBS③ for 5mins.
- At room temperature, take the slides out, wipe the water off the back of the slides. Gently tap at the margin of the slides to remove the residual PBS from the tissue. Place the slides flatwise in the hybridization box. Instill the pre-hybridization solution onto the slide until the tissue is immersed. (Perform the above procedures quickly to keep the sections moist). Cover the lid and pre-hybridize for 1 hour (Figure 3).
- Add the RNA probe into the hybridization solution. The probe’s concentration is 0.1-0.2ng/uL. Treat the solution at 85℃ for 5 minutes using the dry bath incubator, so as to ensure that the probe is linear. Then quickly cool it with ice-water-mixture. Mix well and centrifuge.
- Use the pipette to replace the pre-hybridization solution with hybridization solution. Approximately 100ul hybridization solution is required for each slide. Place the cover glass, or else the solution will be dried during incubation (Figure 4). Incubate overnight (12-16 hours) in the hybridization oven at 65℃.

(A). Lay the slide in the hybridization box. (B). Instill the pre- hybridization solution onto the slide until it immerses the tissue.

(A). Replace the pre-hybridization solution with hybridization solution, and place the cover glass on the top. (B). An enlarged view of (A), showing the position of the cover glass.
C. The second day of ISH
- Prepare 4× (200ml 0.2×SSC) and 2×200ml buffer B1
- Warm 3× (200ml 0.2×SSC) to 65℃. Set the slides upright and let the cover glass fall off. Immerse the slide in the 3 heated 0.2×SSC, each for 20 mins. (Perform the above procedures quickly to keep the sections moist).
- At room temperature, transfer the slide to the fourth 0.2×SSC. Rinse the slide for 5 mins.
- At room temperature, rinse the slide with buffer B1 twice, each for 5 mins.
- Wipe off the remaining solution. Use the hydrophobic barrier pen to draw a circle around the tissue. (Perform the above procedures quickly to keep the sections moist). Lay the slide flatwise. Add approximately 200ul buffer B2 onto the slide and make sure the tissue is immersed. Transfer the slide to the water box and keep the box closed. This blocking process takes 1 hour (Figure 5).
- Use a pipette to remove buffer B2 from the slide. Add buffer B2+anti-DIG-AP(1:5000) onto the slide (about 200 ul for each slide). Put the slide in the water box and keep it closed. Incubate overnight at 4℃.

(A). The hydrophobic circle around the tissue. (B) Add buffer B2 in the hydrophobic circle. Put the slide into the water box.
D. The third day of ISH
- Prepare 3×(200ml buffer B1) and 2×(200ml buffer B3)
- At room temperature, rinse the slide in buffer B1 for 3 times, 20 mins for each time.
- At room temperature, rinse the slide in buffer B3 twice, 5-10 mins for each time.
- Add 200ul buffer B4 onto the slide.
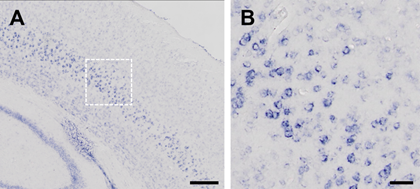
- Place all slides flatwise in the water box. Set the incubator at 28℃ and put the box inside. Time for incubation varies according to the age of the sample, the type of the tissue and the type of the probe. The reaction should be terminated if the signal is distinct and is not being enhanced over time (Figure 6).
- At room temperature, immerse the slide in deionized water to terminate the reaction.
Note: if necessary, the slide can be immersed in 100% methanol for 15-30mins to reduce background. - Mount slides with mounting medium.
(A). The hydrophobic circle around the tissue. (B) Add buffer B2 in the hydrophobic circle. Put the slide into the water box.
Recipes
1. DEPC-treated deionized water (store at room temperature)
Add 0.1%DEPC into deionized water. Mix well, store it in the dark overnight, and sterilize it.
2. DEPC-treated 10×PBS (store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 1L solution requires |
| Sodium chloride | NaCl | 58.44 | 1.54 M | 90g |
| Disodium Phosphate heptahydrate | Na2HPO4·7H2O | 268.07 | 0.03 M | 7.95g |
| Potassium dihydrogen phosphate | KH2PO4 | 136.09 | 0.01 M | 1.44g |
Add 0.1% DEPC, mix well, store in the dark overnight, and sterilize it.
3. 4%PFA (store at -20℃)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 500mL solution requires |
| DEPC-treated 10×PBS | \ | \ | 1× | 50mL |
| PFA | (HCHO)n | (30.03)×n | 4% | 20g |
| DEPC-treated deionized water | H2O | 18 | \ | Fill to 500mL |
Heat it to 65℃. Filter after PFA is completely dissolved. Store at -30℃.
4.1M Triethanolamine (pH=8.0, store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 500mL solution requires |
| Triethanolamine | (HOCH2CH2)3N | 149.19 | \ | 66.5mL |
| DEPC-treated deionized water | H2O | 18 | \ | 413.5mL |
| Hydrochloric acid | HCl | 36.46 | 12 M | 20mL |
5.Hybridization/Pre-hybridization solution (store at -20℃)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 100mL solution requires |
| Formamide | HCONH2 | 45.04 | 50% | 50mL |
| SSC buffer | C6H5O7Na3 | 258.07 | \ | 25mL 20×SSC |
| Yeast RNA | \ | \ | 250g/L | 500uL 50mg/mL yeast RNA |
| Denhardt's solution | \ | \ | \ | 10ml 50×Denhart’s solution |
| Herring sperm DNA | \ | \ | 500g/L | 5mg 10mg/mL herring sperm DNA |
| DEPC-treated deionized water | H2O | 18 | \ | 9.5mL |
6. DEPC-treated 0.5M EDTA (pH=8.0, store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 200mL solution requires |
| EDTA | C10H16N2O8 | 292.24 | \ | \ |
| Sodium hydroxide | NaOH | 40 | \ | Adjust pH to 8.0 |
Add 0.1% DEPC, mix well, store in the dark overnight, and sterilize it.
7. DEPC-treated 1M Tris-HCl (pH=7.4-7.5, store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 500mL solution requires |
| Tris | (HOCH2)3CNH2 | 121.14 | 1M | 60.57g |
| Hydrochloric acid | HCl | 36.46 | \ | Adjust pH to 7.4-7.5 |
| Deionized water | H2O | 18 | \ | Fill to 500mL |
Add 0.1% DEPC, mix well, store in the dark overnight, and sterilize it.
8. 1M Tris-HCl(pH=9.5, store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 500mL solution requires |
| Tris | (HOCH2)3CNH2 | 121.14 | 1M | 60.57g |
| Sodium hydroxide | NaOH | 40 | \ | Adjust pH to 9.5 |
| Deionized water | H2O | 18 | \ | Fill to 500mL |
9.20×SSC (pH=7.0, store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 500mL solution requires | |
| Sodium hydroxide | NaCl | 58.44 | 3M | 87.66g | |
| SSC | C6H5O7Na3 | 258.07 | 0.3 M | 38.71g | |
| Hydrochloric acid | HCl | 36.46 | \ | Adjust pH to 7.0 | |
| Deionized water | H2O | 18 | \ | Fill to 500mL | |
10. 1M magnesium chloride (store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 500mL solution requires |
| Magnesium chloride hexahydrate | MgCl2·6H2O | 203. 3 | 1M | 101.65g |
| Deionized water | H2O | 18 | \ | Fill to 500mL |
11. 5M sodium chloride (store at room temperature)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 500mL solution requires |
| Sodium chloride | NaCl | 58.44 | 5M | 146.1g |
| Deionized water | H2O | 18 | \ | Fill to 500mL |
12.PK buffer (prepared before use)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 200mL solution requires | |
| EDTA | C10H16N2O8 | 292.24 | 0.005 M | 2ml 0.5M EDTA | |
| Tris-HCl | \ | 157.60 | 0.05 M | 10mL 1MTris-HCl | |
| Proteinase | \ | \ | 2mg/mL | 20mL 20mg/mL proteinase | |
| DEPC-treated deionized water | H2O | 18 | \ | 188mL | |
13. 0.1M TEA buffer (prepared before use)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 200mL solution requires | |
| TEA | (HOCH2CH2)3N | 149.19 | 0.1 M | 20mL 1MTEA (pH=8.0) | |
| Acetic anhydride | (CH3CO)2O | 102.09 | \ | 500mL | |
| DEPC-treated deionized water | H2O | 18 | \ | 180mL | |
14.Buffer B1 (prepared before use)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 200mL solution requires | |
| Tris-HCL | \ | \ | 0.1 M | 20ml 1M Tris-HCl (pH=7.4-7.5) | |
| Sodium chloride | NaCl | 58.44 | 0.15 M | 6mL 5M sodium chloride | |
| Deionized water | H2O | 18 | \ | 174mL | |
15.Buffer B2 (prepared before use)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 1mL solution requires | |
| Buffer B1 | \ | \ | \ | 900uL | |
| Sheep serum | \ | \ | \ | 100uL | |
16.Buffer B3 (prepared before use)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 200 mL solution requires | |
| Tris-HCl(pH 9.5) | \ | \ | 0.1 M | 20mL 1M Tris-HCl(pH 9.5) | |
| Sodium chloride | NaCl | 58.44 | 0.1 M | 4mL 5M sodium chloride | |
| Magnesium chloride | MgCl2 | 95.21 | 0.05 M | 10mL 1M magnesium chloride | |
| Tween 20 | \ | 1227.5 | 0.1% | 1mL 20% tween 20 | |
| Deionized water | H2O | 18 | \ | 165mL | |
17.Buffer B4 (prepared before use)
| Specs | Molecular formula | Molecular weight | Concentration for storage | 1mL solution requires | |
| Buffer B3 | \ | \ | \ | 1mL | |
| Nitro blue tetrazolium chloride /5-Bromo-4-Chloro-3-Indolyl Phosphate (NBT/BCIP) | C40H30N10O6C12 /C8H4BrClK2NO4P | 817.6/402.65 | \ | 20mL | |
Acknowledgements
We thank Yongjie Hou for her kind guidance on the experiment.
Reference
- Allen Institute for Brain Science. (2011). Technical white paper: in situ hybridization data production. http://help.brain-map.org/download/attachments/2818169/ABADataProductionProcesses.pdf?version=1&modificationDate=1319477154403&api=v2
- SCHAERENWIEMERS N, GERFINMOSER A. A (1993). A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochem 100(6): 431-40.
- 王雅娜, 田雨, 何苗, 龚玲. (2021). 小鼠脑组织冰冻切片. Bio-101: e1010819. DOI: 10.21769/BioProtoc.1010819.
Appendix 1. Troubleshooting
| Problems | Reasons | Solutions |
| Brain tissue not in good shape | 1.The tissue had insufficient time of exposure to sucrose 2.There’re air bubbles in microscope slides, or some part of the tissue is folded. 3. Insufficient time for drying the slides. 4.The time for proteinase treatment is too long. | Ensure that the tissue is put in 20% sucrose for 1 day as it sinks, and another 1-2 days in 30% sucrose as it sinks. Attach the tissue well to the slide in the first place. Avoid bubbles or folding when applying the cover glass.
Ensure a 2-hour drying time.
Decide the suitable time of treatment according to the mouse’s age. The recommended time is 10mins for adult mice and 1-2 mins for embryonic mice.
|
| Low staining intensity | 1.The tissue sample is not fresh, or it has been degraded by RNase. 2.The probe is too long. / It’s inefficient.
3.The time for chromogenic reaction is too short. 4.The tissue gets dry during the procedures. | Use fresh tissue. All steps before hybridization should be strictly RNase-free.
The optimal length of the probe should be 400-700bp. /Design another probe sequence at another location. Adequately prolong the time of chromogenic reaction (from 30mins to 1 day).
Keep the sample moist during the whole process.
|
| No staining intensity | 1. The tissue sample is not fresh, or it has been degraded by RNase. 2.The probe is not working. | Use fresh tissue. All steps before hybridization should be strictly RNase-free. Use positive control. Design another probe sequence at another location. Some published articles may be referred to.
|
| High background staining | 1. There’re air bubbles in microscope slides./ Some part of the tissue is folded. 2. The time for chromogenic reaction is too long.
3.The probe has low specificity. | Attach the tissue well to the slide in the first place. Avoid bubbles or folding when applying the cover glass.
Terminate the chromogenic reaction and treat the tissue with methanol. Shorten the time of chromogenic reactions. Pay attention to the changes during the chromogenic reaction. Design another probe sequence at another location. Adequately lengthen the sequence. |
Appendix 2. Functions of the reagents.
- DEPC: DEPC is an inhibitor of RNase. It guarantees no RNase in the solution.
- 4% PFA: This reagent is used in tissue fixation. It preserves the morphology of the tissue, as well as maintaining the level of DNA/RNA in cells, thus it enables the probe to get into the tissue more easily.
- PK buffer: With proteinase K inside, PK buffer can degrade proteins. If there’s protein binding to the target nucleic acid through electrostatic interaction, it may interfere with the insertion of the probe. Therefore, proteinase K helps enhance the signals by exposing the target nucleic acid to the probe.
- 0.1M Triethylamine alcohol (TEA): The protein that intrinsically exists in the tissue may be close to the target nucleic acid through electrostatic interaction and interfere with the insertion of the probe. The acetic anhydride in TEA solution contributes to the acetylation of such protein, making its binding to nucleic acid weaker. In this way, it enhances the penetrability of the probe.
- (Pre-)hybridization solution: It contains formamide, Denhardt's solution and Herring sperm DNA. Formamide can lower the Tm value. Every 1% increase in formamide concentration can contribute to about 0.5℃ decrease in the hybridization temperature. Formamide in this hybridization solution can lower the temperature of reaction by about 25℃, thus it relevantly decreases the possibility of unspecific binding between the tissue and the probe. The bovine serum in Denhardt's solution, Yeast RNA, and the herring sperm DNA won’t specifically bind to the target nucleic acid, but will block the potential unspecific binding site of the probe. The pre-hybridization solution has almost the same ingredients as the hybridization solution, except that the latter one has probe inside.
- 0.2×Saline Sodium Citrate (SSC): It is used to rinse the slides after hybridization in order to ensure the specificity. At such low salt concentration, the binding of nucleic acid is unstable, so the unspecific probes can be washed off.
- Buffer B1: It contains Tris-HCl, which is the solvent for nucleic acids and proteins. The buffer pair Tris and HCl sets pH to 7.4-7.5, helping to maintain the pH of the tissue. This buffer is also used in washing off the non-specifically bound probes.
- Buffer B2: It is prepared with buffer B1. With sheep serum inside, this buffer is used in blocking, as well as in dilating the DIG antibody, so as to enhance the signals.
- Buffer B3: It consists of Tween 20, which is used in washing off proteins and non-specifically bound antibodies. The buffer pair Tris-HCl sets pH at about 9.5, which is the optimal temperature for Alkaline phosphatase (AP).
- Buffer B4: It is prepared with buffer B3. The BCIP/NBT inside is responsible for the chromogenic reaction. The AP on anti-DIG-AP catalyzes the hydrolysis of BCIP, producing indole. Indole then reacts with NBT and forms into the insoluble blue/bluish violet sediment. The sediment indicates the expression and reveals the location of the RNA of interest.
Appendix 3. Brief procedures for RNA probe design
- Primer design: Download the cDNA sequence, design a pair of 20-30bp-long primers. Add a restriction enzyme cutting site. A number of additional bases should be added, flanking the recognition sequence.
- Extract Total RNA and reverse transcribe it into cDNA.
- Amplify the targeting fragment by PCR, get the product and perform restriction enzyme digestion overnight.
- Purify the DNA product. Measure its concentration. Perform DNA ligation with vector DNA.
- Transform it to competent cells, incubate overnight.
- Pick monoclonal colonies and plant them in culture medium separately. Shake vigorously by a 37℃-orbital shaker. Meanwhile, perform PCR to ensure the transformation is conducted successfully. Select the correctly connected bacterial solution. Add the same volume of 50% glycerin to part of the bacterial solution and store it at -80℃. Cultivate the rest of the bacterial solution for genome sequencing.
- Select the bacterial solution with the correct sequence and perform plasmid extraction. Use a single restriction endonuclease enzyme to digest overnight, turning the plasmid linear.
- Quantify the linear plasmid, and transcribe in vitro. (Starting from this step, special attention should be paid to avoid RNase contamination.) Store the rest of the linear plasmid at -80℃.
- Use DNase to digest it after the in vitro transcription, and use EDTA to terminate the reaction. Add sodium acetate (NaAc) and isopropanol to promote the aggregation of the nucleic acids. Mix well, centrifuge briefly, store at -20℃ overnight .
- Centrifuge it at 13000rpm, 4℃, and discard the supernatant. Use 75% alcohol to rinse twice. Air dry the RNA pellet, dissolve it in water and determine the RNA yield.
- Add strictly equal amount of formamide. Preserve the RNA probe at -80℃.
Appendix 4. Classification of the probe and its characteristics
| Classify according to types of the nucleic acids | ||
| Type of the nucleic acids | Advantages | Disadvantages |
| cDNA | 1.High sensitivity 2.Stable 3.Various ways of labelling | The connection between the probe and the tissue is weak. The weakly paired strands are easy to be washed off. |
| RNA | 1.High reaction efficiency 2.Ability to form a more stable hybrid molecule | 1.The probe is easy to be degraded by RNase. During the experiment, RNase contamination should be strictly prevented. 2.Relatively more difficult to be made, compared with the cDNA probe. 3.More non-specific binding compared with the cDNA probe. |
| Synthetic oligonucleotide | 1.Short sequence, strong penetration 2.High speed of hybridization 3.Easy to be synthesized | 1.The length of the sequence should be controlled, or else there will be non-specific binding. 2.It carries less markers. 3. The hybrid molecule is relatively not stable. |
Classified according to the type of labelling | ||
| Type of the nucleic acids | Advantages | Disadvantages |
| Radiolabeled | High sensitivity | 1.Risk of radioactive contamination. 2.If the radioactive element has a short half-life, then the quality guaranteed period is short. |
| Biotin labeled | 1.No risk of radioactive contamination 2.It can work as hapten for immunoassay. | 1.Relatively low sensitivity 2.May be interfered by endogenous biotin |
| Digoxin labelled | 1. No risk of radioactive contamination. 2. It can work as hapten for immunoassay | Relatively low sensitivity |
| Fluorescence labelled | 1. No risk of radioactive contamination. 2.Strong signal, high sensitivity 3.High resolution 4.Multiple colors can be used in labelling the same nucleus, so various sequences can be detected at the same time. | The fluorescence signal does not last long, and it is easy to be quenched. |
- Ying, Y, Wu, J and He, M(2021). In-situ hybridization for mouse brain sections. Bio-protocol Preprint. bio-protocol.org/prep1424.
Category
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.