- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Purification of the Active-State G Protein-Coupled Receptor ADGRL4 for Cryo-Electron Microscopy Using a Modular Tag System and a Tethered mini-Gq

Published: Vol 16, Iss 5, Mar 5, 2026 DOI: 10.21769/BioProtoc.5617 Views: 659
Reviewed by: David PaulSrajan KapoorAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2025
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
ADGRL4 is an adhesion G protein-coupled receptor (aGPCR) implicated in tumour progression in multiple malignancies. We recently determined the first cryo-EM structure of active-state ADGRL4, revealing its weak coupling to the heterotrimeric G protein Gq and providing insights into its activation mechanism. Here, we describe a complete modular workflow for purifying active-state ADGRL4 over 2–3 days using a multifunctional tagging strategy incorporating multiple orthogonal detection, purification, and cleavage tags at the N-terminus as well as a tethered mini-Gq at the C-terminus. This configuration enhanced receptor cell-surface expression and stability and allowed different purification strategies to be tested during the development of the purification protocol. Although developed and optimised for ADGRL4, this approach is readily transferable to other weakly coupling aGPCRs or GPCRs where complex stability is a limiting factor for structural analysis.
Key features
• Complete workflow for purifying active-state ADGRL4 for cryo-EM analysis.
• Modular, multifunctional N-terminal tagging strategy supporting multiple orthogonal purification and detection methods without any negative effect on cell surface expression levels.
• Tethered mini-Gq increases stability and receptor cell-surface expression.
• Modular purification tagging configuration provides freedom to change purification methodologies without having to perform additional receptor engineering or cloning.
Keywords: Adhesion GPCRGraphical overview
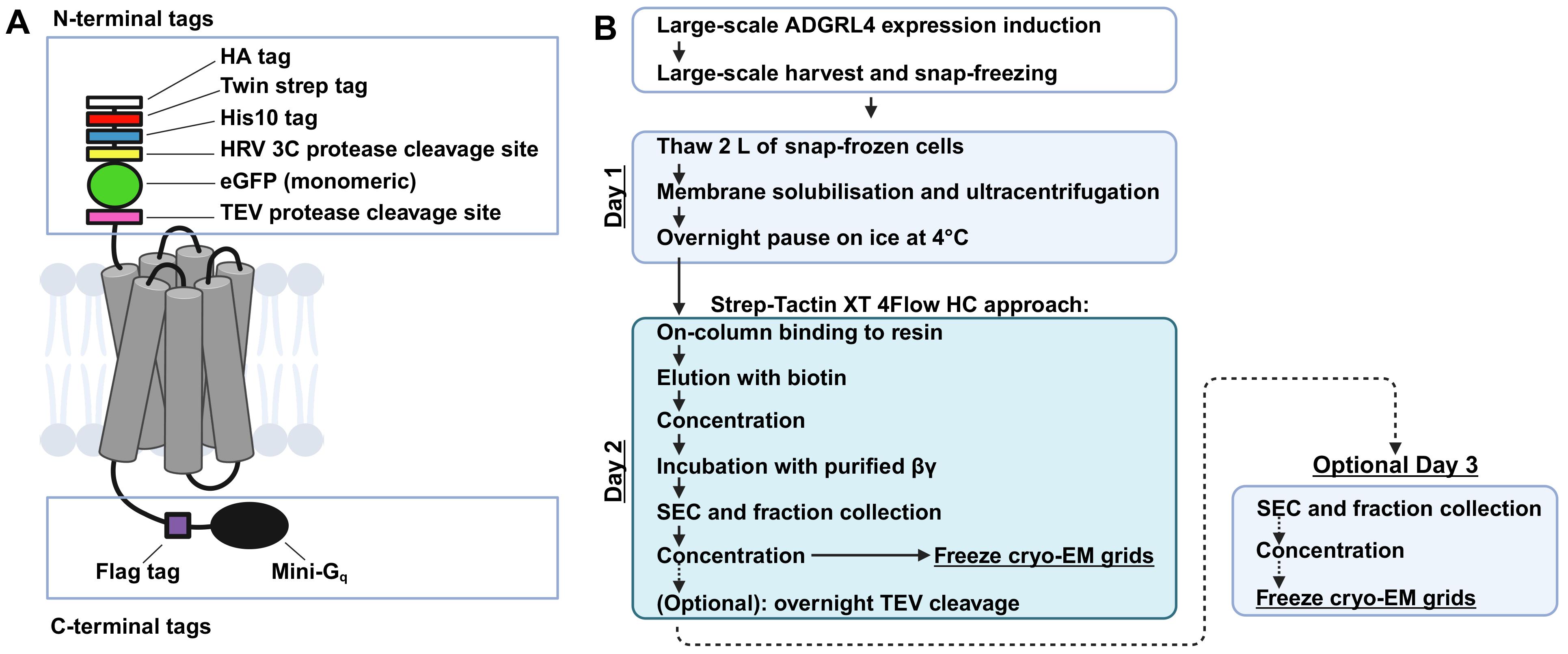
Graphical overview of ADGRL4 purification. (A) Schematic representation of active-state ADGRL4 illustrating the modular multifunctional tagging strategy. The N-terminal tag array provides orthogonal detection, purification, and protease cleavage options (HA, Twin-Strep, His10, HRV 3C cleavage site, monomeric eGFP, and TEV cleavage site). The C-terminus contains a FLAG tag followed by tethered mini-Gq. (B) Overview of the purification procedure. The steps shown for day 2 use Strep-Tactin XT 4Flow high-capacity resin, but any orthogonal purification approach compatible with the tagging strategy may be substituted. Day 3 (optional) outlines purification following overnight TEV cleavage to remove all N-terminal tags. This is optional, as the active-state ADGRL4 structure was determined using a protein that retained its N-terminal tags.
Background
Adhesion G protein-coupled receptors (aGPCRs) are a 32-member GPCR family in humans with diverse cellular roles [1].
ADGRL4 is an aGPCR implicated in tumour progression across multiple malignancies [2–22]. Using cryo-electron microscopy (cryo-EM), we recently determined the first high-resolution structure of active-state ADGRL4 to an overall nominal resolution of 3.1 Å [23], providing the first insights into its mechanism of activation. In the context of ADGRL4, the active state refers to a conformation of the receptor where the tethered agonist has bound to the orthosteric binding pocket, inducing a conformational change on the intracellular surface (outward movements of the intracellular ends of transmembrane helices 5 and 6) to form a cleft where the G protein Gq has coupled. (Subsequent use of the term active state in this protocol implies the above.) Active-state GPCRs are generally purified using detergent solubilisation in combination with engineered G proteins and/or stabilising binders. This has enabled numerous cryo-EM structures to be determined. Many different strategies have been developed, but explicit protocols for GPCR purification are generally unavailable, as they are usually terse descriptions in the methods section of a structural biology paper. An essential aspect of the purification procedure is the maintenance of the receptor complex during purification, which often requires careful choice of the GPCR construct expressed and the exact conditions [type and concentration of detergent(s), buffers, and salts] for the purification. The multi-parameter nature of purifying GPCRs thus makes it particularly challenging, particularly for researchers unfamiliar with the biochemistry of GPCRs; as such, we hope the detailed notes presented here will be useful. This protocol outlines the full workflow for purifying active-state ADGRL4 and can be completed in 2–3 days.
We developed our purification strategy for active-state ADGRL4 after first identifying its G protein coupling partner [23]. Using a NanoBiT split-luciferase complementation system, we found that ADGRL4 couples weakly to Gq. We subsequently observed that making an ADGRL4-mini-Gq chimera by genetically fusing mini-Gq to the C-terminus of active-state ADGRL4 markedly improved both receptor cell-surface expression and stability [23]. We therefore incorporated a C-terminal tethered mini-Gq (version R76 [24,25], capable of binding both βγ subunits of the heterotrimeric G protein complex; sequences in Dataset S1) to the constructs used for structural studies. Recruitment of βγ by the tethered mini-G increased the size of the receptor complex and introduced a distinct structural feature that served as a fiducial marker during cryo-EM particle alignment.
To maximise purification flexibility, we designed a modular system of N- and C-terminal detection, purification, and protease-cleavage tags, which did not adversely affect ADGRL4 cell-surface expression [23]. Our goal was to create a tagging architecture providing multiple orthogonal purification and detection options without requiring additional construct engineering. Most tags were positioned at the N-terminus immediately downstream of the native signal peptide sequence. The N-terminal cassette (Graphical overview; sequences in Dataset S1) consisted of (1) an HA tag for flow cytometry detection and HA-Trap purification, (2) a Twin-Strep tag for Strep-Tactin XT 4Flow high-capacity purification, (3) a His10 tag for nickel-nitrilotriacetic acid (Ni-NTA) purification, (4) an HRV 3c protease site to remove upstream tags, (5) monomeric eGFP (A206K) for flow cytometry detection and GFP-Trap purification, selected for its inability to self-dimerise [26], and (6) a TEV cleavage site to remove the entire N-terminal tag module. N-terminal tags were separated by GSG linkers. The TEV cleavage site was followed by a GSGGSG linker preceding the ADGRL4 sequence.
The C-terminus contained a FLAG tag for optional flow cytometry detection and FLAG-Trap purification, followed by the tethered mini-Gq (Graphical Overview; sequences in Dataset S1). To ensure adequate flexibility for the tethered mini-Gq, two GGSGSG linkers flanked the eight-residue FLAG tag. The C-terminal tethering of the mini-Gq was found to increase surface expression and improve stability of active-state ADGRL4 [23].
For structural studies, the ADGRL4 construct was packaged into lentivirus and used to generate an inducible HEK293 GnTI- TetR-based expression cell line, which was subsequently expanded at large scale [23]. The protocol for generating the ADGRL4 inducible HEK293 GnTI- TetR expression cell line is described in a companion Bio-protocol manuscript [27]. The use of a stable inducible cell line provides higher expression and better scalability compared with transient transfection and prevents the expression of intracellular, misfolded, and inactive receptor [28]. For purification, we selected the Twin-Strep tag in combination with Strep-Tactin XT 4Flow high-capacity resin, although any of the alternative orthogonal purification tags included in the design could be used interchangeably [23].
This protocol yields a high-purity active-state ADGRL4-mini-Gq-βγ complex suitable for cryo-EM grid preparation. Although optimised for ADGRL4, the tagging and mini-G protein tethering strategy and purification workflow are broadly applicable to other active-state aGPCRs and to weakly coupling or unstable GPCR-G protein complexes, particularly in cases where receptor instability has hindered structural characterisation.
Materials and reagents
Biological materials
1. Active-state ADGRL4 (version CTF2B) HEK293 GnTI- TetR stable cell line (stable suspension cell line with tetracycline inducible expression of the CTF2B active-state variant of ADGRL4. Available on request from Dr David Favara). Plasmids used to generate this stable inducible cell line (along with the detailed workflow for cell line development) are available on request from Dr David Favara.
2. Purified Gβγ dimer (69 mg/mL) [23] comprising human Gβ1 and Gγ2 (incorporating a C68S mutation to prevent post-translational lipidation). Expression and purification steps are detailed in the supplementary data section of [25]. Store at -80 °C. Available on request from Dr David Favara.
Reagents
1. Roche cOmplete, EDTA-free protease inhibitor cocktail tablets (Roche, catalog number: 11873580001); store at 4 °C
2. Phosphate-buffered saline (PBS), pH 7.4, 500 mL (Gibco, catalog number: 10010015)
3. Fetal bovine serum (FBS) tetracycline-free, 500 mL (Biosera, catalog number: FB-1001T/500); store at -20 °C
4. Trypan Blue solution, 0.4% (Gibco, catalog number: 15250061)
5. Anti-HA monoclonal antibody conjugated to APC, 200 μL (Miltenyi Biotec, catalog number: 130-123-553); store at 4 °C
6. HEPES buffer (molecular biology grade, ≥99%), 1 kg (Fisher BioReagents, catalog number: BP3101-1)
7. 4 N NaOH, 4 L (Supelco, catalog number: 1115845000)
8. 1 M NaOH, 500 mL (Supelco, catalog number: 79724-500ML)
9. PES membrane filter, 0.22 μm pore size (Millipore, catalog number: GPWP04700)
10. NaCl (Molecular biology grade, ≥99%), 500 g (Sigma-Aldrich, catalog number: S3014-500G)
11. MgCl2 hexahydrate (molecular biology grade, ≥99%), 500 g (Fisher BioReagents, catalog number: BP214500)
12. Glycerol (molecular biology grade, ≥99%), 1 L (Sigma-Aldrich, catalog number: G5516-1L)
13. Tris-(2-Carboxyethyl)phosphine, hydrochloride (TCEP) (molecular biology grade, ≥98%), 1 g (Sigma-Aldrich, catalog number: 75259-1G)
14. Apyrase (500 units/mL), 50 units (NEB, catalog number: M0398L); store at -20 °C
15. Phenylmethylsulfonyl fluoride (PMSF) protease inhibitor, 5 g (Thermo Scientific, catalog number: 36978)
16. Isopropanol (molecular biology grade, 99.5%), 1 L (Acros, catalog number: 10215331)
17. Lauryl maltose neopentyl glycol (LMNG), 25 g (Anatrace, catalog number: NG310 25 GM); store at -20 °C
18. Cholesteryl hemisuccinate tris salt (CHS), 5 g (Sigma-Aldrich, catalog number: C6013-5G); store at -20 °C
19. Benzonase nuclease, 25 KU (Millipore, catalog number: E1014-25KU); store at -20 °C
20. Standard SDS-PAGE reagents and pre-stained protein ladder (any suitable supplier)
21. Iba Lifesciences Strep-Tactin XT 4Flow high-capacity resin, 50 mL, 50% suspension (Iba Lifesciences, catalog number: 2-5030-025); store at 4 °C
22. Iba Lifesciences biotin, 5 g (Iba Lifesciences, catalog number: 2-1016-005); store at 4 °C
23. Iba Lifesciences buffer XT-R, 250 mL (Iba Lifesciences, catalog number: 2-1045-250); store at 4 °C
24. Purified TEV protease (4.4 mg/mL); expression and purification steps detailed in [29]. Alternatively, this can be purchased from NEB (NEB, catalog number: P8112S)
Solutions
1. Flow cytometry buffer (see Recipes)
2. 3 M NaCl (see Recipes)
3. 1 M MgCl2 (see Recipes)
4. 300 mM HEPES (see Recipes)
5. 50% glycerol (see Recipes)
6. 0.5 M TCEP (see Recipes)
7. 500 units/mL apyrase (see Recipes)
8. 0.5 M biotin (see Recipes)
9. 200 mM PMSF (see Recipes)
10. 5% LMNG (see Recipes)
11. 5% LMNG and 0.5% CHS (see Recipes)
12. Cell harvest buffer (see Recipes)
13. Solubilisation buffer (see Recipes)
14. Wash buffer (see Recipes)
15. Elution buffer (see Recipes)
16. SEC running buffer without glycerol (see Recipes)
17. SEC running buffer with 10% glycerol (see Recipes)
Recipes
1. Flow cytometry buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| PBS | 98% | 19.6 mL |
| Tetracycline-free FBS | 2% | 0.4 mL |
| Total | n/a | 20 mL |
Note: Make up fresh and store at 4 °C. Keep on ice when in use.
2. 3 M NaCl
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| NaCl (≥99% pure) | 3 M | 175.32 g |
| Ultrapure water | see below | |
| Total | n/a | 1,000 mL |
a. Weigh 175.32 g of NaCl and dissolve in 700 mL of ultrapure water.
b. Bring to a total volume of 1,000 mL with ultrapure water.
c. Filter through a 0.22 μm pore size PES membrane filter into a sterile bottle and store at room temperature.
3. 1 M MgCl2
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| MgCl2 hexahydrate (≥99% pure) | 1 M | 20.33 g |
| Ultrapure water | see below | |
| Total | n/a | 100 mL |
a. Weigh 20.33 g of MgCl2 hexahydrate and dissolve in 50 mL of ultrapure water.
b. Bring to a total volume of 100 mL with ultrapure water. Store at room temperature.
4. 300 mM HEPES
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| HEPES (≥99% pure) | 300 mM | 71.5 g |
| Ultrapure water | see below | |
| NaOH used to titrate to pH 7.5 at 4 °C | see below | |
| Total | n/a | 1,000 mL |
a. Weigh 71.5 g of HEPES and dissolve in 900 mL of 4 °C ultrapure water. Keep on ice.
b. Measure pH and titrate carefully with 4 N NaOH (coarse titration) and then 1 M NaOH (fine titration) to pH 7.5 at 4 °C. Before titration, calibrate the pH meter with chilled buffers.
c. Bring to a total volume of 1,000 mL with ultrapure water.
d. Filter through a 0.22 μm pore size PES membrane filter into a sterile bottle and store at 4 °C.
5. 50% glycerol
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Glycerol (≥99% pure) | 50% | 500 mL |
| Ultrapure water | 50% | 500 mL |
| Total | n/a | 1,000 mL |
a. Measure 500 mL of glycerol and mix with 500 mL of ultrapure water.
b. Autoclave at 121 °C for 15 min and store at 4 °C.
6. 0.5 M TCEP
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| TCEP (≥98% pure) | 0.5 M | 2.87 g |
| Ultrapure water | see below | |
| NaOH for titration to pH 7 at room temperature | see below | |
| Total | n/a | 20 mL |
a. Weigh 2.87 g of TCEP and dissolve it in 10 mL of ultrapure water.
b. Measure pH and titrate carefully with 4 N NaOH (coarse titration) and then 1 M NaOH (fine titration) to pH 7 at room temperature.
c. Bring to a total volume of 20 mL with ultrapure water.
d. Filter through a 0.22 μm pore size PES membrane filter into a sterile bottle.
e. Make up 1 mL aliquots and store at -20 °C.
Note: TCEP is a reducing agent that prevents the formation of non-native disulphide bonds. It is used to prevent deactivation of the TEV cysteine protease used for receptor cleavage.
7. 500 units/mL apyrase
Aliquoting for storage:
a. Source: apyrase 500 units/mL, 100 μL total, in 20 mM MES, 50 mM NaCl, 0.1 mM CaCl2, 1 mM DTT, 0.1% Tween-20, 50% glycerol.
b. Dispense 10 μL into ten 0.2 mL PCR tubes. Each aliquot contains 5 units apyrase at 500 units/mL.
c. Store at -20 °C.
Note: Apyrase degrades nucleoside triphosphates (ATP, GTP) and diphosphates (ADP, GDP) to their corresponding monophosphates and inorganic phosphate. This enzymatic activity depletes nucleoside tri- or diphosphates that could otherwise destabilise the active-state ADGRL4-mini-Gq-βγ complex. Activity typically requires Ca2+ or Mg2+. In this protocol, we use 1 mM MgCl2.
8. 0.5 M biotin
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Biotin | 0.5 M | 5 g |
| Ultrapure water | see below | |
| NaOH and HCl to adjust pH | see below | |
| Total | n/a | 40.94 mL |
a. Place a glass beaker (≥50 mL) with a magnetic stir bar on a stir plate. Add 12 mL of ultrapure water and start stirring.
b. Weigh 5 g of biotin powder and add gradually while stirring to form a suspension. At this concentration, expect poor solubility below a pH of 7.5–8.
c. Measure pH and carefully titrate pH upward with 1 M NaOH in 0.5 mL increments. Switch to 100–200 μL increments of 1 M NaOH when the suspension becomes less thick and clearer. Allow time for each addition to fully mix before adding the next.
d. The suspension should become clear at pH 8–9.
e. Once suspension is completely clear, measure pH. The final target pH is between 8.2 and 8.4 If required, carefully titrate downward with 1 M HCl in 50–100 μL increments.
f. Once pH has settled at 8.2–8.4, bring to a final total volume of 40.94 mL with ultrapure water.
g. Filter through a 0.22 μm pore size PES membrane filter into a sterile bottle and store at 4 °C.
9. 200 mM PMSF
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| PMSF | 200 mM | 3.484 g |
| Isopropanol | to a 100 mL total volume | |
| Total | n/a | 100 mL |
a. In a fume hood, weigh 3.484 g of PMSF into a beaker.
b. Add 80 mL of isopropanol and mix by swirling to completely dissolve all PMSF.
c. Once fully dissolved, bring to a total volume of 100 mL with additional isopropanol.
d. Aliquot into 10 mL portions in 15 mL tubes and store at -20 °C, protected from light.
Caution: PMSF is toxic and corrosive and should be prepared in a fume hood. Ensure that it is always carefully handled with gloves.
Note: Only add PMSF to aqueous buffers immediately before use due it being unstable in water.
10. 5% LMNG
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| LMNG | 5% | 1 g |
| Ultrapure water | to a 20 mL total volume | |
| Total | n/a | 20 mL |
a. In a clear beaker or 50 mL tube, weigh 1 g of LMNG, mix with 10 mL of ultrapure water, and mix gently until dissolved. Avoid vigorous shaking to minimise bubble formation.
b. Bring to a final volume of 20 mL with additional ultrapure water.
c. Filter through a 0.22 μm pore size PES membrane filter and aliquot into 1.5 mL microcentrifuge tubes.
d. Store at -20 °C.
11. 5% LMNG and 0.5% CHS
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| LMNG | 5% | 2 g |
| CHS | 0.5% | 0.2 g |
| Ultrapure water | to a 40 mL total volume | |
| Total | n/a | 40 mL |
a. In a clear beaker or 50 mL tube, weigh 2 g of LMNG, mix with 20 mL of ultrapure water, and mix gently until dissolved. Avoid vigorous shaking to minimise bubble formation.
b. Weigh 0.2 g of CHS and add this to the LMNG solution.
c. Bring to a final volume of 40 mL with additional ultrapure water.
d. Gently mix overnight at 4 °C in a 50 mL tube using a tube roller.
e. Filter through a 0.22 μm pore size PES membrane filter into a sterile 50 mL tube.
f. Store at -20 °C.
12. Cell harvest buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 300 mM HEPES NaOH pH 7.5 | 10 mM | 3.33 mL |
| cOmplete EDTA-free protease inhibitor tablets (1 tablet for 50 mL) | 1× | 2 tablets |
| Ultrapure water | 1× | 96.67 mL |
| Total | n/a | 100 mL |
Note: Make up fresh on the day of harvesting and keep chilled at 4 °C. Dissolving cOmplete EDTA-free protease inhibitor tablets will require a stir bar for approximately 10 min at 4 °C.
13. Solubilisation buffer
| Reagent | Final concentration in 100 mL | Quantity or volume |
|---|---|---|
| 300 mM HEPES pH 7.5 | 20 mM | 6.67 mL |
| 3 M NaCl | 100 mM | 3.33 mL |
| 1 M MgCl2 | 10 mM | 1 mL |
| 50% glycerol | 20% | 40 mL |
| 0.5 M TCEP | 100 μM | 20 μL |
| 250 units/μL benzonase | 25 units/mL | 10 μL |
| 500 units/mL apyrase | 0.025 units/mL | 5 μL |
| cOmplete EDTA-free protease inhibitor tablets (1 tablet for 50 mL) | 1× | 2 tablets |
| 200 mM PMSF | 2 mM | 1 mL |
| Volume of cell pellet* | ~25 mL for 2 L of cells* | |
| Ultrapure water* | 3.965 mL | |
| Total | n/a | 80 mL |
* Include the volume of the thawed cell pellet in your calculation. When snap-freezing cells in liquid nitrogen, make a note of the volume of the pellet in the 50 mL centrifuge tube. For 2 L of HEK293 GnTI- TetR cells, this amounts to a ~25 mL volume.
Note: Make up fresh before use and keep on ice at 4 °C. Only add the benzonase and PMSF immediately before use. During solubilisation, 20 mL of 5% LMNG, 0.5% CHS is added to the above mixture, bringing the total volume to 100 mL (final detergent concentration being 1% LMNG, 0.1% CHS).
14. Wash buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 300 mM HEPES pH 7.5 | 20 mM | 6.67 mL |
| 3 M NaCl | 100 mM | 3.33 mL |
| 1 M MgCl2 | 10 mM | 1 mL |
| 50% glycerol | 20% | 40 mL |
| 0.5 M TCEP | 100 μM | 20 μL |
| 5% LMNG, 0.5% CHS | 0.02% LMNG, 0.002% CHS | 400 μL |
| cOmplete EDTA-free protease inhibitor tablets (1 tablet for 50 mL) | 1× | 2 tablets |
| 200 mM PMSF | 2 mM | 1 mL |
| Ultrapure water | 47.58 mL | |
| Total | n/a | 100 mL |
Note: Make up fresh before use and keep on ice at 4 °C.
15. Elution buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 300 mM HEPES pH 7.5 | 20 mM | 2.67 mL |
| 3 M NaCl | 100 mM | 1.33 mL |
| 1 M MgCl2 | 10 mM | 400 μL |
| 50% glycerol | 20% | 16 mL |
| 0.5 M TCEP | 100 μM | 8 μL |
| 5% LMNG, 0.5% CHS | 0.02% LMNG, 0.002% CHS | 160 μL |
| 0.5 M biotin | 50 mM | 4 mL |
| cOmplete EDTA-free protease inhibitor tablets (1 tablet for 50 mL) | 1× | 1 tablet |
| 200 mM PMSF | 2 mM | 400 μL |
| Ultrapure water | 15.032 mL | |
| Total | n/a | 40 mL |
Note: Make up fresh before use and keep on ice at 4 °C.
16. SEC running buffer without glycerol
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 300 mM HEPES pH 7.5 | 20 mM | 13.33 mL |
| 3 M NaCl | 150 mM | 10 mL |
| 1 M MgCl2 | 10 mM | 2 mL |
| 0.5 M TCEP | 100 μM | 40 μL |
| 5% LMNG | 0.03% LMNG | 1.2 mL |
| Ultrapure water | 173.43 mL | |
| Total | n/a | 200 mL |
Note: Make up fresh before use and keep on ice at 4 °C.
17. SEC running buffer with 10% glycerol
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 300 mM HEPES pH 7.5 | 20 mM | 3.33 mL |
| 3 M NaCl | 150 mM | 2.5 mL |
| 1 M MgCl2 | 10 mM | 500 μL |
| 50% glycerol | 10% | 10 mL |
| 0.5 M TCEP | 100 μM | 10 μL |
| 5% LMNG | 0.03% LMNG | 300 μL |
| Ultrapure water | 33.36 mL | |
| Total | n/a | 50 mL |
Note: Make up fresh before use and keep on ice at 4 °C.
Laboratory supplies
1. 96-well round (U) bottom plates (Thermo Scientific, catalog number: 163320)
2. 1 L polypropylene bottle assembly (Beckman Coulter, catalog number: C31597)
3. 50 mL centrifuge tubes (Corning, catalog number: CLS430828-100EA)
4. Ultracentrifuge tubes: Beckman Coulter PC tube with aluminium cap, 26.3 mL volume (Beckman Coulter, catalog number: 355618)
5. Econo-column chromatography column, 2.5 × 10 cm (Bio-Rad, catalog number: 7372512)
6. Microcentrifuge tubes, 1.5 mL (Pierce, catalog number: 69715)
7. Amicon Ultra-15 centrifugal filter 100 kDa MWCO (15 mL sample volume) (Millipore, catalog number: UFC910024)
8. Amicon Ultra-4 centrifugal filter 100 kDa MWCO (4 mL sample volume) (Millipore, catalog number: UFC810024)
9. Amicon Ultra-0.5 centrifugal filter 100 kDa MWCO (0.5 mL sample volume) (Millipore, catalog number: UFC510096)
10. Direct Detect assay-free cards, 50 cards (Merck, catalog number: DDAC00010)
11. UltrAuFoil R1.2/1.3 300 mesh Au grids (Quantifoil, catalog number: Q350AR13A)
Equipment
1. Flow cytometer (Sony, model: ID7000 Spectral Cell Analyzer)
2. Temperature-controlled floor-standing centrifuge for 1 L bottles (Beckman Coulter, Avanti JXN-26 Refrigerated Centrifuge, model: JXN-26)
3. Ultrapure water system (Sartorius, model: Arium Pro Ultrapure Water System)
4. Beckman Coulter Type 70 Ti fixed-angle rotor (Beckman Coulter, catalog number: 337922)
5. Temperature-controlled floor-standing ultracentrifuge (Beckman Coulter Optima L-100XP Ultracentrifuge, model: L-100XP)
6. End-over-end rotator (Elmi Rotamix RM1, catalog number: ROTAMIX RM1)
7. Shimadzu HPLC system (Shimadzu Corporation) comprising the following components: Shimadzu degasser module (DGU-20A), Shimadzu solvent delivery module (LC-20AD), Shimadzu autosampler module (SIL-20AC), Shimadzu column oven module (CTO-20AC), Shimadzu UV-detector module (SPD-20A), Shimadzu fluorescence detector module (RF-20A), and Shimadzu fraction collector module (FRC-40)
8. HPLC guard column, Agilent Bio SEC-5 guard column (300 Å, 7.8 × 50 mm, 5 μm) (Agilent, catalog number: 5190-2530)
9. HPLC column, Agilent Bio SEC-5 column (300 Å 4.6 × 300 mm, 5 μm) (Agilent, catalog number: 5190-2528)
10. Temperature-controlled benchtop 1.5 mL tube centrifuge (Eppendorf, model: 5418 R)
11. Protein detection system, Direct Detect infrared spectrometer (Merck, catalog number: DDHW00010)
12. Automated vitrification system for cryo-EM (FEI, model: Vitrobot Mark IV)
13. Ethane cryostat and temperature controller (MiTeGen, catalog number: ML-TCCS-001); design and operation are described in [30]
14. Glow-discharge plasma system (Fischione, catalog number: Model 1070)
Procedure
A. Tag design for structural studies
1. Confirm that the aGPCR construct has the following N-terminal tags (see Dataset S1 for sequences and linker arrangements):
a. HA tag: Enables quantification of cell-surface expression by flow cytometry in live cells; also supports purification using HA-Trap agarose resin.
b. Twin-Strep tag: Enables purification with Strep-Tactin XT 4Flow high-capacity resin.
c. His10 tag: Enables purification with Ni-NTA agarose resin.
d. HRV 3c protease site: Allows removal of upstream N-terminal tags (HA, Twin-Strep, His10)
e. eGFP (A206K): Enables assessment of whole-cell expression by flow cytometry in live cells; also supports purification using GFP-Trap agarose resin. The A206K mutation prevents self-dimerisation.
f. TEV protease site: Allows removal of the entire N-terminal tag cassette.
2. Confirm that the aGPCR construct has the following C-terminal tags (see Dataset S1 for sequences and linker arrangements):
a. FLAG tag: Optional quantification using flow cytometry; also supports purification using FLAG-Trap agarose resin.
b. Tethered mini-Gq: Binds to ADGRL4’s intracellular loops only when the receptor is in its active-state conformation. Activation requires exposure of the N-terminal tethered agonist, which binds to the orthosteric binding pocket. This is achieved by expressing the C-terminal fragment (CTF) version of ADGRL4.
Note: The modular N- and C-terminal tagging strategy described here was validated for active-state ADGRL4 and did not adversely affect receptor expression, stability, or activation. If adapting this protocol to other active-state aGPCRs or GPCR targets, it is important to empirically assess tag compatibility and confirm that protein stability and function are not adversely affected.
B. Harvesting large-scale expression
1. Perform large-scale ADGRL4 expression using an inducible suspension HEK293-based mammalian system for high-level expression [23]. A control flask (non-expressing cells) should be grown as a negative control for flow cytometry assessment of cell-surface ADGRL4 expression at the time of harvesting. We used 500 mL of medium in 2 L roller bottles stood on their end and placed in a shaking incubator, with a total of 3–6 L of cultures required. This is described in detail within a separate companion Bio-Protocol submission, along with detailed instructions for stable HEK293 GnTI- TetR cell line generation [27].
2. On the day of large-scale cell harvesting, prepare the required volume of cell harvest buffer containing HEPES and protease inhibitors (see Recipes for details). Keep on ice at 4 °C. Recommended cell harvest buffer volumes for different culture sizes are shown in Table 1. These contain an excess.
Table 1. Recommended cell harvest buffer volumes
| Culture volume | Cell harvest buffer volume |
|---|---|
| 1 L | 20 mL |
| 2 L | 40 mL |
| 4 L | 80 mL |
| 6 L | 120 mL |
| 8 L | 160 mL |
| 12 L | 240 mL |
3. Perform quality control (QC) checks before starting large-scale harvesting: assess cell count, viability, and ADGRL4 surface expression.
Note: This QC step serves as a decision point to confirm cell viability and that sufficient ADGRL4 has been expressed for purification. Cultures exhibiting a low percentage of cells expressing ADGRL4 at the plasma membrane after induction (<10%) typically yield insufficient protein and should not be processed further.
a. Collect ~0.5 mL from each ADGRL4 expression suspension culture flask and control flask.
b. Determine cell count and viability using trypan blue.
c. Assess surface ADGRL4 expression by flow cytometry:
i. The assay requires an N-terminal HA tag.
ii. Prepare 20 mL of chilled flow cytometry buffer (see Recipes).
iii. Transfer 200 μL of each sample to a standard 96-well U-bottom plate.
iv. Centrifuge cells at 500× g for 5 min at room temperature.
Critical: Use U-bottom plates to retain cell pellets after centrifugation.
v. During centrifugation, prepare the antibody master mix (Table 2):
Table 2. Antibody master mix
| Reagent | Final dilution | 1 well | 5 wells |
|---|---|---|---|
| Anti-HA-APC antibody (see Reagents) | 1:50 | 1 μL | 5 μL |
| Flow cytometry buffer | 49 μL | 245 μL | |
| Total | 50 μL | 250 μL |
vi. After centrifugation, carefully discard the supernatant.
vii. Add 50 μL of antibody master mix to each sample and control well.
viii. Wrap the 96-well plate in foil to protect from light and incubate at room temperature for 15 min.
ix. After incubation, add 200 μL of flow cytometry buffer and centrifuge cells at 500× g for 3 min. Discard the supernatant.
x. Wash the pellet with an additional 200 μL of flow cytometry buffer and repeat centrifugation (500× g for 3 min). Discard the supernatant.
xi. Resuspend cell pellets in 200 μL of flow cytometry buffer for analysis.
xii. Acquire samples using a Sony ID7000 Spectral Cell Analyzer or equivalent. Use control (non-expressing) cells to define negative gating. APC excitation wavelength: 633 nm; emission wavelength: 660 nm.
4. Proceed with large-scale harvesting:
a. After QC, discard control cultures. Sterility is no longer required. Ensure pre-chilled harvest buffer is ready on ice (Table 1).
b. Centrifuge cultures at 500× g for 15 min at 4 °C in 1 L centrifuge bottles. Carefully decant medium without disturbing the cell pellet.
Caution: Decant slowly and carefully as cell pellets may be loose.
c. Resuspend each cell pellet (corresponding to 1 L of cell culture volume) in 20 mL of 4 °C cell harvest buffer.
d. Transfer resuspended cells into pre-chilled 50 mL centrifuge tubes (one per 1 L culture) and centrifuge at 500× g for 10 min at 4 °C.
e. After centrifugation, carefully remove the supernatant, leaving ~2 mL above the cell pellet. A 1 L culture of suspension HEK293 GnTI- TetR cells typically yields a ~10 mL-sized cell pellet.
Critical: Keep samples at 4 °C at all times.
f. Snap-freeze cell pellets in liquid nitrogen for storage.
C. Purification day 1
1. This protocol describes the purification of active-state ADGRL4 (from 2 L of harvested cell pellets) using Strep-Tactin XT 4Flow high-capacity resin (Figure 1). The specifics of the tethered G protein and all purification variables are specific for purifying active-state ADGRL4 and would need modification if applied to another aGPCR.
Note: Any orthogonal purification strategy compatible with the tagging strategy may be used instead.
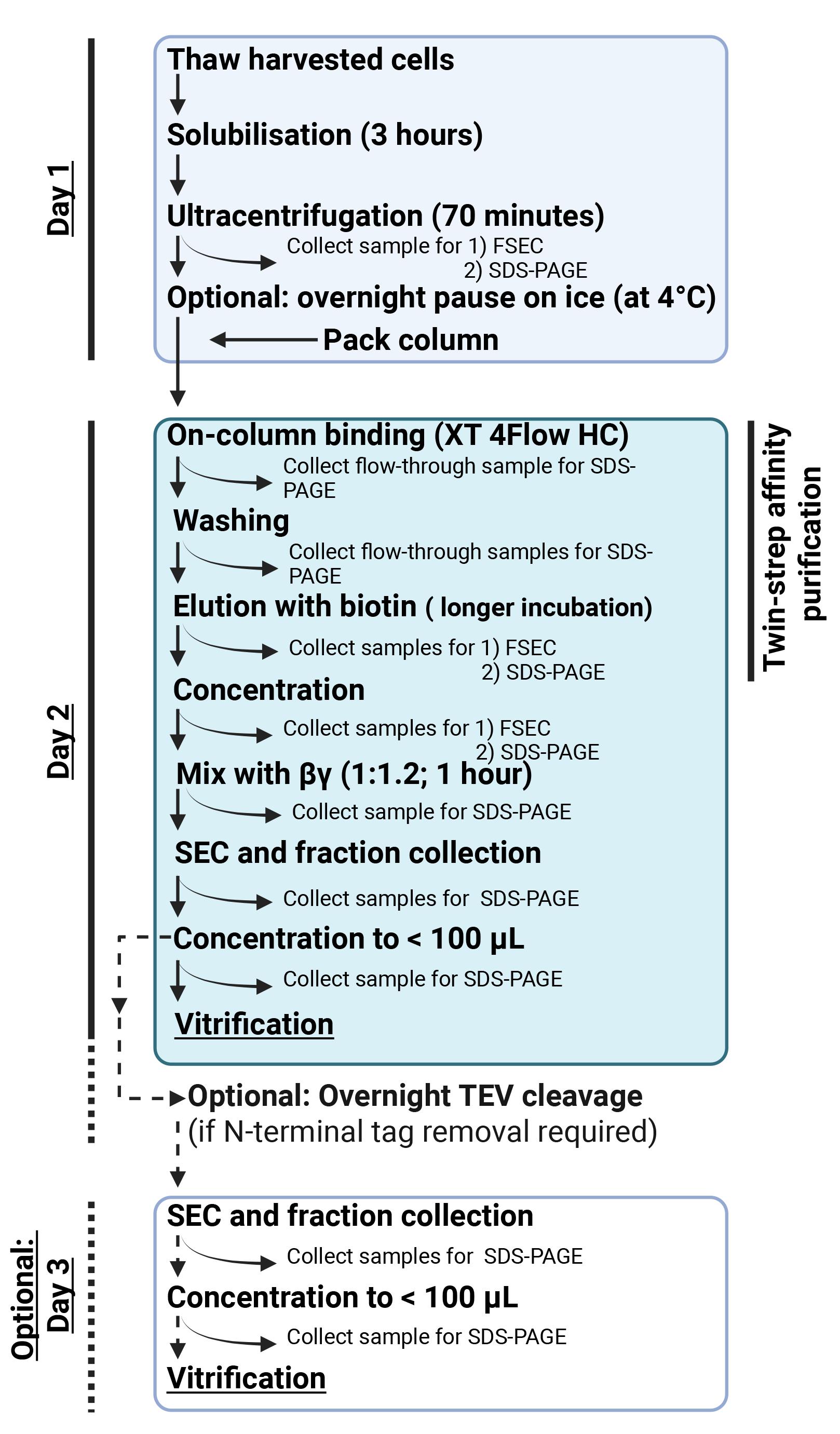
Figure 1. Overview of the active-state ADGRL4 purification workflow using Strep-Tactin XT 4Flow high-capacity resin. Day 1 includes cell thawing, detergent solubilisation, and clarification of the extracted membranes. Day 2 comprises affinity purification, complex assembly, SEC polishing, and vitrification. Day 3 is optional and is only required when removing the N-terminal tag array by TEV protease. Our previously reported active-state ADGRL4 structure was determined using a non-cleaved protein, and TEV digestion was not necessary. The affinity purification step may be substituted with any orthogonal purification approach compatible with the tagging strategy.
2. During purification, collect samples for SDS–PAGE analysis as directed in the protocol (see Figure 2 for an example of this). This is essential for troubleshooting purification issues. If required, these samples can also be assessed by western blotting.
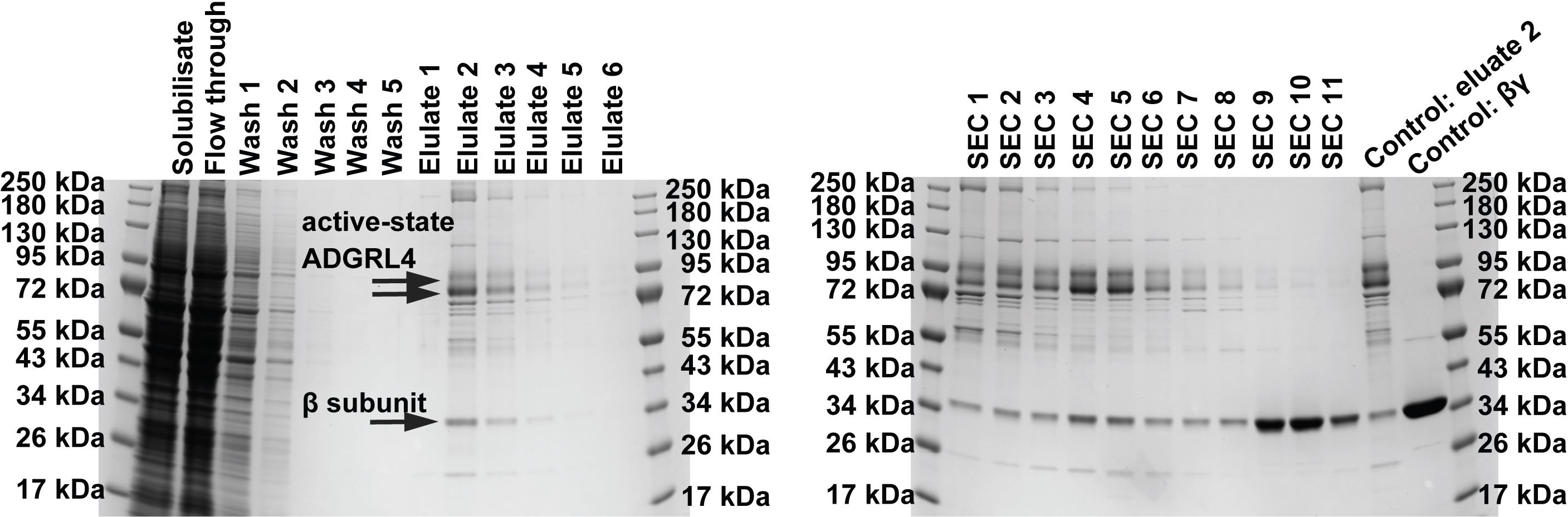
Figure 2. Representative SDS-PAGE analysis of purified active-state ADGRL4 (version CTF2B), reproduced from [23]. Samples should be collected at multiple stages of purification to monitor protein integrity and purity and to aid troubleshooting. Top arrows indicate active-state ADGRL4, while the bottom arrow indicates GNB1 (G protein subunit β1 endogenously expressed in the HEK293 GnTI- TetR stable cell line).
3. Prepare fresh purification buffers (see Recipes) and keep these at 4 °C. For purifying 2 L of HEK293 GnTI- TetR cells, prepare:
a. 55 mL of solubilisation buffer (without detergent). This starting volume is calculated so that the addition of the cell pellet and the later addition of detergent stock bring the total solubilisation mixture to 100 mL. When diluted to this final volume, the buffer contains 20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 20% glycerol, 100 μM TCEP, 25 units/mL benzonase, 1× complete protease inhibitors, and 2 mM PMSF. When applied to the 2 L cell pellet, the buffer will initially bring the slurry to ~80 mL. Add benzonase and PMSF to the buffer immediately before applying it to the pellet.
b. 20 mL of 5% LMNG and 0.5% CHS detergent stock. During solubilisation, add this to the ~80 mL slurry to bring the total volume to 100 mL, yielding a final detergent concentration of 1% LMNG, 0.1% CHS.
c. 100 mL of wash buffer (20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 20% glycerol, 100 μM TCEP, 0.02% LMNG, 0.002% CHS, 1× complete protease inhibitors, and 2 mM PMSF). Add PMSF immediately before use.
d. 40 mL of elution buffer (20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 20% glycerol, 100 μM TCEP, 0.02% LMNG, 0.002% CHS, 50 mM biotin, 1× complete protease inhibitors, and 2 mM PMSF). Add PMSF immediately before use.
e. 200 mL of SEC running buffer without glycerol (20 mM HEPES, 150 mM NaCl, 10 mM MgCl2, 100 μM TCEP, and 0.03% LMNG).
4. Thaw cell pellets. Remove a 2-L equivalent of frozen cell pellets from liquid nitrogen storage and thaw in lukewarm water for 20 min until a thick semi-thawed slurry forms.
Critical: All subsequent purification steps must be performed at 4 °C.
5. Initial resuspension: Transfer the semi-thawed slurry (~25 mL) into a pre-chilled beaker with a stir-bar. Add 55 mL of solubilisation buffer (without detergent) to bring the total volume to 80 mL. Stir the slurry for 45 min at 4 °C.
6. Start a 3-h detergent solubilisation: Stop the stirring and gently add 20 mL of 5% LMNG and 0.5% CHS detergent to reach a 100 mL total volume (final detergent concentration: 1% LMNG, 0.1% CHS). Use the pipette to gently stir the mixture whilst avoiding bubble formation. Divide the 100 mL slurry into four pre-chilled 50 mL centrifuge tubes (~25 mL each). Place each tube in an end-over-end rotator (we use an Elmi Rotamix RM1 rotator) and mix gently (maximum 8 rpm) for 3 h at 4 °C. Excessive agitation produces bubbles, which are to be avoided.
7. During the 3-h solubilisation period, prepare equipment and resin:
a. Cool down the ultracentrifuge and rotor to 4 °C.
b. Prepare the Strep-Tactin gravity-flow column. Use a Bio-Rad 2.5 × 10 cm gravity flow Econo-Column or equivalent (see Laboratory supplies).
i. Gently resuspend the Iba Lifesciences Strep-Tactin XT 4Flow high-capacity resin. This is a 50% slurry and requires a few minutes of gentle rolling to resuspend.
ii. Once fully resuspended, pipette 10 mL of resin into the gravity flow column (stopcock closed).
iii. Gently mix to ensure that the resin has been uniformly spread across the column’s base and remove any bubbles using a pipette. Cover the top of the column and leave it at 4 °C to settle.
8. Ultracentrifugation:
a. Once solubilisation is complete, transfer the solubilised slurry to pre-chilled ultracentrifuge tubes. We use Beckman Coulter 26.3 mL thick-wall ultracentrifuge tubes (see Laboratory supplies).
Note: Do not underfill ultracentrifuge tubes. We apply ~25 mL to each ultracentrifuge tube (maximum volume 26.3 mL).
b. Clarify the solubilisate by ultracentrifugation at 136,000× g for 70 min at 4 °C. We use a Beckman Coulter Type 70 Ti rotor and a Beckman Coulter Optima L-100XP Ultracentrifuge (see Equipment).
9. Immediately after ultracentrifugation, carefully collect and store the clarified solubilisate supernatant whilst avoiding the white precipitates at the surface and the pellet at the bottom. Aliquot 10 μL of the clarified solubilisate and approximately 5 μL of the pellet for downstream SDS-PAGE analysis (the pellet can be collected with a spatula). The remainder of the cell pellet and solubilisate containing white precipitates can be discarded.
Note: The collected clarified solubilisate supernatant can be stored overnight at 4 °C, with affinity purification commencing the following morning.
Critical: For each subsequent purification step, collect samples for SDS-PAGE analysis. This is critical for troubleshooting purification problems. If required, these samples can also be assessed by western blotting.
10. Assess solubilisate quality by fluorescence-detection size-exclusion chromatography (FSEC):
a. Confirm that the saved solubilisate contains a monodispersed fluorescent peak corresponding to the expected molecular weight of active-state ADGRL4 and the LMNG detergent micelle.
b. We use a Shimadzu HPLC system (equipped with a fluorescence detector) with an Agilent Bio SEC-5 guard column (300 Å, 7.8 × 50 mm, 5 μm) and an Agilent Bio SEC-5 column (300 Å 4.6 × 300 mm, 5 μm) (see Equipment).
c. Run the SEC running buffer without glycerol (see Recipes) through the HPLC system at 0.35 mL/min for 60 min before injecting the solubilisate sample. Fluorescence detector settings: excitation 475 nm, emission 515 nm.
d. Inject 20 μL of solubilisate and record the monodispersed fluorescent peak.
11. Equilibrate the Strep-Tactin column with wash buffer (see Recipes):
a. Open the stopcock and wash through with 2 × 5 mL of wash buffer.
b. Add a final 5 mL of wash buffer and close the stopcock when the meniscus reaches the resin surface.
c. Allow the column to equilibrate overnight at 4 °C.
12. Overnight pause point.
D. Purification day 2
1. Load the clarified solubilisate onto the Strep-Tactin column in 20–30 mL batches with the stopcock open. Allow it to pass fully through the column. Save 10 μL of the total flowthrough for SDS-PAGE.
Note: Gravity flow may take up to 2 h.
2. Wash the column with five consecutive 5 mL volumes of wash buffer (see Recipes). Collect 10 μL from the flowthrough of each wash fraction for SDS-PAGE.
3. Elute the bound protein by applying elution buffer (see Recipes) according to the volumes and incubation times listed in Table 3 below. The majority of ADGRL4 protein elutes in elution 2. Save 10 μL from each elution for SDS-PAGE.
Table 3. Elution details
| Elution fraction | Volume of elution buffer | Incubation time |
|---|---|---|
| Elution 1 | 3 mL | 0 min |
| Elution 2 | 8 mL | 45 min |
| Elution 3 | 4 mL | 15 min |
| Elution 4 | 4 mL | 15 min |
| Elution 5 | 4 mL | 15 min |
| Elution 6 | 4 mL | 15 min |
4. Analyse each elution fraction by SEC:
a. Inject 10 μL of each eluate into the Shimadzu HPLC with both the ultraviolet (UV; 280 nm) and fluorescent detectors in line at 0.35 mL/min using the same guard column and SEC columns used previously.
Note: If the HPLC system was off overnight, run SEC running buffer (without glycerol) for 60 min at 0.35 mL/min before injecting samples.
b. Expect a small 280 nm peak (corresponding to the expected molecular weight of active-state ADGRL4 and the LMNG detergent micelle) and a high-intensity fluorescence peak. Elution 2 should give the strongest signal.
5. Concentrate elution 2 (~8 mL volume) using four 100 kDa Amicon Ultra-15 centrifugal filters (see Laboratory supplies).
Critical: For active-state ADGRL4, concentrating the eluate using a single centrifugal filter markedly increases the total concentration time and reproducibly leads to increased aggregation and loss of monodispersity. Distributing the sample across multiple filters reduces the time required for concentration and limits aggregation. The use of multiple Amicon Ultra-15 filters followed by Ultra-4 filters is essential to prevent aggregation.
a. Pre-chill a swing-bucket centrifuge to 4 °C (see Equipment).
b. Rinse each 100 kDa Amicon Ultra-15 centrifugal filter with 10 mL of SEC buffer and centrifuge at 4,000× g for 5 min. Discard the flowthrough.
c. Add 2 mL of elution 2 to each centrifugal filter and centrifuge at 4,000× g in 5 min intervals at 4 °C. Discard the flowthrough between spin cycles.
Critical: After each 5-min spin cycle, gently pipette the retentate over the filter membranes to minimise local detergent concentrations and reduce protein aggregation. Do not touch the membrane with the pipette tip.
d. When the retentate volume in each filter decreases to ~500 μL, transfer all four samples into pre-chilled 100 kDa Amicon Ultra-4 centrifugal filters and continue concentrating at 4,000× g in 5-min intervals.
e. Stop concentrating when the retentate volume in each filter decreases to ~50 μL. Combine all four retentates gently (total volume ~200 μL) and store at 4 °C. Save 5 μL for SDS-PAGE.
Critical: Avoid bubble formation.
6. Whilst concentrating eluate 2, regenerate the Strep-Tactin affinity resin. Strep-Tactin XT 4Flow high-capacity resin can be regenerated three times before needing to be replaced.
a. Wash the Strep-Tactin column (stopcock open) with 30 mL of Iba Lifesciences Buffer XT-R (see Reagents).
b. Immediately after washing is completed, remove buffer XT-R by adding 20 mL of wash buffer (see Recipes) and wash through.
c. Store the Strep-Tactin column with 10 mL of wash buffer at 4 °C until the next purification.
7. Post-concentration SEC:
a. Inject 2 μL of the concentrated eluate into the Shimadzu HPLC system under the same conditions as above (rate: 0.35 mL/min; UV and fluorescent detectors in line; SEC running buffer without glycerol).
b. Expect a larger 280 nm protein peak corresponding to the expected molecular weight of active-state ADGRL4 and the LMNG detergent micelle. The fluorescence detector may saturate due to the higher concentration.
8. Measure the protein concentration of the concentrated eluate using a Direct Detect infrared spectrometer (see Equipment).
a. Use a new Direct Detect assay-free card.
b. Designate a negative control spot and apply 2 μL of elution buffer (not containing any eluted protein) to the reference spot.
c. Designate a sample spot and apply 2 μL of concentrated eluate to this spot.
d. Air dry for 15 min at room temperature.
e. Insert the Direct Detect assay-free card into the Direct Detect instrument card holder and open the Direct Detect software on the associated computer. Define the control spot and input the sample spot details. Ensure that the option “dry sample card” is selected. Measure the card and quantify the eluate concentration relative to the negative control.
9. Complex formation with purified Gβγ dimer.
a. Thaw an aliquot of purified Gβγ dimer (see Reagents; expression and purification detailed in the supplementary data section of [25]).
b. Mix the concentrated eluate with purified Gβγ dimer at a 1:1.2 molar ratio and incubate for 1 h at 4 °C.
10. Perform SEC to isolate the assembled ADGRL4-mini-Gq-βγ complex:
a. Inject up to 99 μL per run (Shimadzu HPLC system maximum) using the same HPLC setup as above (rate: 0.35 mL/min; UV and fluorescent detectors in line; SEC running buffer without glycerol). If there is >99 μL of sample, multiple SEC runs will be required.
b. Collect fractions every 30 s at a flow rate of 0.35 mL/min (175 μL per fraction) around the expected UV peak. Save 5 μL of each SEC fraction for SDS-PAGE.
c. For the active-state ADGRL4 structure paper [23], SEC fractionation at this point in the protocol was performed using a buffer containing 10% glycerol. This required that the flow rate be halved to prevent excessive system pressure. We have since determined that SEC fractionation without added glycerol yields stable fractions. For completeness, both buffer options are provided in Recipes. The presence of glycerol alters surface tension and affects downstream cryo-EM sample vitrification parameters. The vitrification conditions reported here were optimised using the SEC buffer without glycerol; vitrification parameters require re-optimisation when glycerol is present.
11. Concentrate the SEC fraction corresponding to the monodispersed peak of the ADGRL4-mini-Gq-βγ complex:
a. Concentrate using a single pre-chilled (4 °C) 100 kDa Amicon Ultra-4 centrifugal filter in a swing-bucket centrifuge at 4,000× g for 5-min intervals. Dispose of the flowthrough between spin cycles.
Critical: Gently mix retentate over the membrane after each 5-min spin and avoid touching the membrane with the pipette tip.
b. When the retentate volume decreases to ~50 μL, switch to a chilled 100 kDa Amicon Ultra-0.5 centrifugal filter and centrifuge in a fixed-angle rotor centrifuge (see Equipment) at 5,000× g for a maximum of three 10-min intervals at 4 °C. The final target retentate volume is ~20–30 μL.
c. Determine the protein concentration using the Direct Detect method (as described above at steps D8a–e).
12. At this stage, the purified ADGRL4-mini-Gq-βγ complex may be vitrified directly with its N-terminal tags intact, as these tags do not affect cryo-EM particle quality or reconstruction. Alternatively, an optional overnight TEV protease digestion may be carried out to remove all N-terminal tags (see step D14). The previously reported active-state ADGRL4 structure [23] was determined using a protein that retained its N-terminal tags and did not undergo TEV cleavage.
13. Vitrify the purified ADGRL4-mini-Gq-βγ complex on UltrAuFoil R1.2/1.3 300 mesh Au cryo-EM grids (see Laboratory supplies):
a. Equilibrate the Vitrobot automated plunge-freezing system (see Equipment) to 4 °C and 100% relative humidity for 30 min before use. Install fresh blotting paper prior to starting.
b. Prepare liquid ethane using the precision cryostat (see Equipment). Assemble the cryostat and connect the temperature-controller system. Fill both the outer ring and the ethane cup with clean, ice-free liquid nitrogen and allow the system to cool below 100 K. Switch the controller to Run mode and set the cryostat to -181 °C. Once the ethane cup is cold and free of residual liquid nitrogen, briefly purge the ethane line and slowly introduce ethane gas into the cup, allowing it to condense to form liquid ethane. Fill the cup to just below the rim.
Critical: Handle liquid ethane with extreme caution and wear cryogenic PPE, including safety goggles.
c. Glow-discharge the grids. Using a plasma chamber (see Equipment), glow-discharge all UltrAuFoil gold grids in a 9:1 ratio argon/oxygen gas mixture (forward power 38 W; reflected power 2 W; duration: 1 min).
d. Mount the glow-discharged grid into the Vitrobot tweezers.
e. Apply 3 μL of purified ADGRL4-mini-Gq-βγ complex (minimum 1 mg/mL) to the grid, blot for 3 s at a blot force setting of 5, and then plunge the grid rapidly into liquid ethane held at -181 °C to vitrify the specimen.
f. Transfer vitrified grids to cryo grid boxes under liquid nitrogen and store them in liquid nitrogen until cryo-EM imaging.
14. (Optional) Steps for TEV cleavage prior to vitrification:
a. Thaw purified TEV protease (see Reagents) on ice. TEV cleavage reactions are performed in buffer supplemented with 100 μM TCEP to maintain the protease in an active, reduced state.
b. Add TEV protease to the purified ADGRL4-mini-Gq-βγ complex in a 1:20 (TEV:substrate, w/w) ratio in a microcentrifuge tube. For example, for 33 μL of purified ADGRL4 complex at 3.3 mg/mL (108.9 μg total), add 1.24 μL of TEV protease at 4.4 mg/mL (5.45 μg total TEV protein).
c. Add TCEP to reach a final TCEP concentration of 100 μM.
d. Gently mix and incubate overnight (16 h) at 4 °C to allow for complete TEV cleavage.
E. Purification day 3 (Optional: following overnight TEV cleavage)
1. Day 3 is only required if N-terminal tag removal is required. If vitrifying the non-cleaved complex, this section can be omitted.
2. After overnight TEV cleavage, perform SEC to isolate TEV-cleaved ADGRL4-mini-Gq-βγ complex:
a. Inject the entire TEV-digested sample into the Shimadzu HPLC system with both UV and fluorescent detectors in line. Use the same guard and SEC column as per day 2 with the same settings [SEC running buffer without glycerol (see Recipes), flow rate: 0.35 mL/min]. If the HPLC was off overnight, run 60 min of SEC running buffer without glycerol before use.
b. Collect fractions every 30 s at a flow rate of 0.35 mL/min (175 μL per fraction) around the expected UV peak. Save 5 μL of each SEC fraction for SDS-PAGE.
Note: After TEV cleavage, the peak will shift to the right.
3. Concentrate the peak SEC fraction(s).
a. Pool the fractions corresponding to the monodispersed ADGRL4-mini-Gq-βγ complex peak and transfer to a pre-chilled 100 kDa Amicon Ultra-4 centrifugal filter (see Equipment). Concentrate at 4 °C in a swing-bucket centrifuge at 4,000× g for 5-min intervals. Dispose of the flowthrough between spin cycles.
Critical: After every spin, gently resuspend the retentate by pipetting over the membrane surface to prevent local detergent build-up and protein aggregation. Avoid touching the membrane with the pipette tip.
b. When the retentate volume decreases to ~50 μL, switch to a chilled 100 kDa Amicon Ultra-0.5 centrifugal filter and centrifuge in a fixed-angle rotor centrifuge (see Equipment) at 5,000× g for a maximum of three 10-min intervals at 4 °C. The final target retentate volume is ~10–30 μL.
4. Measure the concentration of the concentrated SEC fraction using the Direct Detect method, following the same procedure described for Day 2 (see steps D8a–e).
5. Vitrify SEC fraction onto UltrAuFoil R1.2/1.3 300 mesh Au cryo-EM grids using the same procedure outlined for Day 2:
a. Equilibrate the Vitrobot automated plunge-freezing system (see Equipment) to 4 °C and 100% relative humidity for 30 min before use. Install fresh blotting paper prior to starting.
b. Prepare liquid ethane using the precision cryostat (see Equipment). Assemble the cryostat and connect the temperature-controller system. Fill both the outer ring and the ethane cup with clean, ice-free liquid nitrogen and allow the system to cool below 100 K. Switch the controller to Run mode and set the cryostat to -181 °C. Once the ethane cup is cold and free of residual liquid nitrogen, briefly purge the ethane line and slowly introduce ethane gas into the cup, allowing it to condense to form liquid ethane. Fill the cup to just below the rim.
Critical: Handle liquid ethane with extreme caution and wear cryogenic PPE, including safety goggles.
c. Glow-discharge the grids. Using a plasma chamber (see Equipment), glow-discharge all UltrAuFoil gold grids in a 9:1 ratio argon/oxygen gas mixture (forward power 38 W; reflected power 2 W; duration: 1 min).
d. Mount the glow-discharged grid into the Vitrobot tweezers.
e. Apply 3 μL of purified ADGRL4-mini-Gq-βγ complex (minimum 1 mg/mL) to the grid, blot for 3 s at a blot force setting of 5, and plunge the grid rapidly into liquid ethane held at -181 °C to vitrify the specimen.
f. Transfer vitrified grids to cryo grid boxes under liquid nitrogen and store them in liquid nitrogen until cryo-EM imaging.
Validation of protocol
The purification strategy described in this protocol for active-state ADGRL4 was validated experimentally in the following open-access research publication [23], which determined the active-state structure of ADGRL4 to an overall nominal resolution of 3.1 Å.
The following list identifies relevant figures from the published paper relevant to the validation of each component of this purification protocol:
• Supplementary Figure 3h–j. Figure 3h shows Coomassie-stained SDS-PAGE gel analysis of purified ADGRL4-CTF2B. Collecting samples during purification for SDS-PAGE gel analysis is critical for troubleshooting. Figure 3i–j shows size-exclusion chromatography (SEC) traces of eluted ADGRL4-CTF2B. Running SEC traces on collected eluted purified protein is also essential for troubleshooting: if the protein begins to aggregate, this typically appears as an increased peak at or near the void volume, accompanied by a reduction in the expected monodispersed peak.
• Figure 2a–b
• Figure 3a–d
• Figure 4a–c
• Figure 5a–c
• Figure 6a–c
• Supplementary Figure 4a–f
General notes and troubleshooting
Troubleshooting
Problem 1: Minimal eluted protein.
Possible cause: Insufficient biotin incubation duration; under-loading of solubilisate; incorrect elution buffer composition; resin exhaustion after multiple regenerations.
Solution: Adhere to the 45-min incubation for elution 2; ensure that the elution buffer contains fresh biotin at 50 mM; confirm that the resin used has been regenerated no more than three times; run the resin on an SDS-PAGE gel to ensure that sufficient receptor is bound to the resin.
Problem 2: Aggregation or loss of monodisperse peak during concentration in Amicon Ultra centrifugal filters.
Possible cause: Local increase in LMNG concentration at the filter membrane surface; spinning for too long between resuspension steps; over-concentration of the sample.
Solution: Strictly follow the 5-min centrifugation intervals at 4 °C, gently mix the retentate over the membrane after each 5-min spin without touching the membrane, and avoid reducing the volume too far. If aggregation persists, reduce centrifugal force to 3,000× g with shorter centrifugation cycles.
Problem 3: Loss of protein at an unidentified step.
Possible cause: Multi-step losses during binding, washing, elution, concentration, or SEC; proteolysis; LMNG micelle instability.
Solution: Follow the protocol’s guidance and save samples for SDS-PAGE analysis at all specified purification steps. Comparing these samples will identify the point of maximal loss.
Problem 4: Unexpected extra bands or degradation on SDS-PAGE.
Possible cause: Insufficient protease inhibitors or PMSF; delays during thawing and solubilisation; handling of solubilisate and eluted samples at temperatures above 4 °C.
Solution: Add PMSF immediately before use to all buffers; maintain all steps at 4 °C; minimise interruptions to purification workflow once the solubilisate is applied to the column.
Supplementary information
The following supporting information can be downloaded here:
1. Dataset S1
Acknowledgments
D.M.F. designed and conducted the original study [23] underlying this protocol and wrote this protocol. C.G.T. provided guidance and contributed to writing. This work was supported by funding from Cancer Research UK (CRUK) (grant holder D.M.F.; grant number: RCCPOB-May22\100008), from the Medical Research Council, and from the Department of Oncology, University of Cambridge (fellowship holder: D.M.F.). C.G.T. was supported by core funding from the Medical Research Council (MRC U105197215). This protocol is based on the original research article in which the methodology was first described and validated [23]. For the purpose of open access, the MRC Laboratory of Molecular Biology has applied a CC BY public copyright licence to any Author Accepted Manuscript version arising.
Competing interests
The authors declare no conflicts of interest.
References
- Hamann, J., Aust, G., Araç, D., Engel, F. B., Formstone, C., Fredriksson, R., Hall, R. A., Harty, B. L., Kirchhoff, C., Knapp, B., et al. (2015). International Union of Basic and Clinical Pharmacology. XCIV. Adhesion G Protein–Coupled Receptors. Pharmacol Res. 67(2): 338–367. https://doi.org/10.1124/pr.114.009647
- Dieterich, L. C., Mellberg, S., Langenkamp, E., Zhang, L., Zieba, A., Salomäki, H., Teichert, M., Huang, H., Edqvist, P., Kraus, T., et al. (2012). Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFβ2 in vascular abnormalization. J Pathol. 228(3): 378–390. https://doi.org/10.1002/path.4072
- Towner, R. A., Jensen, R. L., Colman, H., Vaillant, B., Smith, N., Casteel, R., Saunders, D., Gillespie, D. L., Silasi-Mansat, R., Lupu, F., et al. (2013). ELTD1, a Potential New Biomarker for Gliomas. Neurosurgery. 72(1): 77–91. https://doi.org/10.1227/neu.0b013e318276b29d
- Masiero, M., Simões, F. C., Han, H. D., Snell, C., Peterkin, T., Bridges, E., Mangala, L. S., Wu, S. Y., Pradeep, S., Li, D., et al. (2013). A Core Human Primary Tumor Angiogenesis Signature Identifies the Endothelial Orphan Receptor ELTD1 as a Key Regulator of Angiogenesis. Cancer Cell. 24(2): 229–241. https://doi.org/10.1016/j.ccr.2013.06.004
- Favara, D. M., Banham, A. H. and Harris, A. L. (2014). A review of ELTD1, a pro-angiogenic adhesion GPCR. Biochem Soc Trans. 42(6): 1658–1664. https://doi.org/10.1042/bst20140216
- Favara, D. M., Banham, A. H. and Harris, A. L. (2019). ADGRL4/ELTD1 is a highly conserved angiogenesis-associated orphan adhesion GPCR that emerged with the first vertebrates and comprises 3 evolutionary variants. BMC Evol Biol. 19(1): 143. https://doi.org/10.1186/s12862-019-1445-9
- Favara, D. M., Zois, C. E., Haider, S., Pires, E., Sheldon, H., McCullagh, J., Banham, A. H. and Harris, A. L. (2019). ADGRL4/ELTD1 Silencing in Endothelial Cells Induces ACLY and SLC25A1 and Alters the Cellular Metabolic Profile. Metabolites. 9(12): 287. https://doi.org/10.3390/metabo9120287
- Favara, D. M., Liebscher, I., Jazayeri, A., Nambiar, M., Sheldon, H., Banham, A. H. and Harris, A. L. (2021). Elevated expression of the adhesion GPCR ADGRL4/ELTD1 promotes endothelial sprouting angiogenesis without activating canonical GPCR signalling. Sci Rep. 11(1): 8870. https://doi.org/10.1038/s41598-021-85408-x
- Li, J., Shen, J., Wang, Z., Xu, H., Wang, Q., Chai, S., Fu, P., Huang, T., Anas, O., Zhao, H., et al. (2019). ELTD1 facilitates glioma proliferation, migration and invasion by activating JAK/STAT3/HIF-1α signaling axis. Sci Rep. 9(1): 13904. https://doi.org/10.1038/s41598-019-50375-x
- Wang, Q., Zhang, C., Pang, Y., Cheng, M., Wang, R., Chen, X., Ji, T., Yang, Y., Zhang, J., Zhong, C., et al. (2024). Comprehensive analysis of bulk, single-cell RNA sequencing, and spatial transcriptomics revealed IER3 for predicting malignant progression and immunotherapy efficacy in glioma. Cancer Cell Int. 24(1): 332. https://doi.org/10.1186/s12935-024-03511-1
- Sun, B. and Zhong, F. J. (2021). ELTD1 Promotes Gastric Cancer Cell Proliferation, Invasion and Epithelial–Mesenchymal Transition Through MAPK/ERK Signaling by Regulating CSK. Int J Gen Med. 14: 4897–4911. https://doi.org/10.2147/ijgm.s325495
- Wang, L., Xu, Y., Jiang, L. and Liu, J. (2025). The elevated expression of ADGRL4 indicates poor prognosis in gastric cancer. Asian J Surg. 48(4): 2503–2505. https://doi.org/10.1016/j.asjsur.2024.07.355
- Sun, J., Zhang, Z., Chen, J., Xue, M. and Pan, X. (2021). ELTD1 promotes invasion and metastasis by activating MMP2 in colorectal cancer. Int J Biol Sci. 17(12): 3048–3058. https://doi.org/10.7150/ijbs.62293
- Abdul Aziz, N. A., Mokhtar, N. M., Harun, R., Mollah, M. M. H., Mohamed Rose, I., Sagap, I., Mohd Tamil, A., Wan Ngah, W. Z. and Jamal, R. (2016). A 19-Gene expression signature as a predictor of survival in colorectal cancer. BMC Med Genomics. 9(1): 58. https://doi.org/10.1186/s12920-016-0218-1
- Sheldon, H., Bridges, E., Silva, I., Masiero, M., Favara, D. M., Wang, D., Leek, R., Snell, C., Roxanis, I., Kreuzer, M., et al. (2021). ADGRL4/ELTD1 Expression in Breast Cancer Cells Induces Vascular Normalization and Immune Suppression. Mol Cancer Res. 19(11): 1957–1969. https://doi.org/10.1158/1541-7786.mcr-21-0171
- Wang, Z. and Zhang, Z. (2024). Single-cell analysis reveals ADGRL4+ renal tubule cells as a highly aggressive cell type in clear cell renal cell carcinoma. Sci Rep. 14(1): 2407. https://doi.org/10.1038/s41598-024-52928-1
- Kan, A., Le, Y., Zhang, Y. f., Duan, F. t., Zhong, X. p., Lu, L. h., Ling, Y. h. and Guo, R. P. (2018). ELTD1 Function in Hepatocellular Carcinoma is Carcinoma-Associated Fibroblast-Dependent. J Cancer. 9(14): 2415–2427. https://doi.org/10.7150/jca.24406
- Mao, D., Wang, H., Guo, H., Che, X., Chen, M., Li, X., Liu, Y., Huo, J. and Chen, Y. (2024). Tanshinone IIA normalized hepatocellular carcinoma vessels and enhanced PD-1 inhibitor efficacy by inhibiting ELTD1. Phytomedicine. 123: 155191. https://doi.org/10.1016/j.phymed.2023.155191
- Li, Z., Nguyen Canh, H., Takahashi, K., Le Thanh, D., Nguyen Thi, Q., Yang, R., Yoshimura, K., Sato, Y., Nguyen Thi, K., Nakata, H., et al. (2024). Histopathological growth pattern and vessel co-option in intrahepatic cholangiocarcinoma. Med Mol Morphol. 57(3): 200–217. https://doi.org/10.1007/s00795-024-00392-1
- Guihurt Santiago, J., Burgos-Tirado, N., Lafontaine, D. D., Mendoza Sierra, J. C., Camacho, R. H., Vecchini Rodríguez, C. M., Morales-Tirado, V. and Flores-Otero, J. (2021). Adhesion G protein-coupled receptor, ELTD1, is a potential therapeutic target for retinoblastoma migration and invasion. BMC Cancer. 21(1): 53. https://doi.org/10.1186/s12885-020-07768-3
- Geng, G., Zhang, L., Yu, Y., Guo, X., Li, Q. and Ming, M. (2025). ADGRL4 Promotes Cell Growth, Aggressiveness, EMT, and Angiogenesis in Neuroblastoma via Activation of ERK/STAT3 Pathway. Curr Mol Med. 25(1): 45–55. https://doi.org/10.2174/0115665240254765231117122210
- Tian, Y., Bi, Z., Ge, S., Ye, B. and Han, W. (2022). STAT5A modulated EndMT via upregulation of ELTD1 expression in diabetic nephropathy. Clin Exp Pharmacol Physiol. 49(6): 686–695. https://doi.org/10.1111/1440-1681.13644
- Chen, Q., Gusach, A., Diamante, A., Patel, J. C., Edwards, P. C., Tate, C. G. and Favara, D. M. (2025). Structure of the Gq-coupled adhesion receptor ADGRL4. Nat Commun. 17(1): e1038/s41467–025–67629–0. https://doi.org/10.1038/s41467-025-67629-0
- Nehmé, R., Carpenter, B., Singhal, A., Strege, A., Edwards, P. C., White, C. F., Du, H., Grisshammer, R. and Tate, C. G. (2017). Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS One. 12(4): e0175642. https://doi.org/10.1371/journal.pone.0175642
- Carpenter, B. and Tate, C. G. (2016). Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng Des Sel. 29(12): 583–594. https://doi.org/10.1093/protein/gzw049
- Zacharias, D. A., Violin, J. D., Newton, A. C. and Tsien, R. Y. (2002). Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science. 296(5569): 913–916. https://doi.org/10.1126/science.1068539
- Favara, D.M., Tate C.G. (2026). aGPCR-HEK: A stable high-expression inducible mammalian cell expression system for adhesion GPCR structural biology applications. Bio Protoc. 16(5): e5621. https://doi.org/10.21769/BioProtoc.5621
- Thomas, J. A. and Tate, C. G. (2014). Quality Control in Eukaryotic Membrane Protein Overproduction. J Mol Biol. 426(24): 4139–4154. https://doi.org/10.1016/j.jmb.2014.10.012
- Tropea, J. E., Cherry, S. and Waugh, D. S. (2009). Expression and Purification of Soluble His6-Tagged TEV Protease. Methods Mol Biol. 498: 297–307. https://doi.org/10.1007/978-1-59745-196-3_19
- Russo, C. J., Scotcher, S. and Kyte, M. (2016). A precision cryostat design for manual and semi-automated cryo-plunge instruments. Rev Sci Instrum. 87(11): e4967864. https://doi.org/10.1063/1.4967864
Article Information
Publication history
Received: Dec 12, 2025
Accepted: Jan 26, 2026
Available online: Feb 3, 2026
Published: Mar 5, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Favara, D. M. and Tate, C. G. (2026). Purification of the Active-State G Protein-Coupled Receptor ADGRL4 for Cryo-Electron Microscopy Using a Modular Tag System and a Tethered mini-Gq. Bio-protocol 16(5): e5617. DOI: 10.21769/BioProtoc.5617.
Category
Biochemistry > Protein > Structure
Biophysics > Electron cryotomography
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.