- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Step-by-Step Protocol for In Situ Profiling of RNA Subcellular Localization Using TATA-seq
Published: Vol 16, Iss 4, Feb 20, 2026 DOI: 10.21769/BioProtoc.5611 Views: 591
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2025
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Membrane-less organelles play essential roles in both physiological and pathological processes by compartmentalizing biomolecules through phase separation to form dynamic hubs. These hubs enable rapid responses to cellular stress and help maintain cellular homeostasis. However, a straightforward and efficient method for detecting and illustrating the distribution and diversity of RNA species within membrane-less organelles is still highly sought after. In this study, we present a detailed protocol for in situ profiling of RNA subcellular localization using Target Transcript Amplification and Sequencing (TATA-seq). Specifically, TATA-seq employs a primary antibody against a marker protein of the target organelle to recruit a secondary antibody conjugated with streptavidin, which binds an oligonucleotide containing a T7 promoter. This design enables targeted, in situ reverse transcription of RNAs with minimal background noise, a key advantage further refined during data analysis by subtracting signals obtained from a parallel IgG control experiment. The subsequent T7 RNA polymerase-mediated linear amplification ensures high-fidelity RNA amplification from low-input material, which directly contributes to optimized sequencing metrics, including a duplication rate of no more than 25% and a mapping ratio of approximately 90%. Furthermore, the modular design of TATA-seq provides broad compatibility with diverse organelles. While initially developed for membrane-less organelles, the protocol can be readily adapted to profile RNA in other subcellular compartments, such as nuclear speckles and paraspeckles, under both normal and pathogenic conditions, offering a versatile tool for spatial transcriptomics.
Key features
• This protocol provides subcellular spatial resolution by targeting and sequencing RNA from specific membrane-less organelles.
• A T7-based linear amplification step ensures high sensitivity and yield from low-input samples (<10,000 cells).
• The method is adaptable for profiling diverse organelles under various biological conditions.
Keywords: Membrane-less organelles profilingGraphical overview
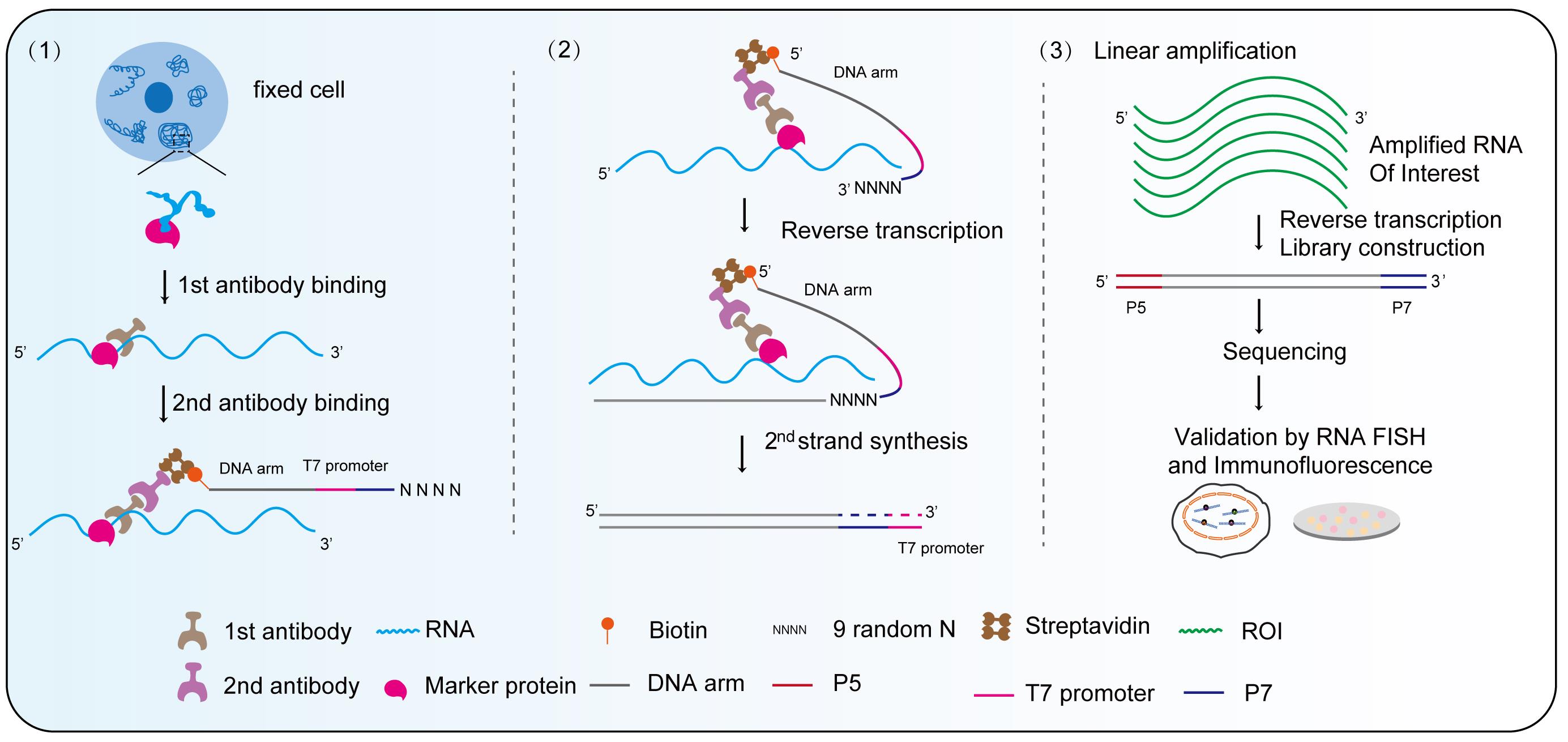
Background
Organelles serve as the fundamental compartments of eukaryotic cells, arising from the spatial organization of intracellular biomacromolecules. This compartmentalization is achieved either by biological membranes, defining classical membrane-bound organelles, or through liquid–liquid phase separation, which gives rise to membrane-less organelles (MLOs) that lack a delimiting membrane [1,2]. MLOs are dynamic hubs critical for diverse cellular functions, including gene transcription, RNA metabolism, translation, protein modification, and signal transduction [3]. They exist in a steady state (homeostasis) and exhibit dynamic responses to fundamental physiological processes, as well as various forms of stress, altered metabolic conditions, and changes in cellular signaling. Membrane-less organelles include the nucleoli, Cajal bodies (CSs), nuclear speckles, paraspeckles, processing bodies (P-bodies), and stress granules (SGs) [4–7]. Notably, P-bodies and SGs are implicated in disease pathogenesis: P-bodies are elevated in leukemia and support acute myeloid leukemia maintenance [8], and dysfunctional SGs are linked to neurodegenerative disorders [9]. RNA is essential for maintaining the integrity of RNA-protein assembly (RNP) granules, whether located in the nucleus or the cytosol [10,11]. Consequently, there is a pressing need for precise methods to map the subcellular distribution and abundance of RNAs within MLOs under both normal and pathological conditions.
Traditional methods for mapping RNA localization, such as GFP-based tagging and organelle fractionation, are hampered by their reliance on overexpression, which complicates application in primary cells and introduces the risk of contamination. Although the subsequent development of APEX-seq enabled the profiling of endogenous RNAs in numerous subcellular compartments, it also requires the time-consuming and complex overexpression of APEX2-fusion proteins, which can disrupt native cellular conditions and limit its use in primary cells [12–14]. Current methods for spatially resolved transcriptomics, such as RT&Tag and ARTR-seq, leverage antibody targeting to profile RNAs near specific proteins. RT&Tag utilizes an oligo(dT) primer for in situ transcription, capturing poly(A) mRNAs but omitting noncoding RNAs crucial for MLOs [15–17]. ARTR-seq addresses this by employing a protein A/G-fused reverse transcriptase and a random-primed strategy, enabling the detection of both poly(A) and non-poly(A) RNAs [18]. However, its multi-step incubation and streptavidin enrichment procedure is cumbersome and increases background noise. Furthermore, library construction from ultra-low cDNA input often yields suboptimal data quality. To overcome these limitations, we developed Target Transcript Amplification and Sequencing (TATA-seq) [19]. Based on these principles, TATA-seq combines antibody targeting with in situ transcription, thereby streamlining the workflow and minimizing nonspecific signals. Importantly, we incorporated a linear amplification step to enhance cDNA yield prior to library construction. This modification substantially improves sequencing quality by decreasing PCR duplication rates and increasing the mapping ratio.
Materials and reagents
Biological materials
1. HeLa cell (Cell Bank/Stem Cell Bank of the Chinese Academy of Sciences)
Reagents
1. DMEM high-glucose medium (BasalMedia, catalog number: L110KJ)
2. Fetal bovine serum (VivaCell, catalog number: C04001)
3. Penicillin-streptomycin (Gibco, catalog number:15140163)
4. Sodium arsenite (NaAsO2) (Sigma-Aldrich, catalog number: S7400)
5. 4% paraformaldehyde (PFA) (Servicebio, catalog number: G1101)
6. Normal guinea pig serum (Jackson ImmunoResearch, catalog number: 006-000-001)
7. Anti-G3BP1 primary antibody (Abcam, catalog number: ab181150)
8. Guinea pig anti-rabbit IgG (Antibodies Online, catalog number: ABIN101961)
9. IgG (Abclonal, catalog number: AC005)
10. Anti-TNRC6A (Abclonal, catalog number: A6115)
11. Streptavidin Conjugation kit (Abcam, catalog number: ab102921)
12. Alexa Fluor 488-labeled goat anti-rabbit IgG(H+L) (Beyotime, catalog number: A0423)
13. CoraLite® Plus 488-conjugated anti-G3BP1 (Proteintech, catalog number: CL488-13057)
14. CoraLite® Plus 488-conjugated anti-IgG (Proteintech, catalog number: CL488-98136)
15. RNase inhibitor (Vazyme, catalog number: R301)
16. SuperScript III (Thermo Fisher Scientific, catalog number: 18080093)
17. DNA polymerase I (E. coli) (NEB, catalog number: M0209)
18. E. coli DNA ligase (NEB, catalog number: M0205)
19. RNase H (NEB, catalog number: M0297)
20. T4 DNA polymerase (NEB, catalog number: M0203)
21. Proteinase K (Thermo Fisher Scientific, catalog number: 25530049)
22. T7 High Yield RNA Transcription kit (Vazyme, catalog number: TR101-01)
23. Dynabeads MyOneTM silane (Thermo Fisher Scientific, catalog number: 37002D)
24. T4 polynucleotide kinase (NEB, catalog number: M0201L)
25. T4 RNA ligase 2 truncated KQ (NEB, catalog number: M0373)
26. M-MLV reverse transcriptase (Promega, catalog number: M1705)
27. NEBNext Ultra II Q5 master mix (NEB, catalog number: M0544)
28. AMPure XP beads (Beckman, catalog number: A63881)
29. Low-melting agarose gel (Solarbio, catalog number: A8350)
30. Gel DNA Recovery kit (Zymo Research, catalog number: D4008)
31. Mold incubator (Changzhou Enpei Instrument Manufacturing Co., Ltd, catalog number: MJX-70BE)
32. Trypsin (BasalMedia, catalog number: S310JV)
33. Poly-L-lysine (Solarbio, catalog number: P8140)
34. PBS (BasalMedia, catalog number: B320KJ)
35. BSA (Sigma, catalog number: SRE0098)
36. RLT (Qiagen, catalog number:79216)
37. Guinea pig serum (absin, catalog number: ABS948)
38. Tween-20 (Life sciences, catalog number: T8220)
39. dNTP (Vazyme, catalog number: P031-01)
40. Second-strand buffer (5×) (Thermo Fisher Scientific, catalog number: 10812-014)
41. 1 M Tris-HCl, pH 7.5 (Invitrogen, catalog number: 15567-027)
42. NaCl (SCR, catalog number: 10019318)
43. Triton X-100 (Urchem, catalog number: 30188983)
44. Protease inhibitor (MCE, catalog number: HY-K0010)
45. Yeast carrier tRNA (Sigma, catalog number: R6750)
46. White egg avidin protein (Sigma, catalog number: 189725)
47. HEPES (Sigma, catalog number: H3375)
48. Spermidine (Solarbio, catalog number: S8030)
49. 0.5 M EDTA (Psaitong, catalog number: E70003)
50. NP-40 (Life sciences, catalog number: N8030)
51. ATP (Sigma, catalog number: A26209)
52. 20× SSC (Beyotime, catalog number: R0227)
53. Formamide (Macklin, catalog number: F810079)
54. Salmon sperm DNA (Biosharp, catalog number: BS191)
55. Pyrophosphatase, Inorganic (Vazyme, catalog number: DD4103-PC-01)
Solutions
1. Permeabilization buffer (see Recipes)
2. Blocking buffer (see Recipes)
3. Antibody buffer (see Recipes)
4. Primary antibody wash buffer (see Recipes)
5. Wash buffer I (high salt + detergent + ATP) (see Recipes)
6. Wash buffer II (low salt + formamide) (see Recipes)
7. Denaturation buffer (see Recipes)
8. Hybridization buffer (see Recipes)
9. Washing buffer (see Recipes)
Recipes
1. Permeabilization buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 1 M Tris-HCl pH 7.5 | 10 mM | 500 μL |
| 5 M NaCl | 10 mM | 100 μL |
| 10% NP-40 | 0.5% | 2.5 mL |
| 10% Triton X-100 | 0.3% | 1.5 mL |
| 10% Tween 20 | 0.1% | 500 μL |
| Protease inhibitor (100×) | 1× | 500 μL |
| 1× PBS | - | To 50 mL |
Store at 4 °C for one week. Add protease inhibitor just before use.
2. Blocking buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 10% BSA | 5% | 25 mL |
| Normal guinea pig serum | 10% | 5 mL |
| Yeast carrier tRNA (10 mg/mL) | 0.01 μg/μL | 50 μL |
| White egg avidin protein (10 mg/mL) | 0.005 μg/μL | 50 μL |
| 1× PBS | - | To 50 mL |
Store at -20 °C for one month.
3. Antibody buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 10% BSA | 3% | 15 mL |
| 1 M HEPES-KOH pH 7.5 | 20 mM | 1 mL |
| 5 M NaCl | 150 mM | 1.5 mL |
| 2 M spermidine | 0.5 μM | 12.5 μL |
| 0.5 M EDTA | 20 mM | 2 mL |
| Protease inhibitor (100×) | 1× | 500 μL |
| Nuclease-free water | - | To 50 mL |
Store at 4 °C for one week. Add spermidine and protease inhibitor just before use.
4. Primary antibody wash buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 1 M HEPES-KOH pH 7.5 | 20 mM | 1 mL |
| 5 M NaCl | 150 mM | 1.5 mL |
| 2 M spermidine | 0.5 μM | 12.5 μL |
| Protease inhibitor (100×) | 1× | 500 μL |
| Nuclease-free water | - | To 50 mL |
Store at 4 °C for one week. Add spermidine and protease inhibitor just before use.
5. Wash buffer I (high salt + detergent + ATP)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 10% NP40 | 0.5% | 500 μL |
| 10% Triton-X 100 | 0.1% | 100 μL |
| 10% Tween 20 | 0.1% | 100 μL |
| 10% BSA | 0.3% | 300 μL |
| Protease inhibitor (100×) | 1× | 100 μL |
| 1 M ATP (pH 7.0) | 10 mM | 100 μL |
| 5 M NaCl | 750 mM | 1.5 mL |
| 1× PBS | - | to 10 mL |
Store at 4 °C for one week. Add protease inhibitor just before use.
6. Wash buffer II (low salt + formamide)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 20× SSC | 0.5× | 250 μL |
| 100% formamide | 15% | 1.5 mL |
| Nuclease-free water | - | to 10 mL |
Store at 4 °C for one week.
7. Denaturation buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Cy3 probe | 10 ng/μL | |
| Salmon sperm DNA (10 mg/mL) | 100 μg/mL | 10 μL |
| Nuclease-free water | - | 1 mL |
Prepare immediately before use.
8. Hybridization buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Yeast tRNA (10 mg/mL) | 100 μg/mL | 100 μL |
| Salmon sperm DNA (10 mg/mL) | 100 μg/mL | 100 μL |
| 20× SSC | 2× SSC | 1 mL |
| Formamide | 10% | 1 mL |
| RNase inhibitor (40 U/μL) | 1 U/μL | 250 μL |
| Protease inhibitor (100×) | 1× | 500 μL |
| Nuclease-free water | n/a | 10 mL |
Store at 4 °C for one week. Add RNase inhibitor and protease inhibitor just before use.
9. Washing buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 20× SSC | 2× SSC | 1 mL |
| Formamide | 10% | 1 mL |
| Nuclease-free water | n/a | 10 mL |
Store at 4 °C for one week.
Laboratory supplies
1. 1.5 mL microcentrifuge tubes (Axygen, catalog number: MCT-150-C)
2. 1.5 mL low-adhesion tube (USA Scientific, catalog number:1415-2600)
3. Amicon Ultra centrifugal filter with 100 kDa MWCO (Millipore, catalog number: UFC8100)
4. 0.2 mL, clear 8-strip tubes (Manufacturer, catalog number: PCR-0208-C)
5. 0.1 mL, PCR 8-strip tubes (NEST Biotechnology, catalog number: 403102)
6. 96-well cell culture plate, 0.1 mL (yueyibio, catalog number: YB96)
7.15 mL centrifuge tube (yueyibio, catalog number: YB0019-15)
8. 50 mL centrifuge tube (yueyibio, catalog number: YB0010-50)
9. 25 cm2 cell culture flasks (yueyibio, catalog number: 1030000)
10. DynaMagTM-2 (Thermo Fisher, catalog number: 12321D)
11. DNA Clean & Concentrator (Zymo Research, catalog number: D4014)
Equipment
1. Centrifuge 5810 (Eppendorf, catalog number: 5810000491)
2. T100 Thermo Cycler (Bio-Rad, catalog number: 1861096)
3. C1000 Touch Thermo Cycler (Bio-Rad, catalog number: 1851148)
4. LightCycler 96 instrument (Roche, catalog number: 13068)
5. QubitTM 4 fluorometer (Invitrogen, catalog number: Q33226)
6. Freezer (-20 °C)
7. Refrigerator (2–8 °C)
Software and datasets
1. Trim Galore (v.0.6.10) (https://github.com/FelixKrueger/TrimGalore)
2. BBMap (https://github.com/BioInfoTools/BBMap)
3. STAR (v2.8.10b) (https://github.com/alexdobin/STAR)
4. macs3 (https://macs3-project.github.io/MACS/)
5. clusterProfiler (https://bioconductor.org/packages/devel/bioc/html/clusterProfiler.html)
6. Deeptools (https://deeptools.readthedocs.io/en/latest/)
Procedure
The overall workflow and time commitment for TATA-seq are summarized in Table 1.
Table 1. Key steps and estimated time frame for the TATA-seq protocol.
| Steps | Processing time | Pausable? |
| A. Sample preparation | ~Overnight | No |
| B–C. Fixation, permeabilization, and blocking | 1.5 h | No |
| D–F. Preparation antibody and binding | 5–6 h | No |
| G. In situ reverse transcription and amplification | 6 h | Yes* (after each bead purification) |
| H–I. Library construction and amplification | 1 day | Yes* (after each bead purification) |
| G. NGS library | 2–3 days |
Note: Durations are approximate and may vary depending on sample number, equipment, and operator experience. The asterisk (*) indicates recommended pause points.
A. Sample preparation
1. Cell seeding: 1–2 days before the experiment, detach adherent cells with trypsin. Coat a 96-well cell culture plate with Poly-L-lysine and seed the cells (20,000–40,000 cells/well). Incubate in incubator (37 °C, 5% CO2) until cells reach 50%–60% confluency on the day of the experiment.
2. On the day of the experiment, add 0.5 mM NaAsO2 to the medium and incubate for 1 h under standard culture conditions.
Note: Sodium arsenite is added to induce stress granule formation.
3. After incubation, aspirate the culture medium and wash cells twice with 200 μL of 1× PBS per well.
B. Cell fixation
1. Add 100 μL of 4% PFA (in 1× PBS) to each well. Incubate cells at room temperature (25 °C) for 15 min.
2. Aspirate the PFA solution and wash cells twice with 100 μL of 1× PBS per well.
Note: In this protocol, an incubation temperature of 25 °C refers to standard laboratory room temperature.
C. Permeabilization and blocking
1. Add 100 μL of permeabilization buffer into each well. Incubate on ice for 15 min.
2. Aspirate the permeabilization buffer and wash cells three times with 100 μL of 1× PBS per well.
3. Add 100 μL of blocking buffer to each well. Incubate at 25 °C for 1 h and aspirate the blocking buffer to avoid washing. Proceed directly to the addition of the primary antibody solution.
Note: Use IgG- and protease-free BSA and validated normal guinea pig serum (Absin) to avoid nonspecific antibody binding.
D. Primary antibody binding
1. Prepare primary antibody master mixes. Use 100 μL of antibody buffer with 3% BSA per reaction for each well. Dilute the antibody of interest at 1:150 in antibody buffer with 3% BSA. Supplement with RNase inhibitor (1 U/μL, 1:40 dilution).
Note: Use protease-free BSA for all blocking and antibody dilution steps.
2. Add 50 μL of the primary antibody master mix (or IgG control mix) to each well. Place the 96-well plate on a rotation shaker and incubate at 25 °C for 2 h.
Note: For the negative control, incubate the sample with an isotype-control IgG instead of the primary antibody.
3. First wash: Aspirate the antibody solution and wash each well three times with 100 μL of primary antibody wash buffer.
4. Second wash: Wash each well three times with 100 μL of PBST (0.1% Tween 20 in 1× PBS). Incubate on the shaker for 5 min per wash to remove unbound primary antibody.
E. Preparation of streptavidin-secondary antibody-biotinylated T7 oligo triplex
Note: Assemble the detection triplex by conjugating streptavidin secondary antibody and biotinylated T7 oligonucleotide, following the schematic overview provided in Figure 1.
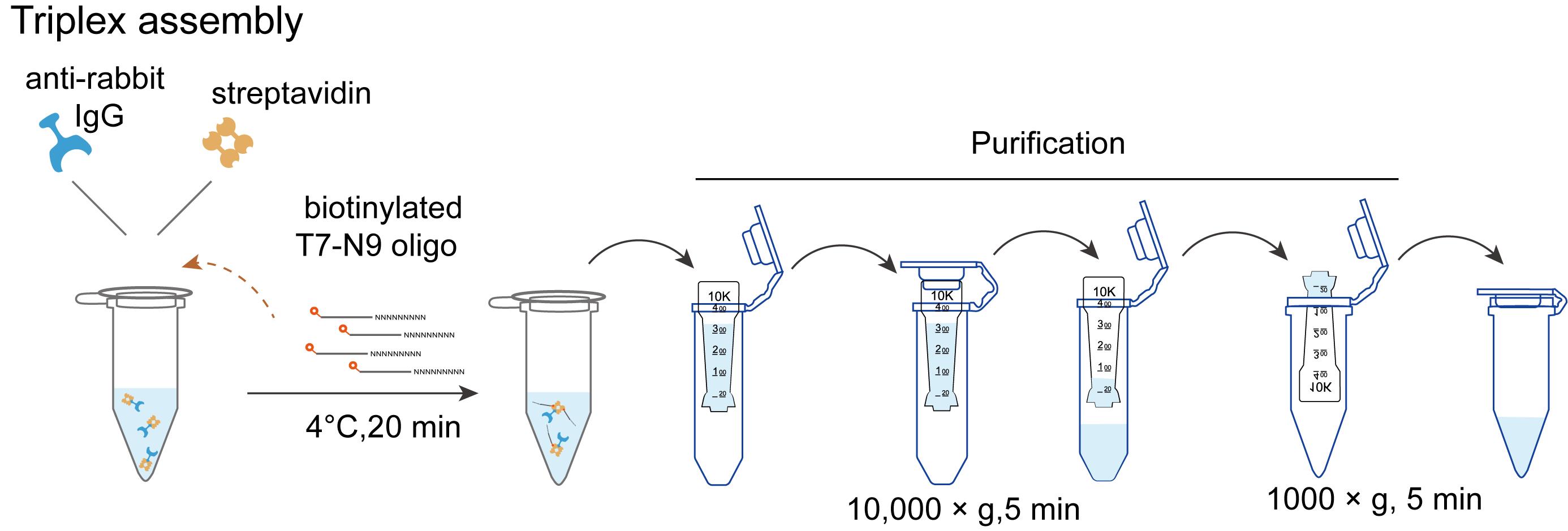
Figure 1. Workflow of triplex assembly
1. Label the 100 μL of guinea pig anti-rabbit IgG (heavy and light chain) antibody with 100 μg of streptavidin using the Streptavidin Conjugation kit.
2. Triplex (streptavidin–secondary antibody–biotinylated T7 oligo) assembly (per well): In a 1.5 mL low-adhesion tube, add 1 μL of 12.5 μM biotinylated T7-N9 oligo and 1 μL of the streptavidin-conjugated secondary antibody (from step E1) to a final volume of 150 μL of primary antibody wash buffer. Incubate on a rotation shaker at 4 °C for 20 min.
Note: This protocol describes the setup for one well. For multiple wells, prepare a scaled-up master mix of the triplex accordingly.
3. Triplex purification: Add 350 μL of primary antibody wash buffer to the tube (from step E2, final volume 500 μL).
4. Load the 500 μL of triplex mixture into the Amicon ultra centrifugal filter (100 kD cutoff) and centrifuge at 10,000× g for 5 min at 4 °C to remove excess biotinylated T7-N9 oligo. Discard the flowthrough.
5. Place the Amicon Ultra filter device upside down in a clean 1.5 mL microcentrifuge tube and spin for 2 min at 1,000× g to transfer the concentrated sample from the device to the tube. Then add primary antibody wash buffer (3% BSA, IgG- and protease-free) to a final volume of 100 μL.
F. Streptavidin–secondary antibody–biotin T7-N9 oligo triplex binding
1. Add 100 μL of triplex into each well of the 96-well plate with the prepared cells from step D4 and incubate the plate on the shaker at 25 °C for at least 45 min. After incubation, aspirate the supernatant carefully.
2. Add 100 μL of primary antibody wash buffer to each well, incubate the plate at 25 °C on the shaker for 5 min, and aspirate the liquid. Repeat this washing step three times.
3. Add 100 μL of wash buffer I to each well and incubate the plate at 25 °C on the shaker for 5 min. Aspirate the liquid.
4. Wash the well with 100 μL of wash buffer II (0.5× SSC/15% formamide) three times. Incubate the plate at 25 °C on a shaker for 5 min each time. Aspirate the liquid.
5. Wash the well with 100 μL of PBST (0.1% Tween 20 in 1× PBS) three times. Incubate the plate on a shaker for 5 min per wash. Then, rinse the well with 100 μL of 1× PBS twice. Aspirate the liquid.
6. Rinse the cells with 100 μL of 1× PBS, then place the 96-well plate in a prewarmed incubator at 37 °C for 10 min. Adjust the incubator setting to 25 °C and incubate for an additional 20 min.
G. RNA reverse transcription and amplification
1. Prepare the master mix according to Table 2:
Table 2. Components for reverse transcription.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 10 mM dNTP | 0.5 mM | 5 μL |
| 5× first-strand buffer | 1× | 20 μL |
| 0.1 M DTT | 10 mM | 10 μL |
| RNase inhibitor | 1U/μL | 4 μL |
| Superscript III | - | 2 μL |
| Nuclease-free H2O | - | 59 μL |
| Total | 100 μL |
2. Add 100 μL of reverse transcription master mix into each well. Incubate the plate in the incubator at 37 °C for 1 h.
3. After reverse transcription, remove the liquid with a pipette. Wash the well once with 10 mM Tris-HCl pH 7.5. Remove the liquid again. Prepare the second strand synthesis master mix according to Table 3.
Table 3. Components for second-strand synthesis.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Second strand buffer (5×) | 1× | 20 μL |
| dNTP (10 mM) | 0.1 mM | 1 μL |
| DNA polymerase I | - | 2.5 μL |
| RNase H | - | 1.5 μL |
| DNA ligase E. coli | - | 1 μL |
| Nuclease free H2O | - | 74 μL |
| Total | 100 μL |
4. Add 100 μL of second strand synthesis master mix into each well. Incubate the plate in the incubator at 16 °C for 2 h.
5. After incubation, add 1 μL of T4 DNA polymerase to each well. Incubate in the incubator for an additional 15 min at 16 °C.
6. Next, add 5 μL of proteinase K and 22 μL of 5× proteinase K buffer to each well. Incubate in the incubator at 55 °C for 30 min.
7. After incubation, gently pipette mix for complete resuspension and transfer the liquid to a new 1.5 mL microcentrifuge tube. Back in the original well, add 86 µL of 1× PBS, pipette to rinse, and transfer the wash to the same tube. Repeat this wash step once. Save the total 300 μL of liquid. Purify the dsDNA product using the DNA Clean & Concentrator.
8. Prepare the linear amplification master mix according to the formulation provided in Table 4.
Table 4. Components for linear amplification.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 10× reaction buffer | 1× | 2.5 μL |
| ATP solution | - | 2 μL |
| GTP solution | - | 2 μL |
| UTP solution | - | 2μL |
| CTP solution | - | 2 μL |
| T7 RNA polymerase mix | - | 2 μL (Vazyme) |
| RNase inhibitor | 1 U/μL | 1 μL |
| dsDNA product (from step G7) | - | 11 μL |
| IPP | 0.002 U/μL | 0.5 μL |
| Total | - | 25 μL |
9. Mix well the linear amplification master mix pipetting up and down at least 10 times with the pipette set to 20 μL. Incubate at 37 °C for 2 h in a thermocycler.
Note: All subsequent incubation steps are performed in a thermocycler.
10. Silane beads clean-up
a. Wash 20 μL of beads in 175 μL of buffer RLT. Then, resuspend them in 175 μL of buffer RLT.
b. Mix the beads with 50 μL of linear amplification master mixture (from step G8) and 300 μL of 100% EtOH. Incubate at 25 °C for 5 min. Separate the beads on magnetic racks (~1 min).
c. Keep the tubes on the magnetic stand. Add 200 μL of freshly-made 70% ethanol to each sample without disturbing the beads to wash away contaminants. Wait for 30 s and carefully pipette out the supernatant (Figure 2).

Figure 2. Bead washing steps
d. Repeat step G10c once. Air-dry with lids open for 4 min.
Note: Over-drying might result in sample loss.
e. Remove the tubes from the magnetic stand and resuspend the beads in 37.5 μL of nuclease-free H2O. Incubate at room temperature for 5 min.
f. Separate the beads on magnetic racks and transfer 37.5 μL of the elute to a new 0.1 mL PCR tube.
Pause point: RNA product could be stored at -80 °C for one week.
H. Library construction
Note: Library construction was performed following the m6A-sac-seq technology protocol [20,21].
1. Assemble the following reaction (Table 5) for end-repair.
Table 5. Components for RNA end-repair.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| RNA | - | 37.5 μL |
| 10× T4 polynucleotide kinase reaction buffer | 1× | 5 μL |
| T4 polynucleotide kinase (10 U/μL) | 1 U/μL | 5 μL |
| SUPERase·In RNase inhibitor (20 U/μL) | 1 U/μL | 2.5 μL |
| Total | - | 50 μL |
2. Mix the end-repair mixture from step H1 well by pipetting up and down at least 10 times with the pipette set at 30 μL. Incubate at 37 °C for 30 min. Purify the product using silane beads as described in step G10 and elute with 10 μL of nuclease-free water to a new 0.1 mL PCR tube.
3. Assemble the following reaction (Table 6) for RNA adaptor ligation.
Table 6. Reaction setup for RNA adapter ligation.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| End-repaired RNA | - | 10 μL |
| 3′ adaptor (50 μM) | 0.8 μM | 1 μL |
| Nuclease-free H2O | 0.04 ng/μL | 1 μL |
| Total | - | 12 μL |
4. Incubate the RNA mixture from step H3 at 70 °C for 2 min. Then, add the following components (Table 7) in the listed order and mix well by pipetting up and down at least 10 times with the pipette set at 30 μL.
Table 7. Composition of the T4 RNA ligase reaction mixture.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| RNA mixture (from step H3) | - | 12 μL |
| 10× T4 RNA ligase reaction buffer | 1× | 2.5 μL |
| SUPERase·In RNase inhibitor (20 U/μL) | 0.8 U/μL | 1 μL |
| 50% PEG 8000 | 15% (w/v) | 7.5 μL |
| Total | - | 23 μL |
5. Add T4 RNA ligase (Table 8) to the mixture from step H4 and mix well by pipetting up and down at least 10 times with the pipette set at 30 μL.
Table 8. Final composition of the T4 RNA ligase reaction mixture.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Mixture from step H4 | - | 23 μL |
| T4 RNA ligase 2, truncated KQ (200 U/μL) | 16 U/μL | 2 μL |
| Total | - | 25 μL |
6. Incubate the ligation mixture from step H5 at 25 °C for 2 h and 16 °C for 12 h; then, hold at 4 °C in a thermocycler.
Note: As an alternative, the ligation reaction can be incubated at 25 °C for 6 h, followed by holding at 4 °C until further processing.
7. Purify the product using silane beads as described in step G10 and elute with 10.5 μL of nuclease-free water to a new 0.1 mL PCR tube.
Pause point: The RNA product could be stored at -80 °C for one week.
8. Add the following components (Table 9) to the RNA elution from step H7.
Table 9. Reaction setup for RNA adapter RT primer annealing.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| RNA elution | 10.5 | |
| 20 μM RT primer | 0.08 μM | 1 μL |
| dNTP solution mix (10 mM) | 1 mM | 2.5 μL |
| Total | - | 14 μL |
9. Incubate the mixture from step H8 in a 0.1 mL PCR tube at 70 °C for 2 min. Hold at 4 °C. Then, add the following components (Table 10) one at a time in the order provided in the table into the PCR tube.
Table 10. Composition of the reverse transcriptase enzyme mixture.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 5× M-MLV reverse transcriptase reaction buffer | 1× | 5 μL |
| RNase OUT (40 U) | 4 mM | 1 μL |
| Promega RT enzyme | 4 U/μL | 1μL |
| Actinomycin D (31.25 μM) | 5 μM | 4 μL |
| Total | - | 11 μL |
10. Mix the reverse mixture (from step H9) well by pipetting up and down at least 10 times with the pipette set at 20 μL and incubate at 42 °C for 1 h. Add 1 μL of RNase H and incubate at 37 °C for 30 min to digest the RNA.
11. Silane beads clean up:
a. Wash 20 μL of beads in 150 μL of buffer RLT. Then, resuspend them in 150 μL of buffer RLT.
b. Mix the beads with 50 μL of reaction mixture (if the volume is less than 50 μL, add nuclease-free H2O to 50 μL) and 75 μL of 100% EtOH. Incubate at room temperature for 5 min.
c. Separate the beads on magnetic racks (~1 min). Aspirate the supernatant.
d. Keep the tubes on the magnetic stand. Add 200 μL of freshly-made 70% ethanol to each sample without disturbing the beads to wash away contaminants. Wait for 30 s and carefully pipette out the supernatant.
e. Repeat step H11c once. Air-dry with lids open for 4 min.
Note: Over-drying might result in sample loss.
f. Remove the tubes from the magnetic stand and resuspend the beads in 21 μL of nuclease-free H2O. Incubate at room temperature for 5 min.
g. Separate the beads on magnetic racks and transfer 21 μL of the elute to a new 0.1 mL PCR tube.
Pause point: cDNA can be stored at -20 °C for one week.
I. Library amplification
1. Take 1 μL (from step H11) of the elute for the qPCR test and add the following components (Table 11) into the PCR tube. Mix well by pipetting up and down at least 10 times with the pipette set at 20 μL.
Table 11. Composition of qPCR.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| cDNA product (from step H11) | - | 1 μL |
| NEBNext Ultra II Q5 master mix | 1 × | 12.5 μL |
| Universal primer (10 μM) | 0.5 μM | 1.25 μL |
| Index primer (10 μM) | 0.5 μM | 1.25 μL |
| Adaptor‐ligated DNA | - | 1 μL |
| Syber green (25×) | 1× | 1 μL |
| Nuclease-free H2O | - | 8 μL |
| Total | - | 25 μL |
2. Recommended thermocycling qPCR conditions are shown in Table 12.
Table 12. Next-generation sequencing (NGS) qPCR.
| Step | Temperature | Time | Cycles |
| Initial denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | 40–50 |
| Annealing | 65 °C | 60 s | |
| Extension | 65 °C | 15 s | |
| Final extension | 65 °C | 5 min | 1 |
| 4 °C | Forever |
3. Determine the optimal PCR cycle number by observing the qPCR amplification plot. Read out the cycle number at the beginning of the transition between the exponential phase and the plateau as N. N should be around 15–17. The final amplification cycle number is N−4 (Figure 3).
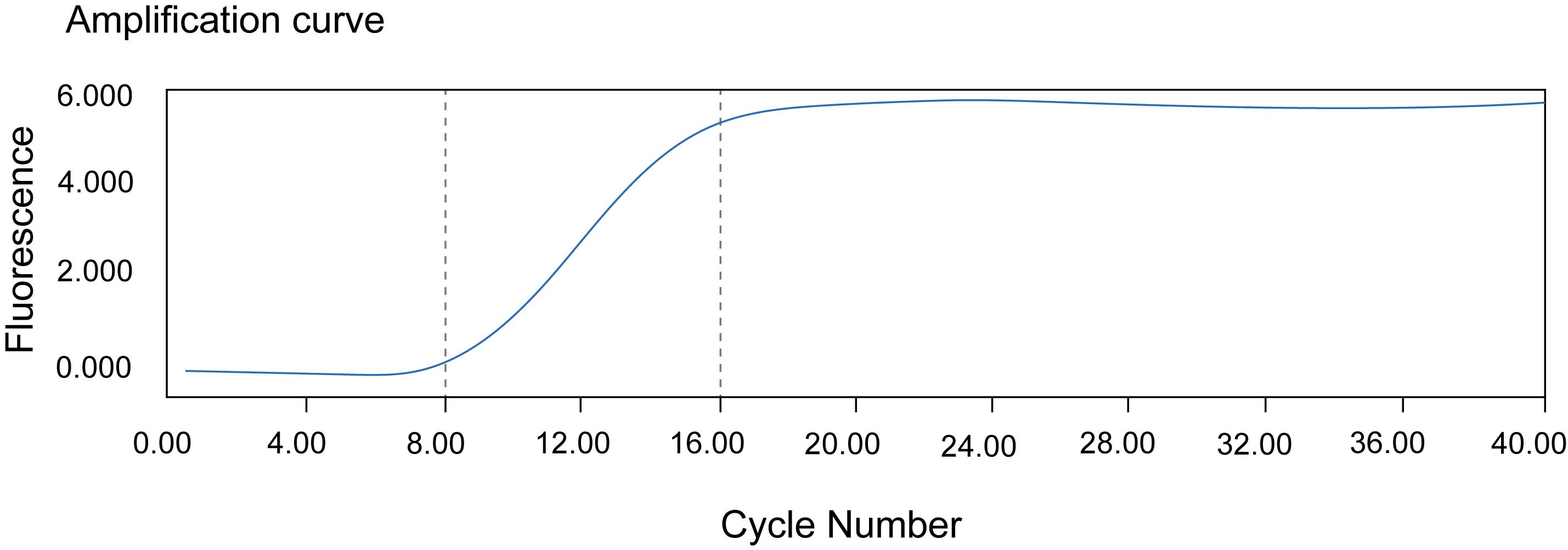
Figure 3. Example of cycles for PCR amplification
4. Use the remaining 20 μL (from step H11) of the elute for library construction and add the following components (Table 13) into the PCR tube. Mix well by pipetting up and down at least 10 times with the pipette set at 30 μL.
Table 13. PCR composition.
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| cDNA product (from step H11) | - | 20 μL |
| NEBNext Ultra II Q5 master mix (2×) | 1× | 25 μL |
| NEBNext Index X primer for Illumina (10 μM) (X = 1–12) | 0.5 μM | 2.5 μL |
| NEBNext Universal PCR primer for Illumina (10 μM) | 0.5 μM | 2.5 μL |
| Adapter ligated cDNA | - | 20 μL |
| Total | - | 50 μL |
5. Recommended thermocycling PCR conditions are shown in Table 14.
Table 14. Next-generation sequencing (NGS) PCR program.
| Step | Temperature | Time | Cycles |
| Initial denaturation | 98 °C | 30 s | 1 |
| Denaturation | 98 °C | 10 s | N−4 |
| Annealing | 65 °C | 60 s | |
| Extension | 65 °C | 15 s | |
| Final extension | 65 °C | 5 min | 1 |
| 4 °C | Forever |
1. Over-amplification will increase PCR duplications. Under-amplification will generate insufficient libraries for sequencing. Choose an appropriate number.
2. Amplified libraries could be left in the PCR block at 4 °C overnight or stored at -20 °C for at least one month.
6. Library purification: Equilibrate the AMPure XP beads to room temperature for 30 min.
a. Fill the PCR product (from step I5) with nuclease-free H2O to a final volume of 100 μL.
b. Add 100 μL (1×) beads to each PCR mixture. Mix well by pipetting up and down at least 10 times with the pipette set at 180 μL. Incubate at room temperature for 5 min.
c. Separate the beads on magnetic racks (~1 min). Aspirate the supernatant.
d. Keep the tubes on the magnetic stand. Add 200 μL of freshly-made 80% ethanol to each sample without disturbing the beads to wash away contaminants. Wait for 30 s and carefully pipette out the supernatant.
e. Repeat step I6d once.
f. After the final ethanol wash and aspiration, briefly centrifuge the tube and place it back on the magnetic rack. Use a low-volume pipette tip to carefully remove any residual droplets from the tube walls and cap. Then, air-dry the open tube at room temperature for 5–10 min or until the bead pellet transitions from a glossy to a matte appearance and no liquid film or droplets are visible.
Note: Use freshly prepared 80% EtOH. Remaining droplets might introduce primer dimers in the purified library.
g. Remove the tubes from the magnetic stand and resuspend the beads in 16 μL of nuclease-free H2O. Incubate at room temperature for 5 min.
h. Separate the beads on magnetic racks and transfer 15 μL of the elute to a new 1.5 mL tube.
i. After purification, run the product on a 2% low-melting agarose gel and cut the DNA fragments between 200 and 500 bp (Figure 4A). Purify the library with the Gel DNA Recovery kit and elute with 15 μL of nuclease-free H2O to a new 1.5 mL tube.
Pause point: Purified libraries could be stored at -20 °C for at least one month.
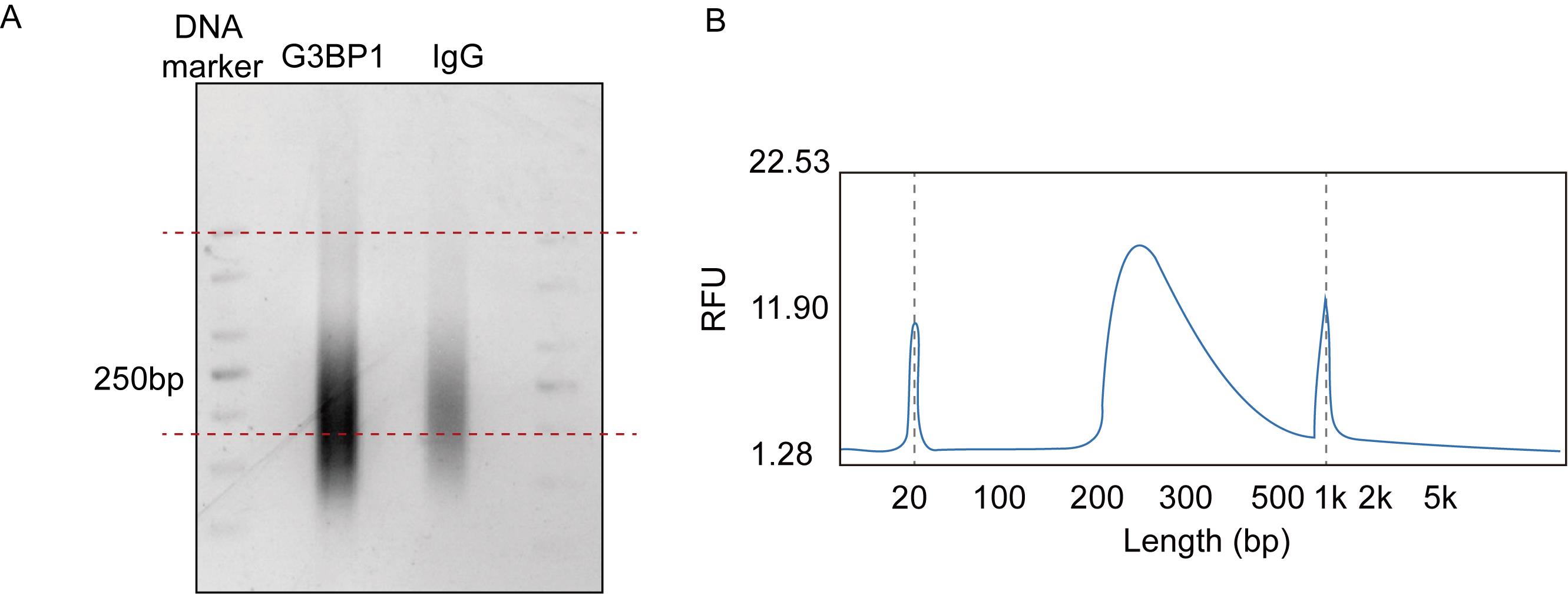
Figure 4. Library quality assessment by agarose gel electrophoresis and Bioanalyzer analysis. (A) Library agarose gel. Red dashed lines indicate the size selection (200–500 bp) used during gel extraction to isolate library fragments of the desired length. (B) Size distribution using Bioanalyzer. Any peak <150 bp should be minimal. The major peak should peak around 200–300 bp.
7. Library quality control:
a. Following bead purification, measure concentration with the Qubit dsDNA HS Assay kit. Follow the manufacturer’s directions. The final concentration should be >2 ng/µL.
b. The library should exhibit a monodisperse peak centered around 200–300 bp on a Bioanalyzer high-sensitivity DNA assay (Figure 4).
8. Validation of TATA-seq results (optional): TATA-seq results can be validated using immunofluorescence and RNA in situ hybridization.
a. Immunofluorescence
i. Plate HeLa cells on a 48-well plate and aim to reach 70%–90% confluency the next day.
ii. Remove medium and wash cells once with 1× PBS. Incubate with 500 μL of 4% PFA at 25 °C for 15 min to fix cells.
iii. Wash cells once with PBS and permeabilize them with 500 μL of 0.2% Triton X-100. Incubate at 25 °C for 30 min.
iv. After incubation, remove 0.2% Triton X-100 and wash cells three times with PBS.
v. Block cells with 500 μL of 10% goat serum at 37 °C for 1 h.
vi. After incubation, remove blocking buffer.
vii. Dilute the primary antibody anti-G3BP1 at a 1:500 ratio and add 100 μL of antibody anti-G3BP1 to each well. Incubate at 4 °C for 6 h.
viii. After washing with 1× PBS, incubate the cells with Alexa Fluor 488-labeled goat anti-rabbit IgG (H+L) (1:500 dilution) at 25 °C for 1 h, followed by counterstaining with DAPI (1:5,000 dilution) at 25 °C for 10 min.
ix. Capture images using the Olympus FLUOVIEW FV4000 confocal laser scanning microscope and perform quantitative analysis using ImageJ with the FIJI plugin.
b. RNA fluorescence in situ hybridization
i. Cell fixation and permeabilization: Wash cells once with 1× PBS. Fix the cells with 4% PFA for 15 min at room temperature. Then, permeabilize the cells with 0.2% Triton X-100 for 15 min at room temperature.
ii. Probe preparation: Prepare the Cy3-labeled DNA probes in denaturation solution. Denature and linearize the probes by incubating at 70 °C for 5 min and then hold at 37 °C before use.
Note: To minimize steric hindrance, the DNA probes were designed with a 30-carbon spacer (C30) at the 5′ end [22].
iii. Hybridization: Incubate the fixed and permeabilized cells with the denatured Cy3-labeled DNA probes in hybridization buffer. Perform the hybridization overnight at 37 °C.
iv. Post-hybridization washes: The following day, wash the cells twice with washing buffer to remove unbound probes, followed by two washes with 1× PBS.
v. Optional consecutive staining: The cells with bound Cy3-labeled probes can subsequently be subjected to standard immunofluorescence staining for protein detection (as described in step I8a).
vi. Image acquisition and analysis: Acquire images using a confocal laser scanning microscope (e.g., Olympus FLUOVIEW FV4000). Perform quantitative analysis of FISH signals using ImageJ software with the FIJI plugin.
c. Microscopy and image acquisition
i. Microscope settings (laser power and filters): Optimal settings are highly dependent on sample fluorescence intensity, antibody-dye conjugate efficiency, and microscope configuration. Therefore, rather than prescribing fixed values, we advise users to perform a preliminary optimization for each new antibody or sample type. The key principle is to set laser power and exposure times to achieve a clear signal while avoiding saturation (pixel intensity 4,095 for 12-bit cameras) and minimizing background. Specific filter sets should match the emission spectra of the fluorophores used (e.g., DAPI, FITC, Cy3).
ii. Number of fields: We recommend acquiring a minimum of 10–15 non-overlapping, randomly selected fields of view per experimental condition to ensure statistical robustness and account for cell-to-cell heterogeneity. For quantitative comparisons, this number should be increased.
Data analysis
Analysis of the sequencing/experimental data was conducted according to the following key steps (Figure 5):
1. Trim adapters and perform quality control on the raw short-read sequences using Trim Galore (v.0.6.10) [23] with the following command:
trim_galore R1.fq.gz --fastqc --length 202. Remove PCR duplicates using the clumpify.sh tool from BBMap with the command:
clumpify.sh qin=auto in=R1_trimmed.fq.gz out=R1_dedup.fq dedupe=t repair=f lowcomplexity=f rcomp=f deleteinput=f deletetemp=t3. Align the reads to the hg38/GRCh38 genome assembly using STAR (v2.8.10b) [24]. A representative command is:
STAR --runThreadN 10 \--genomeDir star_index \--readFilesIn R1_dedup.fq \--limitOutSJcollapsed 2000000 \--alignEndsType Local \--outFilterScoreMinOverLread 0.5 \--outFilterMatchNminOverLread 0.5 \--outFilterMatchNmin 15 \--outFilterMismatchNmax 5 \--outFilterMismatchNoverLmax 0.2 \--outFilterMultimapNmax 50 \--outSAMmultNmax -1 \--outReadsUnmapped Fastx \--outSAMattrRGline ID:glori SM:sample LB:RNA PL:Illumina PU:SE \--outSAMattributes NH HI AS nM NM MD jM jI MC \--limitBAMsortRAM 11441229259 \--outSAMtype BAM SortedByCoordinate \--outFileNamePrefix bam4. Peak calling was performed with MACS3 [25] with default parameters, except for the addition of keepdup broadcutoff 0.01 to account for duplicate reads and refine peak detection. The specific command line is:
macs3 callpeak -t treat.bam -c input.bam -f BAM -g hs -n result -B --keep-dup all --broad --broad-cutoff 0.01 --outdir macs3/5. Pathway enrichment analysis was conducted using clusterProfiler v4.8.1 [26]. Usable BAM files were converted to bigWig format using the bamCoverage tool from the deepTools suite [27].
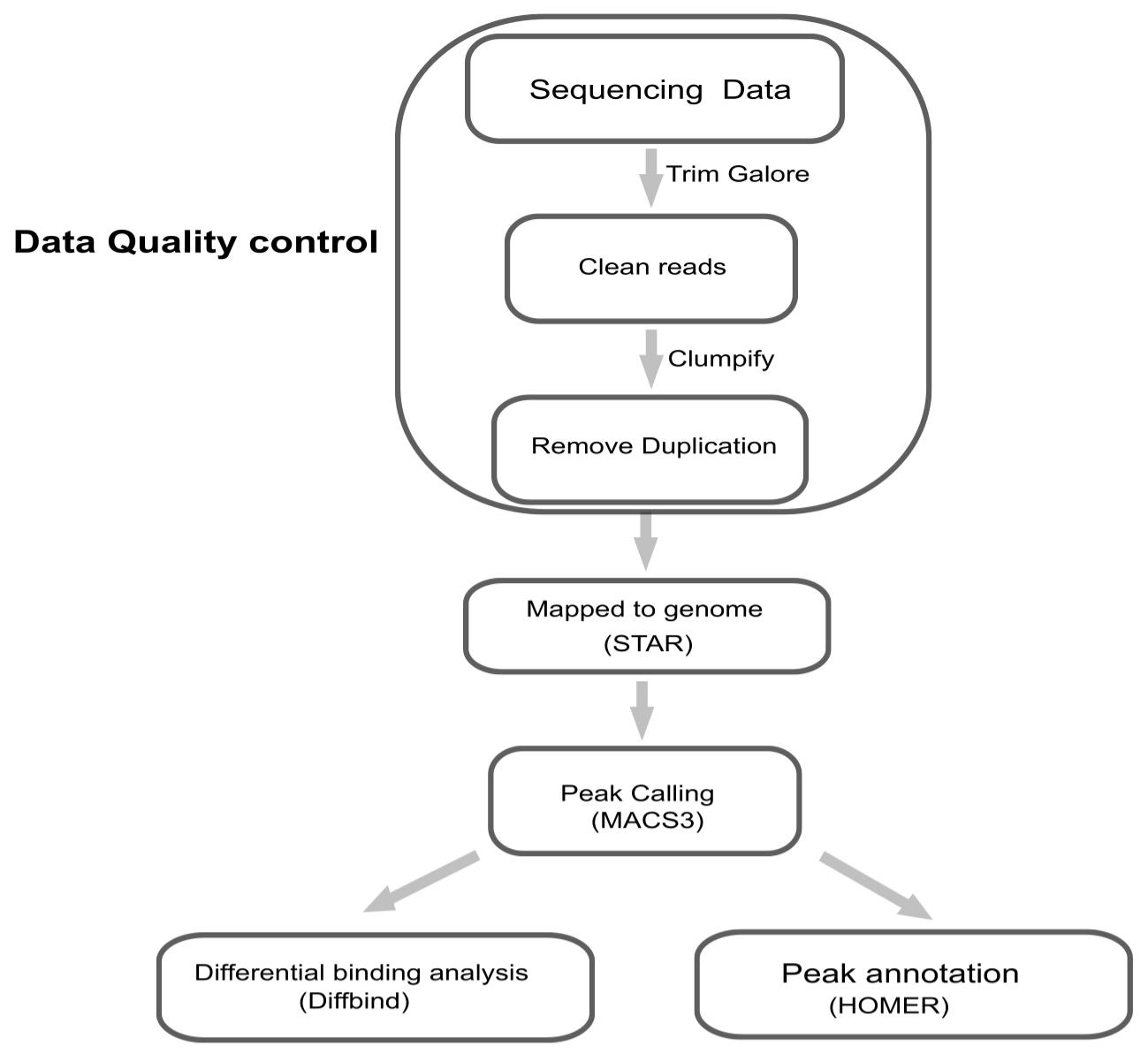
Figure 5. Workflow of bioinformatics analysis
Validation of protocol
This protocol has been used and validated in the following research article:
• Li et al. [19]. Profiling RNA subcellular localization in situ by TATA-seq. RNA. (Figures 1–5, Supplemental S1–S4).
General notes and troubleshooting
General notes
Note: This section outlines critical considerations for successfully implementing the TATA-seq protocol. Adhering to these guidelines will enhance reproducibility, safety, and experimental outcomes.
1. Safety first. This protocol involves the use of hazardous chemicals, including but not limited to paraformaldehyde, formamide, and sodium arsenite. Always consult Safety Data Sheets (SDS) before use. Prepare and handle PFA under a fume hood. Wear appropriate personal protective equipment (PPE): lab coat, gloves, and safety goggles. Dispose of chemical waste according to your institution's environmental health and safety regulations.
2. Key technical considerations
a. RNase inhibition: RNA degradation is a major cause of failure. Add RNase inhibitor to all relevant buffers and reaction mixes immediately after the permeabilization step.
b. Precision in liquid handling: Accurate pipetting is critical, especially during master mix preparation and bead-based cleanups. Calibrate pipettes regularly and use reverse pipetting for viscous solutions.
c. Bead handling: During magnetic bead cleanups, ensure the bead pellet is fully visible and settled before removing supernatant. Always perform a brief, quick spin after elution to collect all liquid from the tube walls.
d. Critical antibodies used in this study, including their vendors, catalog numbers, and dilutions, are listed in Table 15.
Table 15. List of validated antibodies.
| Target/Antigen | Vendor | Catalog | Validation source (e.g., IF) | Dilution used |
|---|---|---|---|---|
| G3BP1 (SG marker) | Abcam | ab181150 | Cited in [19]; in-house IF validation in Figure 1A | 1:200 |
| Anti-TNRC6A | ABclonal | A6115 | Manufacturer's datasheet (IF) | 1:100 |
| IgG Control | ABclonal | AC005 | N/A | 1:100 |
3. Protocol adaptability: This protocol has been optimized for adherent cell lines (e.g., HEK293T, HeLa) and specific MLOs (e.g., stress granules, nuclear speckles). For suspension cells, fix cells first, then adhere to poly-L-lysine-coated coverslips placed in a multi-well plate before proceeding with permeabilization. For primary Cells or tissue sections, titrate fixation and permeabilization conditions. Antibody accessibility and incubation times may require optimization.
Troubleshooting
Problem 1: Cell detachment during washing steps.
Possible cause: Inherently weak adhesion of certain cell types.
Solution: Pre-coat glass slides or culture plates with 0.01% poly-L-lysine to improve cell attachment prior to seeding.
Problem 2: Low RNA yield after linear amplification.
Possible cause: Inadequate starting material or suboptimal amplification time.
Solution: First, verify that the target membrane-less organelles are adequately present. We strongly recommend performing a pilot immunofluorescence experiment using a validated antibody against a core marker (e.g., G3BP1 for stress granules, SC35 for nuclear speckles) to visually confirm granule abundance and morphology under your experimental conditions. A useful benchmark is observing ~5–10 clear granules per cell on average. Induction treatments are recommended if basal levels are low. Strictly include RNase inhibitors in all relevant buffers and enzymatic mixes immediately after cell permeabilization to prevent RNA degradation throughout the procedure. Use only high-affinity, specificity-validated antibodies for the target organelle to ensure efficient in situ capture. If organelles are confirmed to be present but yield remains low, extend the T7-based amplification step (step G9) to ~6 h to increase product yield.
Problem 3: Cell loss or poor adherence when processing suspension cells.
Possible cause: Standard TATA-seq protocol is optimized for adherent cultures.
Solution: For suspension cells, first fix the cells and then adhere them gently onto poly-L-lysine-coated coverslips. Place the coverslip into a 48- or 96-well plate and proceed with the entire protocol from the permeabilization step onward (section C) as described for adherent cells.
Problem 4: High background in immunofluorescence.
Solutions:
a. Titrate antibodies: Perform a dilution series for both primary and secondary antibodies on control samples to identify the concentration that yields optimal specific signal with minimal background.
b. Include rigorous controls: Always run parallel samples with (a) an isotype control antibody (for the primary antibody) and (b) a secondary antibody-only control. This is essential for distinguishing a specific signal from nonspecific binding during image analysis and sequencing data interpretation.
Acknowledgments
Conceptualization, L.H.; Investigation, X.J.; Writing—Original Draft, X.J. and C.X.; Writing—Review & Editing, X.J. and C.X.; Funding acquisition, L.H.; Supervision, L.H.
The authors would like to thank the National Key R&D Program of China (2024YFA1307800, 2021YFA1100400), General Program of National Natural Science Foundation of China (32471340), Natural Science Foundation of Science and Technology Commission of Shanghai Municipality (STCSM, 21ZR1480300), and Fudan University Start-up funding to L.H. We thank the Core Facility of Shanghai Medical College, Fudan University, for providing the instruments used in this work. We thank the Core Instrument Sharing Platform of Shanghai Sycamore Research Institute of Life Sciences for providing the instruments used in this work.
This protocol is based on and was validated in the original research by Li et al. 2025 (RNA) [19].
Competing interests
A patent has been filed with the Shanghai Cancer Center, Fudan University. The authors declare that they have no competing interests.
Ethical considerations
This protocol did not involve human participants, primary human tissues, or animal experiments. All experiments were performed using established human cell lines (e.g., HeLa). Therefore, ethical approval from an institutional review board or ethics committee was not required.
References
- Riback, J. A., Zhu, L., Ferrolino, M. C., Tolbert, M., Mitrea, D. M., Sanders, D. W., Wei, M. T., Kriwacki, R. W. and Brangwynne, C. P. (2020). Composition-dependent thermodynamics of intracellular phase separation. Nature. 581(7807): 209–214. https://doi.org/10.1038/s41586-020-2256-2
- Ruan, K., Bai, G., Fang, Y., Li, D., Li, T., Liu, X., Lu, B., Lu, Q., Songyang, Z., Sun, S., et al. (2024). Biomolecular condensates and disease pathogenesis. Sci China Life Sci. 67(9): 1792–1832. https://doi.org/10.1007/s11427-024-2661-3
- Christopher, J. A., Stadler, C., Martin, C. E., Morgenstern, M., Pan, Y., Betsinger, C. N., Rattray, D. G., Mahdessian, D., Gingras, A. C., Warscheid, B. et al. (2021). Subcellular proteomics. Nat Rev Methods Primers. 1: 32. https://doi.org/10.1038/s43586-021-00029-y
- Gall, J. G. (2003). The centennial of the Cajal body. Nat Rev Mol Cell Biol. 4(12): 975–980. https://doi.org/10.1038/nrm1262
- Kodali, S., Proietti, L., Valcarcel, G., López-Rubio, A. V., Pessina, P., Eder, T., Shi, J., Jen, A., Lupión-Garcia, N., Starner, A. C., et al. (2024). RNA sequestration in P-bodies sustains myeloid leukaemia. Nat Cell Biol. 26(10): 1745–1758. https://doi.org/10.1038/s41556-024-01489-6
- Mao, Y. S., Zhang, B. and Spector, D. L. (2011). Biogenesis and function of nuclear bodies. Trends Genet. 27(8): 295–306. https://doi.org/10.1016/j.tig.2011.05.006
- Pederson, T. (2011). The nucleolus. Cold Spring Harb Perspect Biol. 3. https://doi.org/10.1101/cshperspect.a000638
- Decker, C. J. and Parker, R. (2012). P-Bodies and Stress Granules: Possible Roles in the Control of Translation and mRNA Degradation. Cold Spring Harbor Perspect Biol. 4(9): a012286–a012286. https://doi.org/10.1101/cshperspect.a012286
- Cui, Q., Liu, Z. and Bai, G. (2024). Friend or foe: The role of stress granule in neurodegenerative disease. Neuron. 112(15): 2464–2485. https://doi.org/10.1016/j.neuron.2024.04.025
- Lopez de Heredia, M. and Jansen, R. P. (2004). mRNA localization and the cytoskeleton. Curr Opin Cell Biol. 16: 80–85. https://doi.org/10.1016/j.ceb.2003.11.002
- St Johnston, D. (2005). Moving messages: the intracellular localization of mRNAs. Nat Rev Mol Cell Biol. 6(5): 363–375. https://doi.org/10.1038/nrm1643
- Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. and Zhuang, X. (2015). Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 348(6233): eaaa6090. https://doi.org/10.1126/science.aaa6090
- Fazal, F. M., Han, S., Parker, K. R., Kaewsapsak, P., Xu, J., Boettiger, A. N., Chang, H. Y. and Ting, A. Y. (2019). Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell. 178(2): 473–490.e26. https://doi.org/10.1016/j.cell.2019.05.027
- Weil, T. T., Parton, R. M. and Davis, I. (2010). Making the message clear: visualizing mRNA localization. Trends Cell Biol. 20(7): 380–390. https://doi.org/10.1016/j.tcb.2010.03.006
- Di, L., Fu, Y., Sun, Y., Li, J., Liu, L., Yao, J., Wang, G., Wu, Y., Lao, K., Lee, R. W., et al. (2020). RNA sequencing by direct tagmentation of RNA/DNA hybrids. Proc Natl Acad Sci USA. 117(6): 2886–2893. https://doi.org/10.1073/pnas.1919800117
- Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., Ahmad, K. and Henikoff, S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 10(1): 1930. https://doi.org/10.1038/s41467-019-09982-5
- Khyzha, N., Henikoff, S. and Ahmad, K. (2022). Profiling RNA at chromatin targets in situ by antibody-targeted tagmentation. Nat Methods. 19(11): 1383–1392. https://doi.org/10.1038/s41592-022-01618-9
- Xiao, Y., Chen, Y. M., Zou, Z., Ye, C., Dou, X., Wu, J., Liu, C., Liu, S., Yan, H., Wang, P., et al. (2024). Profiling of RNA-binding protein binding sites by in situ reverse transcription-based sequencing. Nat Methods. 21(2): 247–258. https://doi.org/10.1038/s41592-023-02146-w
- Li, J., Xu, C., Jiang, X., Huang, X., Ye, D. and Hu, L. (2025). Profiling RNA subcellular localization in situ by TATA-seq. RNA. 31: 1523–1535. https://doi.org/10.1261/rna.080670.125
- Ge, R., Ye, C., Peng, Y., Dai, Q., Zhao, Y., Liu, S., Wang, P., Hu, L. and He, C. (2022). m6A-SAC-seq for quantitative whole transcriptome m6A profiling. Nat Protoc. 18(2): 626–657. https://doi.org/10.1038/s41596-022-00765-9
- Hu, L., Liu, S., Peng, Y., Ge, R., Su, R., Senevirathne, C., Harada, B. T., Dai, Q., Wei, J., Zhang, L., et al. (2022). m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat Biotechnol. 40(8): 1210–1219. https://doi.org/10.1038/s41587-022-01243-z
- Huang, N., Fan, X., Zaleta-Rivera, K., Nguyen, T. C., Zhou, J., Luo, Y., Gao, J., Fang, R. H., Yan, Z., Chen, Z. B., et al. (2020). Natural display of nuclear-encoded RNA on the cell surface and its impact on cell interaction. Genome Biol. 21(1): 225. https://doi.org/10.1186/s13059-020-02145-6
- Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1): 10. https://doi.org/10.14806/ej.17.1.200
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T. R. (2012). STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29(1): 15–21. https://doi.org/10.1093/bioinformatics/bts635
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W., et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 9(9): R137. https://doi.org/10.1186/gb-2008-9-9-r137
- Wu, T., Hu, E., Xu, S., Chen, M., Guo, P., Dai, Z., Feng, T., Zhou, L., Tang, W., Zhan, L., et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. The Innovation. 2(3): 100141. https://doi.org/10.1016/j.xinn.2021.100141
- Ramírez, F., Ryan, D. P., Grüning, B., Bhardwaj, V., Kilpert, F., Richter, A. S., Heyne, S., Dündar, F. and Manke, T. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44: W160–W165. https://doi.org/10.1093/nar/gkw257
Article Information
Publication history
Received: Oct 23, 2025
Accepted: Jan 18, 2026
Available online: Jan 30, 2026
Published: Feb 20, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Jiang, X., Xu, C. and Hu, L. (2026). Step-by-Step Protocol for In Situ Profiling of RNA Subcellular Localization Using TATA-seq. Bio-protocol 16(4): e5611. DOI: 10.21769/BioProtoc.5611.
Category
Molecular Biology > RNA > RNA localisation
Molecular Biology > RNA > RNA sequencing
Systems Biology > Spatial transcriptomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.