- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Combining Suction-Pipette Spectral Identification With Single-Cell RT-PCR to Make Differential Analyses of Amphibian Red and Green Rods

Published: Vol 16, Iss 5, Mar 5, 2026 DOI: 10.21769/BioProtoc.5623 Views: 87
Reviewed by: Marquis WalkerAnonymous reviewer(s)
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Amphibian retinas contain “green” rods, which are rod-shaped photoreceptors with a cone-type visual pigment. These rods are considered a potentially transitional photoreceptor type, but their phototransduction cascade’s molecular composition has remained uncertain. Here, we present a streamlined electrophysiology-molecular workflow that enables the rapid spectral identification, physical capture, and targeted single-cell reverse transcription-polymerase chain reaction (RT-PCR) of individual amphibian photoreceptors. After suction-pipette spectral screening under alternating red and green illumination, electrophysiologically identified cells are isolated and processed directly for reverse transcription and PCR. Coupling real-time functional phenotyping with sensitive molecular profiling provides a practical tool for resolving photoreceptor molecular heterogeneity and investigating evolutionary transitions between rod and cone phenotypes.
Key features
• New combined workflow: Rapid electrophysiological spectral typing by suction-pipette, followed by single-cell transfer, reverse transcription, and targeted PCR.
Keywords: Two-cell PCRGraphical overview
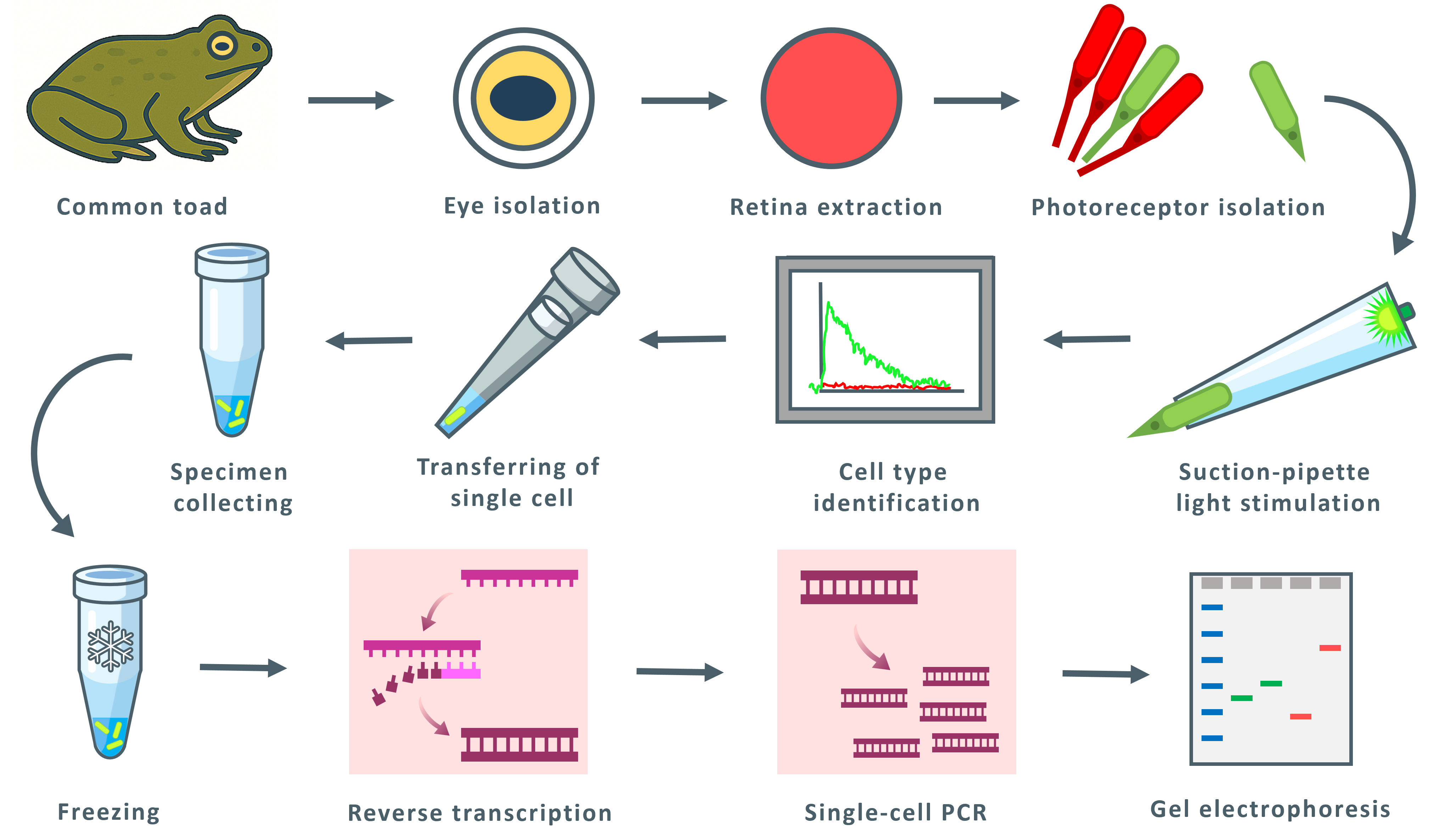
Background
Vertebrate photoreceptors in the retina that drive image-forming vision are divided into two principal types: rods and cones. This fundamental dichotomy, with ancient evolutionary origins, underlies vision across an expansive range of ambient light intensities, spanning approximately ten orders of magnitude [1]. Rods, specialized for scotopic vision, demonstrate maximal light sensitivity; however, they are characterized by relatively low temporal resolution, which limits their ability to discern rapidly changing visual stimuli. Conversely, cones adapted for photopic vision exhibit lower sensitivity but possess a remarkable capacity for adaptation to varying light levels and offer faster kinetic responses. It is noteworthy that no individual photoreceptor type exhibits the remarkable sensitivity of rods and the high velocity and adaptability of cones. However, intermediate forms have been observed, suggesting the possibility of evolutionary transitions between these two specialized cell types.
A compelling example of such an intermediate photoreceptor is the “green” rod, a distinctive and functionally well-characterized cell found in amphibians. Morphologically, the green rod is virtually indistinguishable from the classic amphibian “red” rod, exhibiting a typical rod-like structure, while its visual pigment most closely aligns with cone-type opsins [2–4]. As demonstrated in our previous works, there are significant differences in the phototransduction cascade parameters between green and red rods [5]. Specifically, green rods exhibit a heightened light sensitivity compared to red rods, primarily attributable to a slower turn-off of the phototransduction cascade. This unique combination of features positions the green rod as a transitional photoreceptor, uniting rod morphology, a cone visual pigment, and rod-like photoresponse kinetics, offering valuable insights into the evolutionary diversification of vertebrate vision. It is reasonable to hypothesize that the phototransduction cascade in amphibian green rods consists of a mixture of cone- and rod-specific protein isoforms. The present study aims to determine whether the principal cascade proteins belong to the rod or cone type photoreceptors.
To address this question, a unique experimental approach was developed that permits the selection of sufficient numbers of exclusively green or exclusively red rods. We established a rapid electrophysiological screening protocol that allows unambiguous identification of rod subtypes.
Previous studies have not reported single-cell PCR from individually isolated photoreceptors. We have developed and validated a protocol for electrophysiological identification, isolation, and single-cell PCR of these cells. At an intermediate stage, we revealed the expression of the rod isoform of rhodopsin kinase (GRK1), the rod isoform of the transducin α-subunit (GNAT1), and the rod isoform of GCAP (GUCA1). Furthermore, exclusive expression of the rod isoform of phosphodiesterase (PDE6A) was observed, with no detectable expression of the cone isoform. Although the rod isoforms of the transducin α-subunit and GCAP predominate, trace amounts of the corresponding cone isoforms (GNAT2 and GUCA2) were also detected. It remains unclear whether these signals represent minor fractions of endogenous mRNA within the photoreceptors or contamination from extracellular RNA.
Materials and reagents
Biological materials
1. Adult common toads (Bufo bufo, n = 12) were collected from the wild in northern Russia. Animals were housed in terraria (60 × 40 × 40 cm) at room temperature. Soil was used as substrate to provide natural conditions for locomotion and resting, and access to a water container was provided. Illumination was provided by a visible and an ultraviolet (UV) lamp under 8/16 h light/dark period. The diet consisted of American cockroaches (Periplaneta americana) and mealworm larvae (Tenebrio molitor), offered twice weekly. To prevent calcium deficiency, the diet was additionally supplemented with a specialized mineral supplement. The handling of animals was conducted in accordance with the Council Directive of the European Communities (24 November 1986; 86/609/EEC), and the experimental protocol was approved by the local Institutional Animal Care and Use Committee (protocol #1–7/24 from 31/01/2024).
Reagents
1. Liquid nitrogen
2. Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S7653)
3. Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9333)
4. Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M1028)
5. Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 21115)
6. Sodium bicarbonate (NaHCO3) (Sigma-Aldrich, catalog number: S5761)
7. HEPES (Sigma-Aldrich, catalog number: H3375)
8. Glucose (D-Glucose) (Sigma-Aldrich, catalog number: G8270)
9. Ethylenediaminetetraacetic acid (EDTA) (Sigma-Aldrich, catalog number: E5134)
10. Sodium hydroxide (NaOH) (Sigma-Aldrich, catalog number: 72068)
11. RNase inhibitor RiboCare (Evrogen, catalog number: EK005M)
12. Triton X-100 (Sigma-Aldrich, catalog number: T8787)
13. Ethanol (ethyl alcohol, 96%) (LenReaktiv, catalog number: 000164)
14. MMLV Reverse Transcriptase kit (Evrogen, catalog number: SK021)
15. Fusion 2.0 polymerase (Biolabmix, catalog number: Е-14005)
16. Agarose low-EEO (BioinnLabs, catalog number: bn-1G1A)
17. EtBR 10 mg/mL (Biolabmix, catalog number: EtBr-10)
18. 50× TAE buffer (Evrogen, catalog number: PB122)
19. DNA-ladder Step100 (Biolabmix, catalog number: S-8100)
20. 4× gel loading dye (Evrogen, catalog number: PB020)
21. Forward and reverse gene-specific oligonucleotide primers (Biolabmix)
Solutions
1. Frog Ringer (FR) solution (see Recipes)
2. RNA preserving cell buffer (CB) (see Recipes)
3. Agarose gel (see Recipes)
4. 1× TAE EtBr electrophoresis buffer
Recipes
1. Frog Ringer (FR) solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| NaCl 5 M | 90 mM | 18 mL |
| KCl 1 M | 2.5 mM | 2.5 mL |
| MgCl2 1 M | 1.4 mM | 1.4 mL |
| CaCl2 1 M | 1.05 mM | 1.05 mL |
| NaHCO3 1 M | 5 mM | 5 mL |
| HEPES 1 M | 5 mM | 5 mL |
| Glucose 1 M | 10 mM | 10 mL |
| EDTA 1 M | 0.05 mM | 0.1 mL |
| NaOH 0.5 M | to pH 7.6 | |
| Milli-Q (MQ) water | to 1,000 mL |
2. RNA-preserving cell buffer (CB)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| RNase inhibitor, 40 U/μL | 2 U/μL | 50 μL |
| Triton X-100, 100% | 0.2% | 2 μL |
| MQ water | To 1 mL |
Store at -20 °C and use an icepack while working.
3. Agarose 2.5% gel
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Agarose powder | 2.5% | 2.5 g |
| 50× TAE | 1× | 2 mL |
| EtBR, 10 mg/mL | 1 μg/mL | 10 μL |
| MQ water | To 100 mL |
4. 1× TAE EtBr electrophoresis buffer (EtBr 1 μg/mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 50× TAE | 1× | 20 mL |
| EtBR, 10 mg/mL | 1 μg/mL | 100 μL |
| MQ water | To 1,000 mL |
Laboratory supplies
1. Microcentrifuge tubes, 0.2 and 1.5 mL (SSIbio, catalog numbers: 1110-00 and 1210-00)
2. 3 cm Petri dishes (Jet Biofil, catalog number: TCD000035)
3. Dissecting blades (MedTech, catalog number: Sb-3)
4. Dissecting scissors (MedTech, catalog number: S-1141)
4. Dissecting dressing tweezers (MedTech, catalog number: F-6421)
4. Borosilicate glass capillaries for suction micropipettes (WPI, catalog number: TW150-4)
5. Silver chloride electrodes (WPI)
6. Icepack (Termokont MK)
7. Sterile filter pipette tips (Helicon, catalog number: AT-10-S-F-K)
8. Light-tight container (any small box or jar to prevent light exposure)
9. Mineral supplement for reptiles (Aquamenu)
Equipment
1. Suction pipette system with infrared (IR) observation (custom-made)
2. Milli-Q® water system (Merck Life Science, model: Ultrapure, Type 1)
3. Stereomicroscope (Nexcope, model: NSZ-810)
4. Pipette puller (HEKA Elektronik, Germany, model: PIP6)
5. Microforge (custom-made)
6. Centrifuge for Eppendorf tubes (Eppendorf, Germany, model: MiniSpin Plus)
7. Vortex MultiReax (Heidolph instruments GmbH, Germany, catalog number: 545-10000-00)
8. Veriti 96-well thermal cycler (Applied Biosystems, model: 9902)
9. Gel-documentation imaging system ChemiDoc MP (Bio-Rad, catalog number: 12003154)
Software and datasets
1. Light stimulation, data recording, and processing programs in LabView (National Instruments; see Supplementary Materials)
2. Gene sequence database: National Center for Biotechnology Information (NCBI)
3. Primer design software: Integrated DNA Technologies (IDT) PrimerQuest Tool
Setup of the device used to record the current of a single vertebrate retinal rod:
This device (Figures 1–3) is an intricate piece of hardware that enables:
1. High-resolution visualization of individual retinal photoreceptors in infrared light.
2. Fixation of the photoreceptors inside a suction pipette.
3. Stimulation of a selected cell using a two-channel light stimulation system with a wide range of stimulus duration (starting from 2 ms), stimulus brightness (more than seven orders of intensity), and stimulus color (four LEDs).
4. Recording of photoreceptor responses to light stimulation with low instrumental noise.
5. Primary signal processing of the photoreceptor responses is carried out in real time by a dedicated control software based on LabView.
6. Additional processing and analysis of the photoreceptor responses using a special analytical package based on LabView.

Figure 1. Photograph of the experimental setup. The suction micropipette is mounted in the holder and connected to the electrode and oil-based pressure control system.
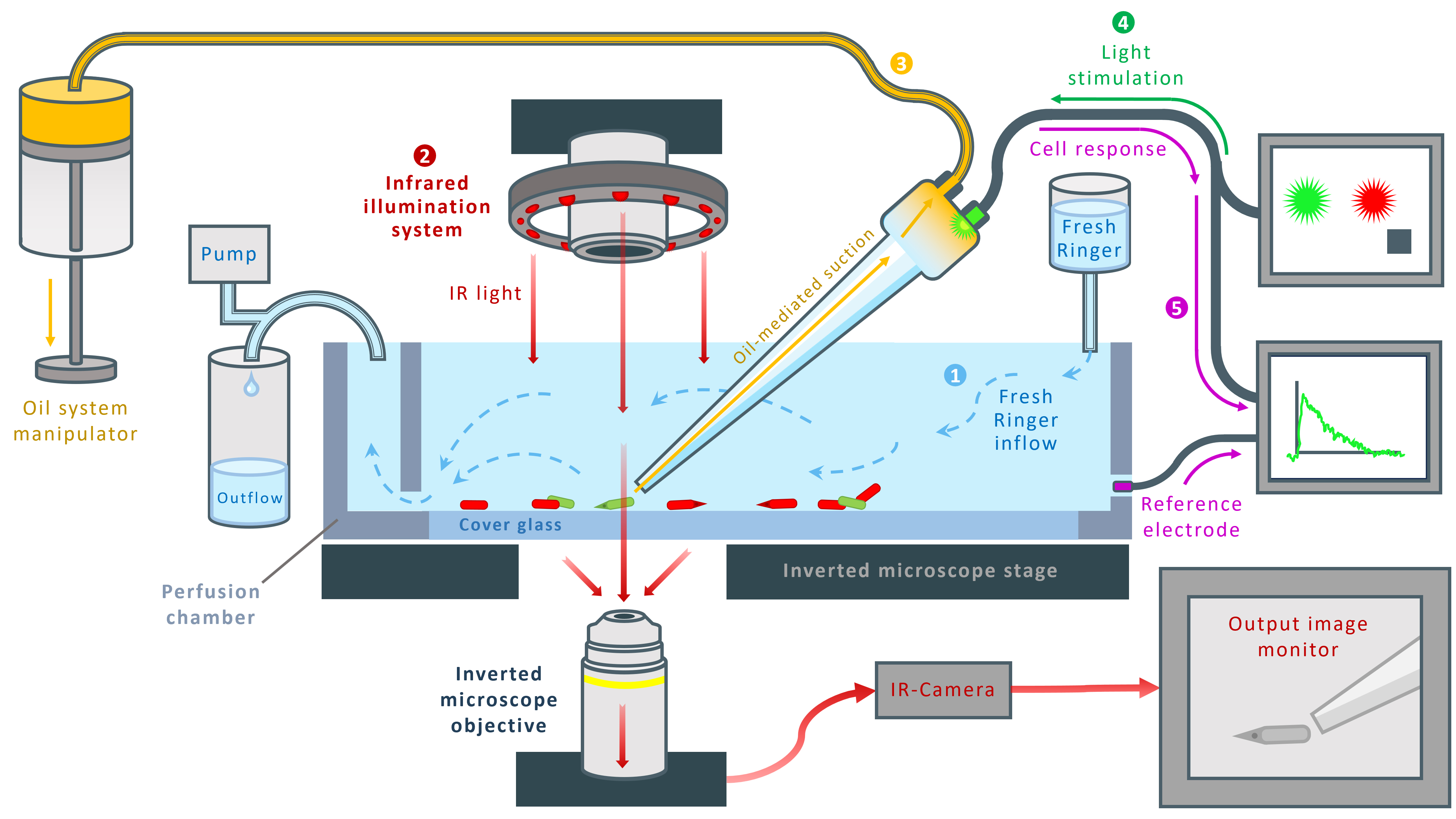
Figure 2. Schematic representation of the custom-made suction-pipette system. 1: Perfusion chamber. The glass micropipette is immersed in a perfusion chamber with a transparent cover glass bottom. The chamber is continuously supplied with fresh Ringer’s solution (blue arrows) and connected to a waste pump for constant outflow. 2: Infrared (IR) illumination system. IR light (red arrows) is directed onto the preparation from above, passes through the chamber bottom, and is collected by the objective of an inverted microscope. An IR-sensitive camera attached to the microscope transmits the live image to the output monitor. 3: Oil-based suction control. Negative pressure applied to the glass pipette is regulated through an oil-filled system operated via micromanipulators (yellow arrows). 4: Light stimulation. The photoreceptor is stimulated with controlled light flashes delivered through a software-regulated stimulation pathway (green arrows). 5: Signal recording. The photocurrent response to the light stimulus is measured through the suction pipette system.
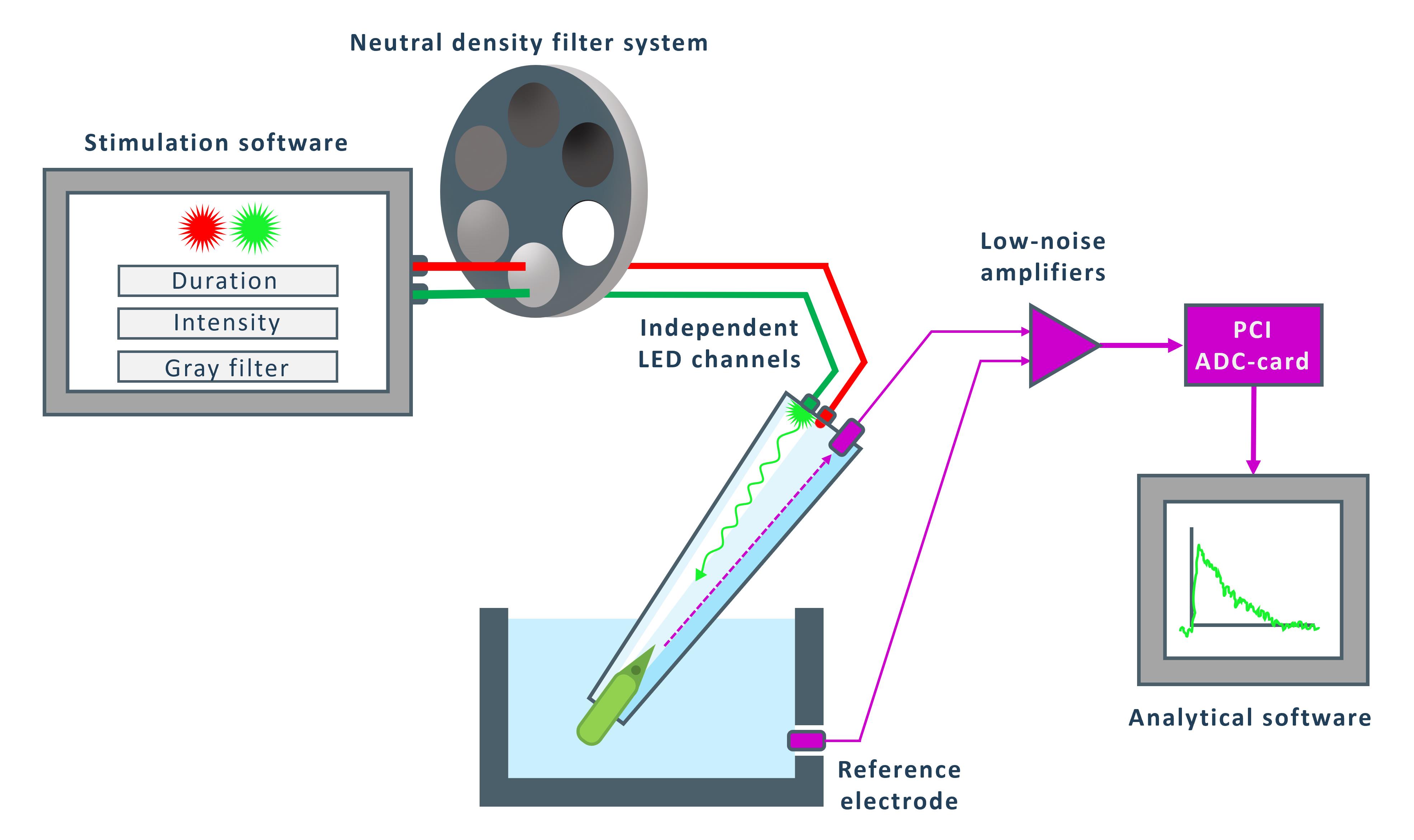
Figure 3. Workflow of the stimulation and recording system. The stimulation software controls two independent LED channels (red and green), allowing adjustment of stimulus duration, intensity, and attenuation via a neutral density filter system. The selected photoreceptor is illuminated, and its photocurrent response is recorded through a reference electrode positioned in the perfusion chamber. The signal is amplified by low-noise preamplifiers and digitized using a PCI-ADC card, after which the data are transferred to the analytical software.
The complex includes:
1. Visualization and stimulation unit comprising a Micromed 1B trinocular inverted microscope with infrared specimen illumination, a high-sensitivity IR CCD camera with image output to a monitor, and an integrated custom-made stimulation system including two independent LED-based optical channels. The stimulation intensity is regulated by the LED current (two orders of adjustment) and by an additional system of grey light filters with a density step of one log unit (five orders of adjustment).
2. Perfusion and cell selection system, including a custom-made chamber for visualizing and selectively collecting cells, glass micropipettes, and manipulators.
3. Unit for recording the electrical response of cells, including low-noise preamplifiers and amplifiers, and a 16-bit NI-6229 ADC card (National Instruments).
The capabilities of the LabView-based software package are as follows:
1. Control package: allows simultaneous stimulation of a selected cell and recording of its electrical response. Stimulation can be performed on one or two channels simultaneously, with any stimulus duration starting from 1 ms and any number of stimuli presented sequentially. The brightness of the stimulation can vary across 7 orders of intensity. Stimulation can be single or multiple to accumulate the signal and improve the signal-to-noise ratio. In the case of multiple stimulation, the experiment operator can view the accumulated signal in the current average mode. After each stimulation, the photoreceptor response is recorded on a hard drive for further processing.
2. The analytical package then reads the stored data in semi-automatic mode. It performs optimal filtering and averaging of rod responses to light stimulation and determines the magnitude and time delay of the maximum response. If necessary, it also determines various other response parameters, including frequency analysis and the kinetics of the rise and fall phases of the response.
Procedure
A. Preparation of retinal sample
Note: Operations from steps A3 to С8 must be performed in a dark room under dim red light or using an infrared observation system to prevent rhodopsin bleaching. Unless otherwise stated, all procedures are performed at room temperature.
1. Dark-adapt the animals overnight.
2. Manufacture a suction micropipette using a puller and a microforge. The inner tip diameter of the micropipette should be approximately 8–9 μm, corresponding to the average diameter of rod photoreceptors in this species.
3. Decapitate the animal with scissors. After decapitation, destroy the brainstem using a thick needle.
4. Use scissors to remove extraocular tissues.
5. While holding the eye on a dry tissue, make an incision in the isolated eye with a scalpel on one side of the eyeball. Use spring scissors to cut along the equatorial line to hemisect the eye. Carefully remove and discard the anterior chamber, lens, and vitreous humor (for details, see [6]).
6. Repeat for the second eye. While processing the first retina, store the second eye semi-cup containing the neural retina in FR solution in a light-tight container at 4 °C until use.
7. Pour several milliliters of FR solution into a 3 cm Petri dish. Place the first eye semi-cup containing the neural retina into the solution, ensuring it is fully submerged. Use scissors to cut the eye semi-cup into two equal segments.
8. Using a stereomicroscope, carefully separate the retina of each segment from the pigment epithelium with forceps. Discard the eye semi-cup segments containing the pigment epithelium. If any pigment epithelium remains on the retina, gently shake the retinal segment in the solution and then remove the remaining epithelium with tweezers, taking care not to damage the retina.
9. Place the small piece of the retina, previously separated from the pigment epithelium, into a drop of FR in the empty Petri dish.
10. Using a pair of forceps, hold the retina on one side and press it against the bottom of the dish. With a second pair of forceps, perform rapid tearing motions across the retina within the drop. During this manipulation, a large number of photoreceptor cells detach from the tissue and become isolated in the solution (for details, see [7]).
11. Using an automatic adjustable pipette, aspirate the suspension containing retinal fragments and isolated photoreceptors and transfer it into the perfusion chamber.
B. Suction pipette
1. Allow retinal fragments and isolated rod photoreceptors to settle to the bottom of the perfusion chamber.
2. Immerse the glass suction pipette in the perfusion chamber.
3. Using the image from the infrared camera attached to the microscope, locate an isolated rod with an intact inner segment.
4. Use suction to pull the rod into the glass pipette, positioning the inner segment inside the pipette.
5. Select the light-stimulation parameters in the control software. For toad rod visual pigments, appropriate red and green light excitation parameters are λmax = 630 nm and λmax = 525 nm, respectively (Figure 4).
6. Stimulate the cell sequentially with red and green light.
7. Examine the recorded response traces. For red rods, the response to green light will be slightly larger than to red light. For green rods, the response to green light will be slightly smaller than the green response of red rods, and there are no responses to red light.
8. Advance the rod slightly further into the suction pipette to protect it from damage when it crosses the aqueous–air interface at the pipette tip.
9. Lift the glass suction pipette containing the cell above the surface of the perfusion chamber solution for cell transferring.
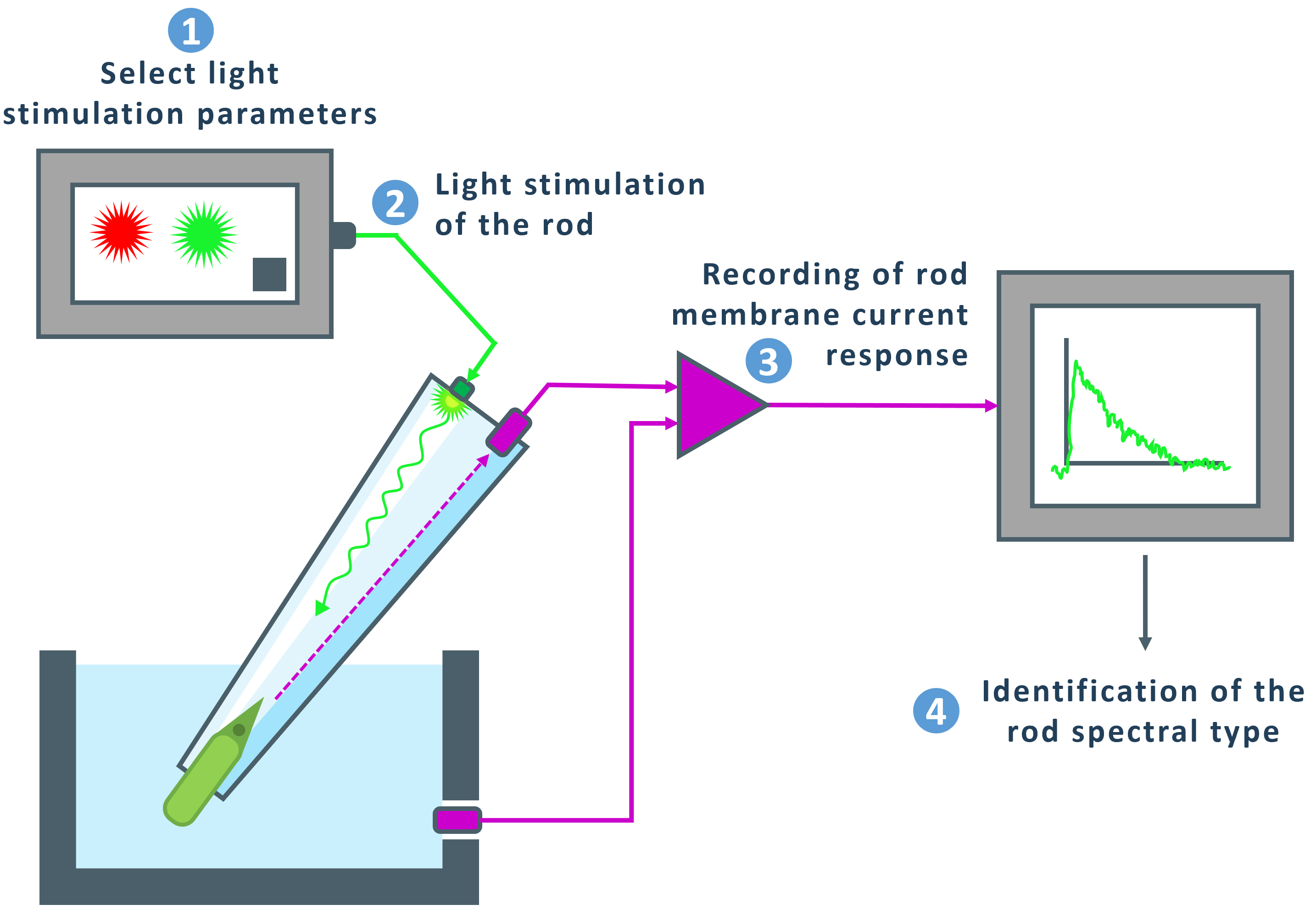
Figure 4. Schematic representation of the suction-pipette technique. A glass suction pipette is immersed in the perfusion chamber. A rod photoreceptor is sucked into a glass pipette with the inner segment facing the pipette. 1: Light stimulation parameters are selected using the software. 2: The cell is sequentially stimulated with red (λmax = 630 nm, ~1057 photons/μm2 per flash) and green (λmax = 525 nm, ~23 photons/μm2 per flash) light. 3: The photoreceptor current is recorded by the software and displayed on the screen. 4: The spectral type of the rod can be identified based on the obtained response traces.
С. Single-cell transferring and collecting (Figure 5)
1. Use sterile scissors to trim approximately 2 mm from the tip of a 10 μL plastic filter pipette tip in order to widen its opening.
2. Using an automatic pipettor, aspirate 0.5 μL of CB into the modified pipette tip.
3. Remove the tip from the pipettor and manually attach it to the end of a glass micropipette.
4. Transfer the cell from the glass micropipette into the drop of buffer held in the plastic tip. Then, remove the plastic tip from the glass micropipette.
5. Using the automatic pipettor, transfer the drop from the plastic tip to the bottom of a 200-μL microcentrifuge tube.
6. Close the tube and store it in an icepack while collecting additional cells (in this protocol, each tube contains two cells).
7. Collect several additional cells of the same type into the same tube by repeating steps C1–6 as required.
8. Freeze the tube in liquid nitrogen and store at -80 °C.
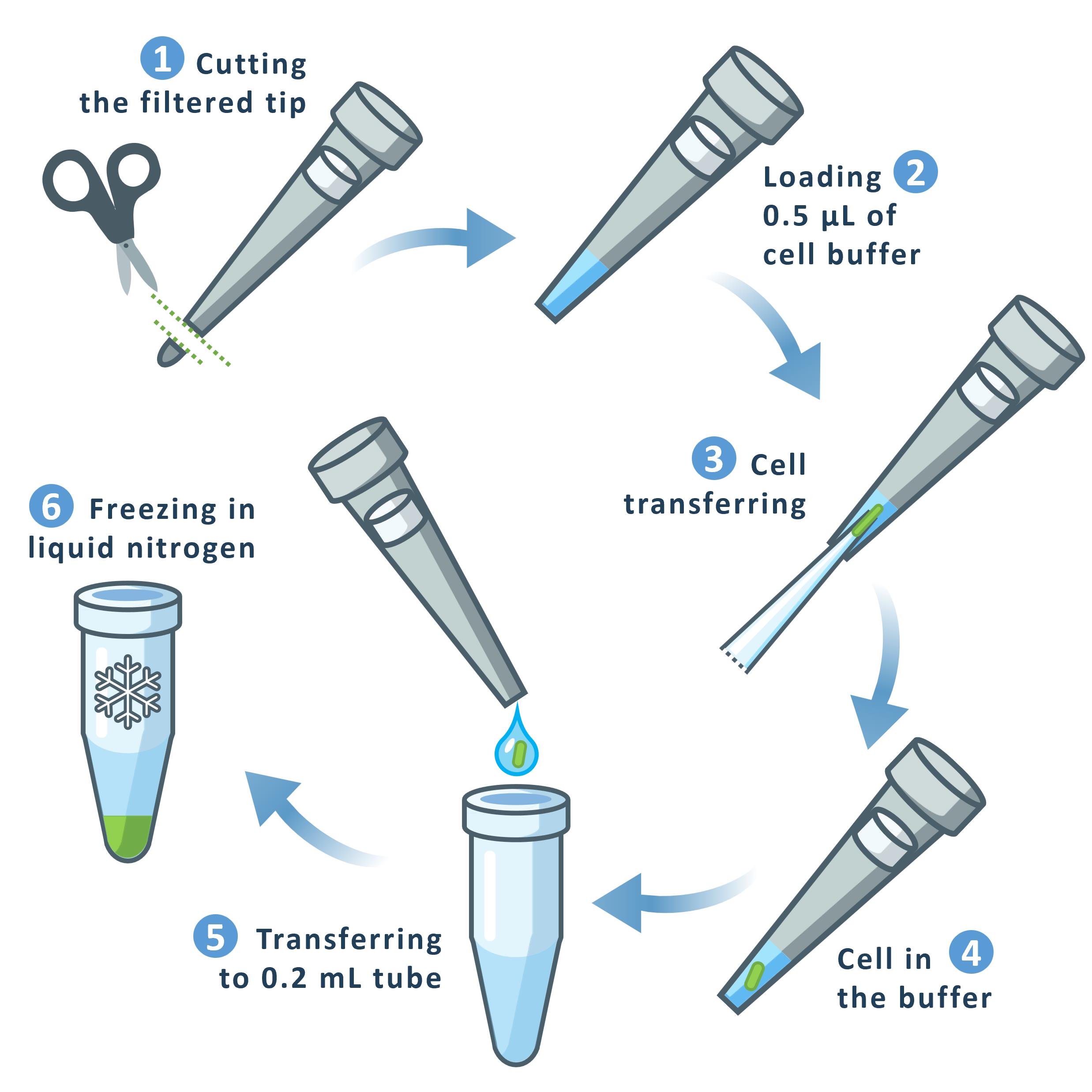
Figure 5. Schematic representation of cell transferring. 1: Approximately 2 mm of a filtered plastic pipette tip is cut off using sterile scissors. 2: The modified plastic tip is loaded with 0.5 μL of cell buffer. 3: The glass pipette containing the cell is inserted into the plastic pipette tip. 4: The cell is expelled from the glass pipette into the buffer within the plastic tip. 5: The buffer droplet containing the cell is transferred to the bottom of a 0.2 mL microcentrifuge tube. 6: The tube containing the cell is frozen in liquid nitrogen.
D. Primer design
The coding sequences of the analyzed Bufo bufo genes were retrieved from the National Center for Biotechnology Information (NCBI). PCR primers (Table 1) were designed using the Integrated DNA Technologies (IDT) PrimerQuest Tool. All gene-specific primers were designed to span intron–exon junctions in order to avoid amplification of genomic DNA during PCR.
Table 1. Primer sets designed and used in this study
| Gene | Primer | Primer sequence | Amplicon size (bp) |
| GNAT1 | Forward | 5′- CTTCGCCACCACTTCCATT -3′ | 138 |
| Reverse | 5′- TTAATGTAGTTCCCAGCGTCTT -3′ | ||
| GNAT2 | Forward | 5′- TGGAGCTAGTGCAGAAGACA -3′ | 132 |
| Reverse | 5′- TGCTCTTTCCAGACTCTCCG -3′ | ||
| GRK1 | Forward | 5′- GAGAGGACTGGTTTATGGACTTC -3′ | 216 |
| Reverse | 5′- GAAATGCATATGCCAGGGAAAC -3′ | ||
| GRK7 | Forward | 5′- AGGACATTGGAGGAAGAGGT -3′ | 234 |
| Reverse | 5′- AACGTCCTTGGCATAGACCA -3′ | ||
| PDE6A | Forward | 5′- TGTGGTCAGCCAACAAGGTT -3′ | 188 |
| Reverse | 5′- GTCCTGAATATGGCGGCACT -3′ | ||
| PDE6C | Forward | 5′- GGTCCCGAAACGGAATACCA -3′ | 207 |
| Reverse | 5′- TGCTTGTCCGCAAAGTCAGA -3′ |
E. Targeted reverse transcription
Note: The RNA extraction step is omitted due to significant material loss at this stage. Reverse transcription is performed using the MMLV Reverse Transcriptase kit with an elevated incubation temperature.
1. To each tube containing two identical cells (i.e., the same 200 μL tubes in which the cells were captured), add 1 μL of forward target-specific primer (20 μM), 1 μL of reverse target-specific primer (20 μM), and 1 μL of dNTP mix (10 mM), and fill with nuclease-free distilled water (dH2O) to a final volume of 5 μL.
2. Mix gently by pipetting and briefly spin down the droplets in a mini centrifuge (700× g for 10 s; hereafter, the same conditions are applied).
3. Incubate the mixture for 3 min at the primer annealing temperature, then immediately place it on ice to cool.
Note: Due to the transfer procedure, a small amount of FR solution remains in the buffer; therefore, the total reaction volume after adding the mix will be approximately 10 μL.
4. Add 0.5 μL of MMLV Reverse Transcriptase to the premix. Vortex to mix thoroughly and spin down briefly.
5. Add the enzyme premix to the samples and incubate at 42 °C for 60 min, followed by 70 °C for 10 min.
6. Upon completion of reverse transcription, freeze the resulting cDNA and store at -80 °C until PCR setup.
F. Two-cell PCR
Note: PCR is performed using Fusion 2.0 polymerase in a 20 μL reaction volume according to the standard protocol, without addition of DMSO. Various polymerases were tested, but due to the limited amount of starting material, only Fusion and Fusion 2.0 polymerases yielded reliable results.
1. Prepare Fusion PCR mix (10 μL) by adding 4 μL of 5× reaction buffer, 0.4 μL of dNTPs (10 mM each), 1 μL of forward and reverse specific primers (20 μM), 0.2 μL of Fusion 2.0 polymerase, and MQ water to 10 μL.
2. Add 10 μL of PCR mix to each tube containing cDNA.
3. Mix gently by pipetting the reaction mix up and down a few times.
4. Run the following PCR program (Table 2):
Table 2. PCR program
| Step | Temperature | Time | Cycles |
| Initial denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | 34 |
| Annealing | 67 °C | 20 s | |
| Extension | 72 °C | 30 s | |
| Final extension | 72 °C | 10 min | 1 |
Note: Fusion 2.0 polymerase is capable of functioning at elevated temperatures. Accordingly, the annealing temperature of primers can be increased by 3–10°C, up to 72 °C, to reduce nonspecific binding.
G. Gel electrophoresis of PCR products
1. Prepare a 2.5% agarose gel (see recipes).
2. Add 1× loading DNA buffer to each tube with the PCR product.
3. Load the total amount of DNA.
4. Add a 100 bp ladder for reference.
5. Run the gel electrophoresis in 1× TAE EtBr buffer for 30 min at 80–100 V.
6. Image gel in the gel-documentation imaging system.
Validation of protocol
Initially, spectral typing was performed by measuring the unsaturated rod responses at three wavelengths: red (λmax = 630 nm), green (λmax = 525 nm), and blue (λmax = 460 nm). We subsequently proved that stimulation with red and green light of a certain intensity alone reliably discriminates between green and red rods (Figure 6).
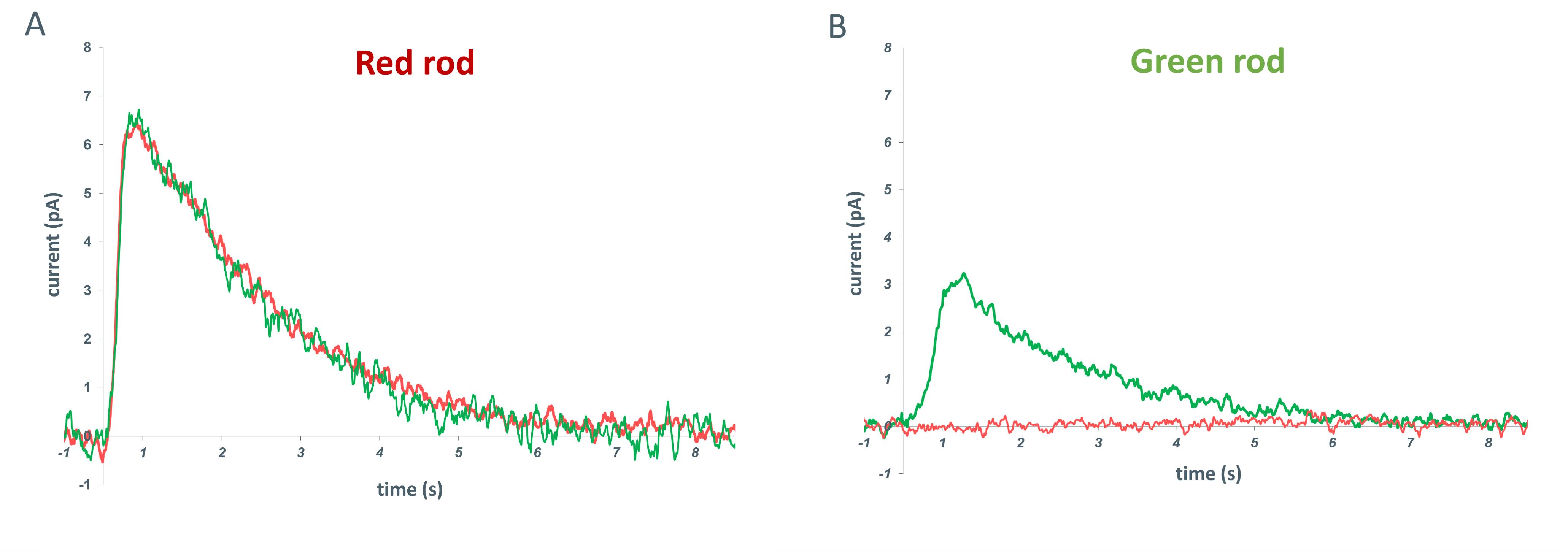
Figure 6. Comparison of photoresponses of red and green rods to light flashes. (A) Representative response of a red rod to red and green flashes. (B) Representative response of a green rod to red and green flashes. The red line indicates the response to a red flash (excitation λmax = 630 nm; 10 ms; ~1057 photons/μm2 per flash), and the green line indicates the response to a green flash (excitation λmax = 525 nm; 10 ms; ~23 photons/μm2 per flash). In total, 192 red rods and 147 green rods were tested.
Results showed that the minimum number of cells required for reliable data is two. However, recent evidence indicates this can be reduced to one for highly expressed genes without loss of accuracy, substantially improving efficiency when sample material is limited.
Results demonstrated robust expression of the rod isoforms of the transducin α-subunit (GNAT1) and rhodopsin kinase (GRK1) (Figure 7). We also observed exclusive expression of the rod isoform of phosphodiesterase (PDE6A) with no detectable expression of the cone isoform.
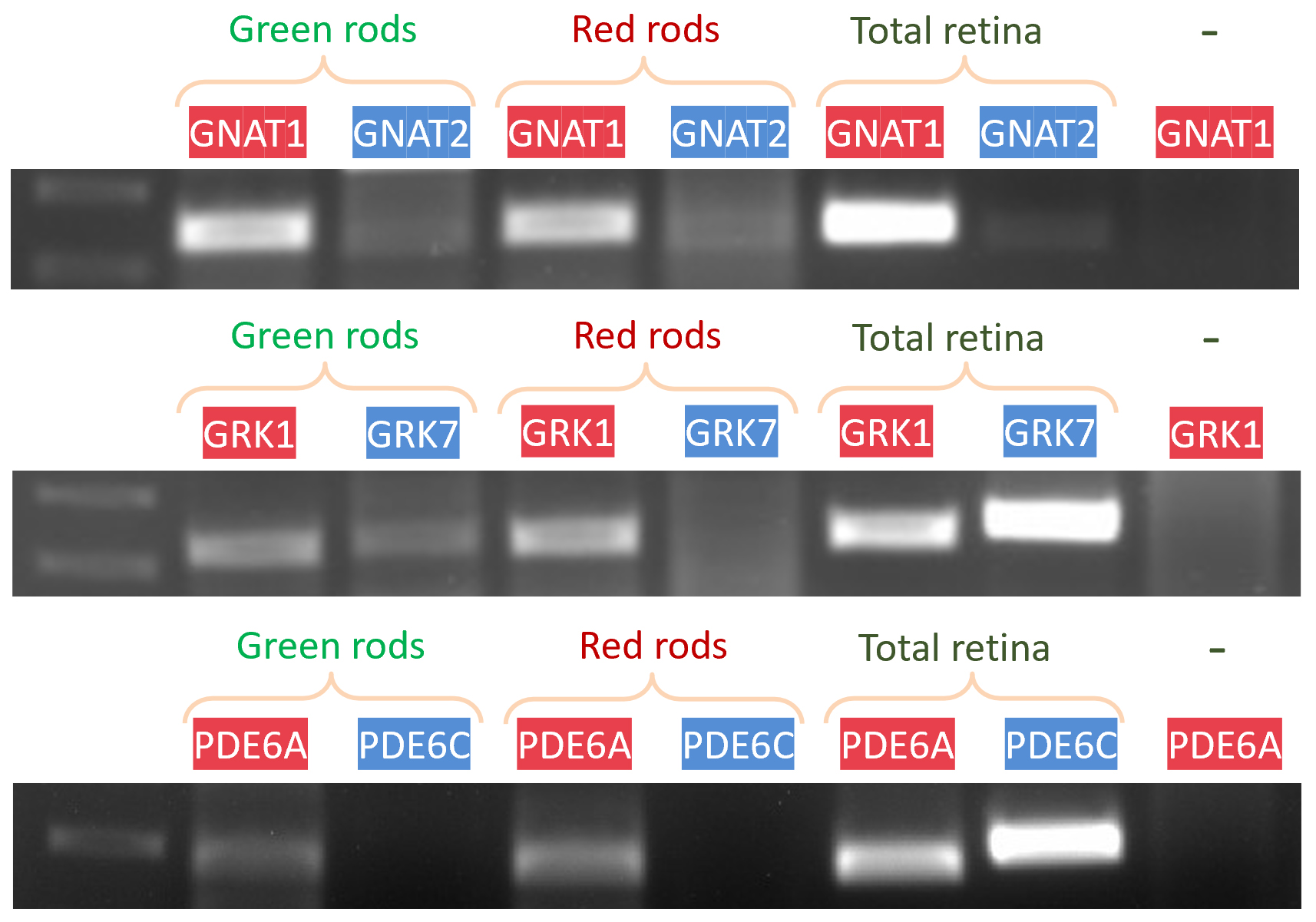
Figure 7. Gel electrophoresis of PCR products from two-cell samples. The expression of rod and cone isoforms of the transducin α-subunit (GNAT1, GNAT2), rhodopsin kinase (GRK1, GRK7), and phosphodiesterase (PDE6A, PDE6C) in homogeneous fractions of green and red rods. Genes listed above each gel band are markers for rods (red) or cones (blue). Each sample contains two cells. “Total retina” represents RNA from the whole retina and serves as a positive control. Raw and normalized intensity values for each lane are presented in Supplementary Table S1.
General notes and troubleshooting
General notes
The protocol presented in this article offers researchers a distinctive approach involving selective testing and material collection, followed by analysis. Although we developed this method for the comparative analysis of protein isoform composition in the phototransduction cascades of red and green rods in amphibian retinas, its range of applications is much broader. It may be useful for the selective comparative analysis of genetic material, proteins, signaling metabolites, and other components of different types of photoreceptors from a single retina. At the same time, the method can be relatively easily adapted to cones as well as rods.
Troubleshooting
Problem 1: One significant difficulty we encountered is the contamination of the environment surrounding the cell by products from neighboring cell destruction.
Possible cause: mRNA released from damaged cells can adsorb onto the surface of the collected photoreceptor, leading to distorted gene-expression measurements and false-positive results.
Solution: Implement measures to reduce extracellular RNA contamination, including controlled treatment with ribonucleases (RNases) and sequential washing of the captured cell in multiple solutions to eliminate residual contaminating RNA.
Acknowledgments
Conceptualization, M.F.; Investigation, O.C., D.N.; Writing—Original Draft, O.C.; Writing—Review & Editing, O.C., M.F., D.N.; Figure creation, O.C.; PCR protocol optimization, D.M., O.C.; Primer design, V.V.; Material procurement, V.V.; Funding acquisition, M.F.; Supervision, M.F.
This research was funded by an RSF grant #24-24-00484 to M.F.
All figures were created using Microsoft PowerPoint.
Competing interests
The authors declare no conflicts of interest.
Ethical considerations
Animals were handled in accordance with the Council Directive of the European Communities (24 November 1986; 86/609/EEC), and the experimental protocol was approved by the local Institutional Animal Care and Use Committee (protocol # 4/22 at 28.01.2022).
References
- Lamb, T. D. (2022). Photoreceptor physiology and evolution: cellular and molecular basis of rod and cone phototransduction. J Physiol. 600(21): 4585–4601. https://doi.org/10.1113/jp282058
- Boll, F. (1977). On the anatomy and physiology of the retina. Vision Res. 17(11–12): 1249–1265. https://doi.org/10.1016/0042-6989(77)90112-2
- Rosenkranz, J. (1977). New Aspects of the Ultrastructure of Frog Rod Outer Segments. Int Rev Cytol. 50(1977): 25–158. https://doi.org/10.1016/s0074-7696(08)60098-4
- Hisatomi, O., Takahashi, Y., Taniguchi, Y., Tsukahara, Y. and Tokunaga, F. (1999). Primary structure of a visual pigment in bullfrog green rods. FEBS Lett. 447(1): 44–48. https://doi.org/10.1016/s0014-5793(99)00209-4
- Astakhova, L. A., Novoselov, A. D., Ermolaeva, M. E., Firsov, M. L. and Rotov, A. Y. (2021). Phototransduction in Anuran Green Rods: Origins of Extra-Sensitivity. Int J Mol Sci. 22(24): 13400. https://doi.org/10.3390/ijms222413400
- Chernyshkova, O., Belyakov, M., Meshalkina, D. and Firsov, M. (2025). Time-Resolved cAMP Level Determination in Frog Retina Samples Using LC–MS/MS. Bio Protoc. 15(17): e5431. https://doi.org/10.21769/bioprotoc.5431
- Astakhova, L. A., Samoiliuk, E. V., Govardovskii, V. I. and Firsov, M. L. (2012). cAMP controls rod photoreceptor sensitivity via multiple targets in the phototransduction cascade. J Gen Physiol. 140(4): 421–433. https://doi.org/10.1085/jgp.201210811
Article Information
Publication history
Received: Nov 4, 2025
Accepted: Jan 26, 2026
Available online: Feb 12, 2026
Published: Mar 5, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Chernyshkova, O. V., Nikolaeva, D. A., Meshalkina, D. A., Veligura, V. I. and Firsov, M. L. (2026). Combining Suction-Pipette Spectral Identification With Single-Cell RT-PCR to Make Differential Analyses of Amphibian Red and Green Rods. Bio-protocol 16(5): e5623. DOI: 10.21769/BioProtoc.5623.
Category
Neuroscience > Sensory and motor systems > Retina
Molecular Biology > DNA > PCR
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.