
- Home
- Protocols

-

EMSA
Last updated date: Jul 18, 2021 Views: 774 Forks: 0
| Electrophoretic mobility shift assay (EMSA) | |||
| Prepared by: | Date: | Performed by: | Date: |
| Stefan Ilic | 12/07/2016 | ||
Preparation of the native polyacrylamide gel:
1. Assemble the glass plates for gel casting
2. Prepare the appropriate gel mix. Note that the concentration of acrylamide we have in the lab (40%, 29:1:0.5) is different from the one in the table (30%). Therefore, the amounts have been recalculated:
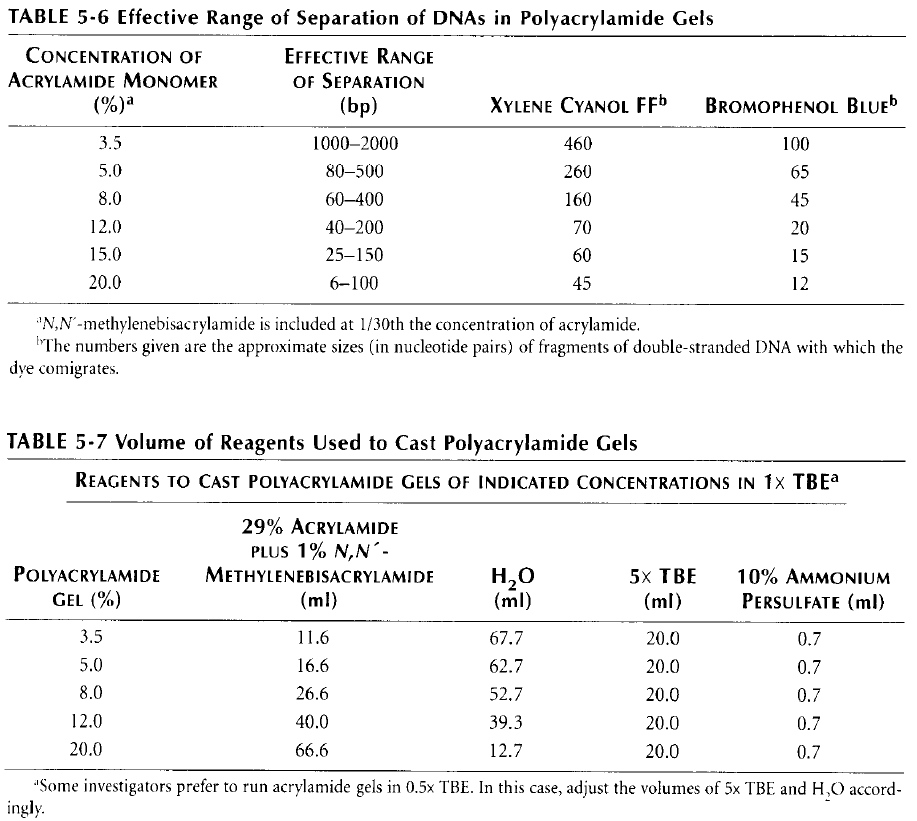
3. Add 3.5μL of TEMED and 70μL of APS for each 10mL of gel-mix
4. Cast the gel and wait until it polymerizes
5. Prepare one of the loading buffers according to the following table:

Preparation of the radioactively labeled template:
| Component: | Volume: |
| 1μM oligo | 5μL |
| [γ-32P] ATP | 3μL |
| 10X PNK buffer | 1μL |
| PNK (10000U/μL) | 1μL |
| Incubate the reaction at 37oC for 30min. | |
Preparation of the columns for template purification (BioRad, Cat. No. 732- 6227):
- One columns is needed for each oligo.
- Mix well the content of the columns to make it homogenous.
- Remove the cap and then break the tip of the column to let the buffer flow out.
- Centrifuge the column (4000rpm, 4min) and discard the flow-though.
Purification of the radioactively labeled oligo:
- Load 10μL of the previously prepared template on the column.
- Wash the tube that contained labeled template two times with DDW (10μL each time) and load each of these washes (check with Geiger counter) on the same column. Be careful not to exceed the maximum volume specified by the column manufacturer.
- Centrifuge the column (4000rpm, 4min) and keep the flow-through (the yield after purifications is about 90% which means we get approx. 150nM DNA in the end).
- To perform successful annealing, complementary strand should be added in a small excess (e.g. 1.2 times more). For example, 30uL of 180nM DNA will be mixed with 30uL of labeled 150nM oligo.
- Anneal the oligos: 3 min at 95°C; then let it cool down gradually to 25°C.
- You can freeze the DNA at -20°C or use it immediately after the purification.
Experimental procedure:
1. Make serial dilutions (e.g. 1:2 or 1:3) of the investigated protein with the appropriate buffer.
2. Prepare the reaction mix:
5X activity buffer | ddWater | 100nM labeled DNA | Investigated protein solution | |
| 1 RXN | 2 | 1 | 1 | 6 |
| Mix (x10) | 20 | 10 | 10 | / |
3. Divide the reaction mix to PCR tubes (4μL to each tube).
4. Add 6μL of protein storage buffer to the first tube (negative control);
5. Add 6μL of serially diluted protein to the rest of the PCR tubes.
6. Mix the content of the tubes and afterwards do spin-down.
7. Incubate the reaction(s). This might require some optimization (try different temperatures and incubation times)!
8. Quench the reaction by adding 2μL of 6X gel-loading buffer.
9. Mix the content of the tubes again and do spin-down.
10. Load the samples on the gel and perform electrophoresis at 4oC and constant voltage (10V/cm of gel length).
11. Dry the gel for 60 min in a specialized gel dryer (https://www.bio-rad.com/en- il/product/model-583-gel-dryers?ID=N3OQZWKSY).
12. Expose phosphorus plate to the radioactive gel (to induce photostimulated luminescence).
13. Detect the signal by autoradiography (use phosphorimager).
- Onn, L, Ilic, S and Toiber, D(2021). EMSA. Bio-protocol Preprint. bio-protocol.org/prep1302.
- Onn, L., Portillo, M., Ilic, S., Cleitman, G., Stein, D., Kaluski, S., Shirat, I., Slobodnik, Z., Einav, M., Erdel, F., Akabayov, B. and Toiber, D.(2020). SIRT6 is a DNA double-strand break sensor. eLife. DOI: 10.7554/eLife.51636
Category
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.