- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Inverse Restriction Site-Associated DNA Sequencing (iRAD-seq)
Published: Vol 16, Iss 2, Jan 20, 2026 DOI: 10.21769/BioProtoc.5599 Views: 320
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 1000
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


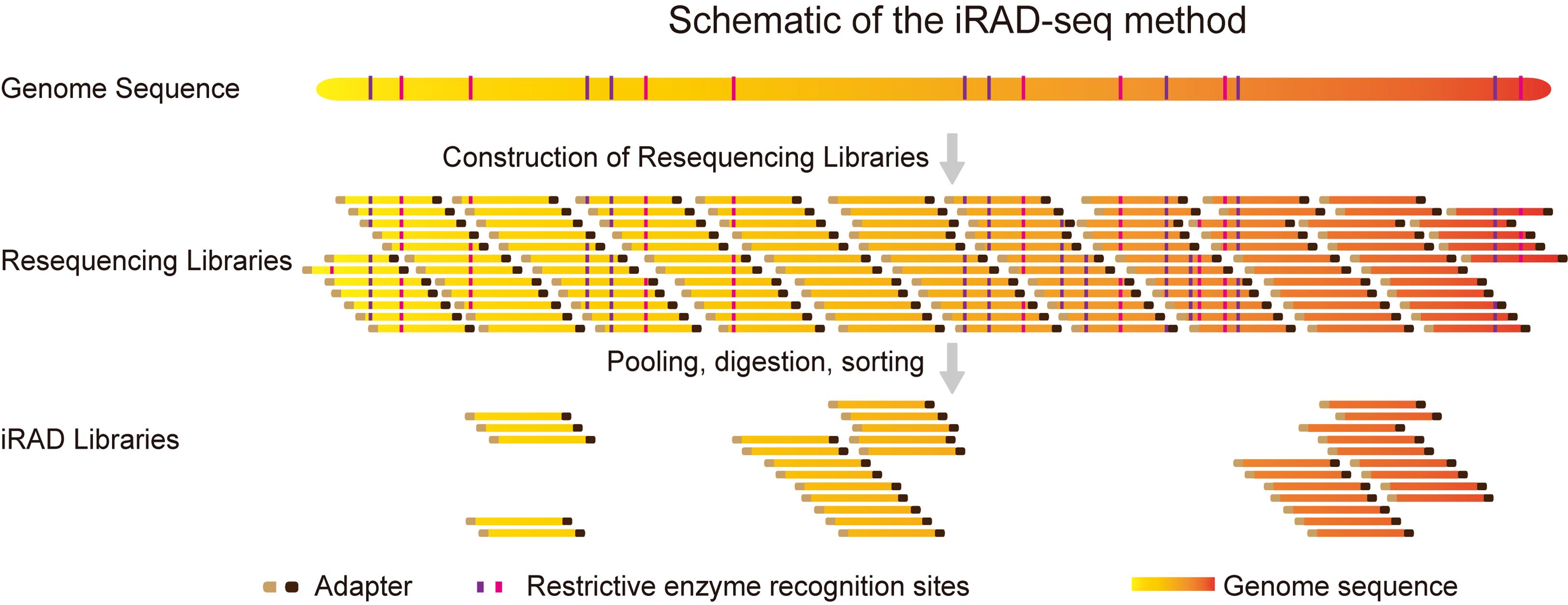
Abstract
Reduced representation sequencing (RRS), particularly through restriction site-associated DNA sequencing (RAD-seq), has been widely adopted for whole-genome genotyping due to its cost-effectiveness and cross-species applicability. Nevertheless, conventional RAD-seq approaches are constrained by intricate workflows and substantial labor intensity. These methods predominantly adhere to a “fragment selection precedes library construction” paradigm, wherein DNA fragments adjacent to restriction enzyme cleavage sites are specifically targeted. In contrast, we present an innovative strategy termed inverse restriction site–associated DNA sequencing (iRAD-seq), which implements a reversed workflow, “library construction precedes fragment selection,” to enable efficient enrichment of DNA fragments not associated with restriction sites for genome-wide genotyping. This approach harnesses Tn5 transposase to concurrently fragment genomic DNA and ligate sequencing adapters, followed by pooled processing of hundreds of libraries under a unified batch restriction digestion step. The iRAD-seq workflow thereby achieves significant simplification and enhances operational efficiency in RAD-seq library preparation.
Key features
• iRAD-seq is a swift and simple RRS method based on Tn5 library construction and restriction enzyme digestion.
• The library preparation for iRAD-seq adopts a strategy of library construction followed by fragment selection.
• iRAD-seq enables effective reduction of genome complexity, with the extent of simplification flexibly tunable by adjusting the combination of restriction enzymes.
Keywords: Reduced representation sequencingGraphical overview
Background
Single-nucleotide polymorphism (SNP) discovery and genotyping are fundamental to breeding research [1,2]. However, whole-genome sequencing for this purpose is often too costly and yields a high number of redundant SNPs. In response, reduced-representation sequencing (RRS) has emerged as a more efficient alternative. This strategy involves selectively sequencing only a small, targeted fraction (typically 1%–10%) of the genome, enabling efficient genome-wide marker detection for breeding programs [3,4].
As a classical RRS method, restriction site-associated DNA sequencing (RAD-seq) follows a multi-step protocol. The workflow starts with restriction enzyme (RE) digestion of genomic DNA and ligation of a barcoded P1 adapter. After pooling individually barcoded samples, the DNA is randomly sheared and size-selected. Finally, a P2 adapter is ligated, followed by PCR amplification and sequencing [5]. While this approach effectively captures partial genomic regions and has been widely applied in genetic and breeding studies across diverse species, its multi-step manual procedures—including fragment shearing, purification, and adapter ligation—render the library preparation process cumbersome, which has spurred ongoing optimization efforts [5,6]. Subsequent derivative methods, such as 2b-RAD, have been developed to simplify the protocol. They utilize type-IIB restriction enzymes, which cleave DNA both upstream and downstream of their recognition sites, producing uniform fragments of approximately 30 bp. This inherent uniformity removes the need for separate shearing and size selection, thereby streamlining library preparation [7,8]. Double-digest RAD (dd-RAD) has introduced improvements by eliminating random shearing and implementing defined size selection, thereby restricting sequencing to regions adjacent to RE recognition sites [9]. Furthermore, ezRAD [10] and MethylRAD [11] have also been developed to further streamline workflows or target specific genomic features. Nevertheless, these all require restriction enzyme digestion first, followed by sequencing library construction through adapter ligation or TA ligation. These methods still involve labor-intensive, multi-step separation and purification, requiring significant time and resources—especially for large sample sets. Furthermore, the single-sample size selection used in ezRAD and ddRAD can cause inter-sample variability in fragment recovery. This variability arises from the randomness of the selection process and can lead to marker loss and reduced reproducibility [6].
We leverage the simplicity and high efficiency of the Tn5 transposase, which performs simultaneous DNA fragmentation and adapter ligation [12,13]. Building on this foundation, we integrated it with our previously developed all-in-one sequencing (AIO-seq) method. AIO-seq enables pooled PCR amplification, size selection, and quantification for hundreds of samples [14]. On this basis, we introduce an innovative RRS strategy termed inverse restriction site–associated DNA sequencing (iRAD-seq). This approach fundamentally reverses the conventional workflow by adopting an inverse sequence: constructing the sequencing library first and subsequently selecting target fragments (Figure 1). iRAD-seq integrates Tn5 transposase to achieve one-step DNA fragmentation and adapter tagging. This innovation dramatically simplifies library construction, boosts operational efficiency, and ultimately delivers a more streamlined, high-throughput, and user-friendly solution for RRS library preparation.
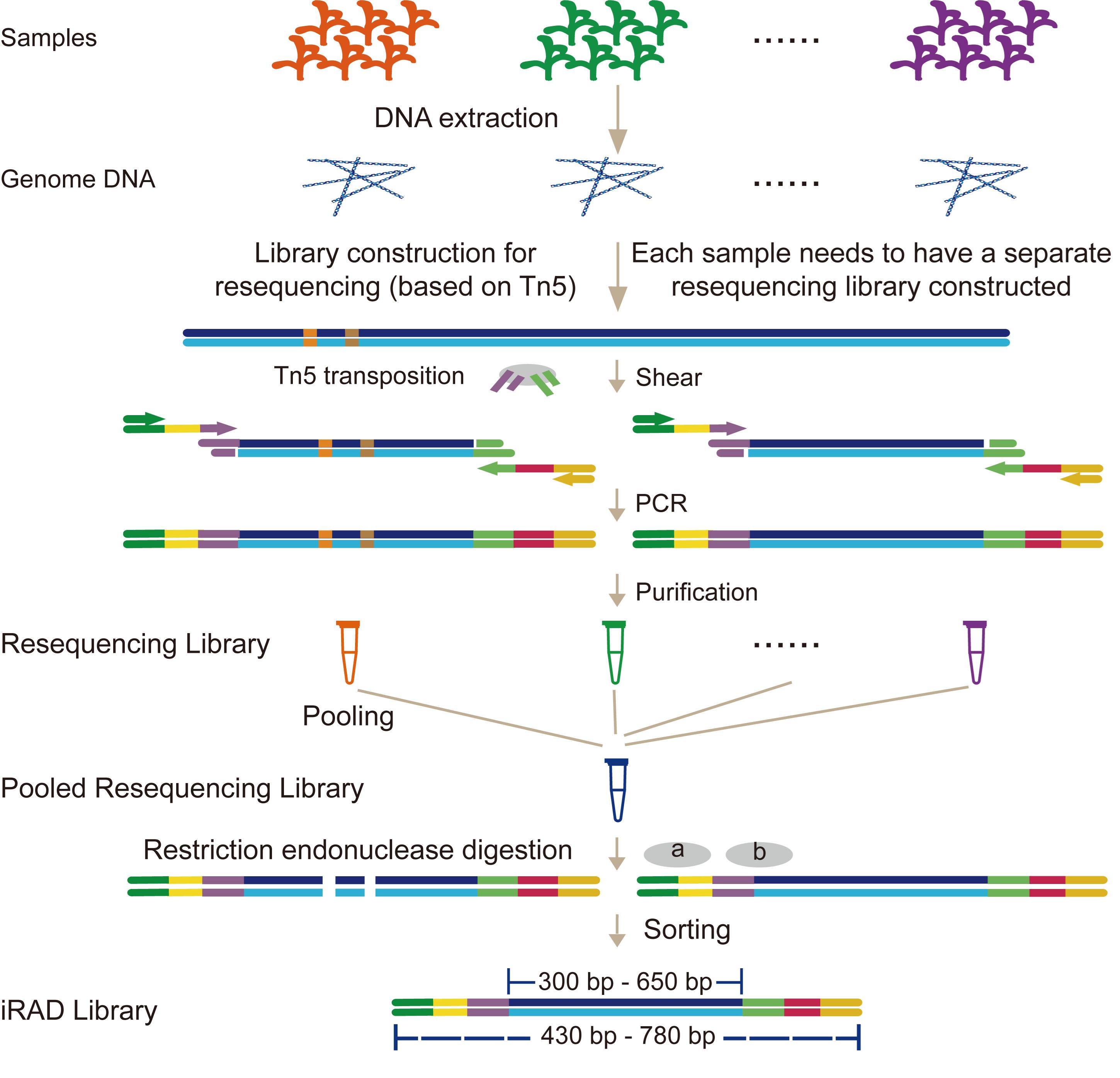
Figure 1. Workflow of the inverse restriction site–associated DNA sequencing (iRAD-seq) protocol
Materials and reagents
1. Pipette tips (Axygen, catalog number: T-200-Y, T-1000-B, T-300)
2. 0.2 mL PCR tube (Sangon Biotech, catalog number: F611542)
3. Magnetic Plant Genomic DNA kit (TIANGEN, catalog number: DP342)
4. 1× dsDNA HS Assay kit for Qubit (YEASEN, catalog number: 15642ES76)
5. 0.2 mL DNase/RNase-free PCR strip tubes with individual caps (Vazyme, catalog number: PCR00832)
6. Nuclease-free water (not DEPC-treated) (YEASEN, catalog number: 60169E)
7. TruePrep® DNA Library Prep kit V2 for Illumina (Vazyme, catalog number: TD501)
8. Stop buffer (0.9% SDS) (GenScript, catalog number: M00138)
9. TruePrep Index kit V2 for Illumina (Vazyme, catalog number: TD202)
10. VAHTS DNA clean beads (Vazyme, catalog number: N411)
11. Ethanol 80% (Macklin, catalog number: E809061)
12. S2 cartridge (BiOptic, catalog number: C105101)
13. MseI 10 U/μL (New England Biolabs, catalog number: R0525S)
14. MspI 20 U/μL (New England Biolabs, catalog number: R0106S)
15. AluI 10 U/μL (New England Biolabs, catalog number: R0137S)
16. 2% agarose (Sage Science, catalog number: HTC2010, 100–600 bp)
Equipment
1. Qubit fluorometer (Thermo Fisher Scientific, catalog number: Qubit 3.0)
2. Vortex (Scientific Industries, catalog number: SI-0256)
3. Mini centrifuge (cubee, catalog number: aqbd)
4. 0.2 mL 8-strip tube magnet (QuaYad, catalog number: QYM07)
5. Metal block heater (SANGSHAI, catalog number: BD300)
6. Thermocycler (analytikjena, catalog number: 846-2-070-301)
7. -20 °C freezer (Haier, catalog number: DW-30L818BPFL)
8. Qsep100 (Bioptic, catalog number: C104250)
9. PippinHT DNA Size Selection System (Sage Science, catalog number: HTP0001)
10. Concentrator Plus (Eppendorf, catalog number: 5305000797)
11. Illumina Sequencing System (Illumina, model: NovaSeq 6000)
Software and datasets
1. Solve-V3.5 (Bionano, 3.5)
2. All code (fa2cmap_multi_color.pl, find_enzyme.pl, and find_RESite_From_Adapter.py) have been deposited to GitHub: https://github.com/chenpeng-allen/iRAD-protocol.git (access date, 11/14/2025)
Procedure
A. DNA extraction and quantification
1. Extract genome DNA from maize leaf tissues using the Magnetic Plant Genomic DNA kit following the manufacturer’s protocol. Typically, 100 mg of leaf tissue yields 20–30 μg of DNA. Then, quantify DNA concentration using a Qubit fluorometer with the 1× dsDNA HS Assay kit.
2. Place the extracted DNA in a -20 °C freezer for later use.
B. In silico restriction digestion
To achieve the desired genome coverage, reference genomes from various species can be subjected to in silico digestion to evaluate the genome reduction efficiency of different restriction enzyme combinations. Figure 2 shows the in silico digestion results of the genome under different restriction enzyme combinations. Different enzyme combinations can achieve varying levels of genome reduction efficiency, allowing researchers to select appropriate restriction enzymes based on experimental needs. The reduction efficiency of custom enzyme combinations can also be evaluated by performing in silico digestion with self-selected enzymes. Based on the current results, achieving a reduction efficiency exceeding 50% generally requires the use of at least two restriction enzymes that recognize four-base cutting sites. In addition, different levels of genome coverage can be obtained by sorting fragments with different size ranges. As shown in Figure 2, longer sorted fragments lead to higher genome reduction efficiency, resulting in fewer detectable genomic regions and consequently less variation information.
1. Use the fa2cmap_multi_color.pl script from the Solve-V3.5 software package to locate REs recognition sites in the reference genomes; it performs in silico cleavage at these sites and generates a comprehensive list of fragment information.
2. The find_enzyme.pl script processes the output from fa2cmap_multi_color.pl and extracts fragments whose lengths exceed a user-specified threshold. Fragment information is obtained using the minimum fragment length as the threshold.
3. Based on the obtained fragment information, the total length of all fragments is calculated to estimate the genome coverage achievable in the experiment. The appropriate restriction enzyme combination can then be selected according to the specific experimental requirements.
Note: Subsequent enzymatic digestion experiments are performed using MseI, MspI, and AluI as examples.
4. Following enzyme selection, use the script find_RESite_From_Adapter.py to exclude library adapter primers (the N5 and N7 primers used in the subsequent PCR step) that contain restriction enzyme recognition sites, as their presence would otherwise lead to library construction failure.
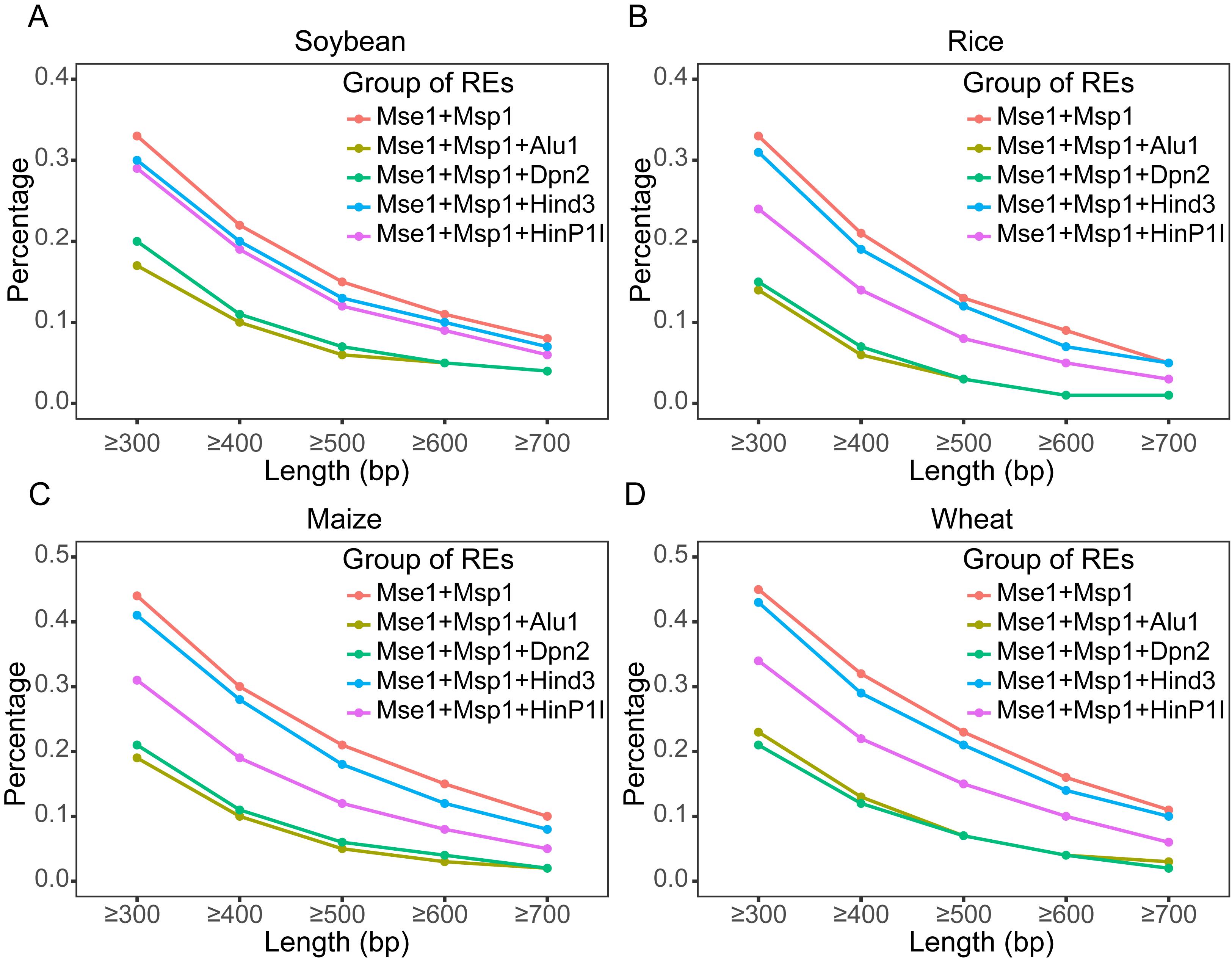
Figure 2. Genome coverage analysis plot of in silico digestion. The genomes are digested in silico according to the recognition sites of the restriction enzyme combination, followed by statistical analysis of the proportion of the total length of fragments within different size ranges relative to the whole genome. Genome coverage analysis of fragments of different lengths in (A) soybean, (B) rice, (C) maize, and (D) wheat.
C. Tn5 transposase fragmentation
The TruePrep® DNA Library Prep kit V2 for Illumina is used with the total reaction volume scaled down to 8 μL to reduce reagent consumption.
Note: The performance of transposase complexes and the accompanying buffer—whether obtained commercially or prepared in-house—may vary between brands or batches. Tn5 also exhibits varying fragmentation efficiency for genomes of different qualities. It is therefore recommended to perform a preliminary optimization to determine the optimal reagent amounts for the specific conditions used.
1. For each sample, dilute 30 ng of DNA with nuclease-free water to a final volume of 4 μL in a 0.2 mL PCR tube.
Note: For high-throughput setups, 0.2 mL 8-strip PCR tubes or 96-well plates can be used.
2. Add 2.4 μL of TTEmix (Tn5 Transposase Complex) and 1.6 μL of TTBL (buffer) to each sample tube. Both components are provided in the TruePrep® DNA Library Prep kit V2 for Illumina.
Note: Mix the reaction thoroughly on a vortex mixer, then centrifuge briefly in a mini centrifuge to collect the mixture at the bottom of the tube.
3. Incubate the fragmentation reaction in a thermocycler for 10 min at 55 °C, with the lid temperature set to 65 °C.
4. Immediately add 1 μL of stop buffer to the reaction mixture after incubation, vortex thoroughly to ensure complete mixing, and let stand for 5 min.
Note: To adapt to the scaled-down 8 µL reaction volume, we employed a stop buffer consisting of 0.9% SDS to terminate the digestion. This stop buffer was not part of the original kit components.
D. PCR with adapter-specific primers
1. Prepare a PCR mix reaction for each sample following the instructions provided in Table 1. N7 primer and N5 primer are provided with the TruePrep Index kit V2 for Illumina. At least one of the indices on the N5 and N7 primers must be different across samples. PPM, TAB, and TAE are provided with TruePrep® DNA Library Prep kit V2 for Illumina.
Note: N7 primer (CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTCTCGTGGGCTCGG) and N5 primer (5′-AATGATACGGCGACCACCGAGATCTACACXXXXXXXXTCGTCGGCAGCGTC-3′) can be synthesized individually, in which the X sequence is the index sequence. Mix the reaction thoroughly on a vortex mixer, then centrifuge briefly in a mini centrifuge to collect the mixture at the bottom of the tube.
Table 1. Library amplification system preparation
| Reagent | Volume for one reaction (μL) |
|---|---|
| Fragmentation products | 9 |
| Nuclease-free water | 21 |
| N7 primer (N7XX) | 3 |
| N5 primer (N5XX) | 3 |
| PPM | 3 |
| TAB | 10 |
| TAE | 1 |
2. Insert the PCR tube into the thermocycler and run under the conditions indicated in Table 2.
Table 2. PCR amplification program
| Step | Temperature (°C) | Time | Cycle (×) |
|---|---|---|---|
| 1: Displacement | 72 | 3 min | 1 |
| 2: Denaturation | 98 | 30 s | 1 |
| 3.1: Denaturation | 98 | 15 s | 5 |
| 3.2: Annealing | 60 | 30 s | |
| 3.3: Extension | 72 | 3 min | |
| 4: Extend | 72 | 5 min | 1 |
| 5: Hold | 4 | Hold | 1 |
E. Bead clean-up (1.2×)
After the PCR step, VAHTS DNA clean beads are used to clean up the reaction products. To improve resuspension of the beads later on, warm up nuclease-free water in a metal block heater at 55 °C.
Note: Prewarming the nuclease-free water to 55 °C aids bead resuspension, particularly if they become over-dried. VAHTS DNA clean beads are usually stored in a -4 °C freezer and need to be taken out in advance and equilibrated to room temperature before use.
1. Add 60 μL of beads (1.2× ratio) to each sample. Mix the reaction thoroughly on a vortex mixer and then centrifuge briefly in a mini centrifuge to collect the mixture at the bottom of the tube.
2. Incubate the mixture for 10 min at room temperature.
3. Place the 0.2 mL tubes on a 0.2 mL 8-strip tube magnet for 5–10 min. At this stage, DNA is bound to the beads until the solution becomes clear.
4. Aspirate and discard the supernatant without disturbing the bead pellet.
5. Wash the beads by adding 100 μL of 80% freshly prepared ethanol. Incubate for 1 min and remove ethanol without disturbing the beads.
6. Repeat step E5 for a second wash.
7. Carefully aspirate the remaining ethanol with a 10 μL tip.
8. Dry the beads for approximately 5–10 min until the beads appear dull.
Note: Over-drying (which leads to cracking) should be avoided, as it will decrease the elution efficiency of the beads.
9. Immediately remove the tube from the magnetic stand and add 20 μL of nuclease-free water preheated to 55 °C. Thoroughly resuspend the beads in the water by flicking the tube. Briefly centrifuge the tube in a microcentrifuge to collect the solution at the bottom. Incubate for 5 min at room temperature (not on the rack).
Note: Avoid excessive centrifugation, as it may pellet the beads and reduce the elution efficiency.
10. Place the tube on the magnetic stand for 5–10 min until the solution becomes clear.
11. Pipette the supernatant carefully and transfer it to a fresh tube.
F. Library quality control and pooling
The aforementioned PCR amplification results in a yield of 300–400 ng (20–30 ng/μL). The Qubit fluorometric with 1× dsDNA HS Assay kit for Qubit and Qsep100 with S2 cartridge are then used to quantify libraries (Figure 3). Table 3 demonstrates the pooling method through a practical example. At the same time, it is important to mix different species in proportion to their genome sizes when pooling, in order to obtain data with uniform genome depth and avoid data wastage.
Note: Since the sequencing employs the PE150 method, selecting an insert size of 300–600 bp maximizes the efficiency of the sequencing data. This approach avoids both the waste of data from over-sequencing and the reduction in library template quantity due to excessively long inserts, thereby minimizing the required sequencing data volume during pooling. Additionally, because sequencing libraries require the addition of sequencing adapters (approximately 150 bp) on both ends of the fragments, fragments of approximately 430–780 bp are selected during size selection.
1. Based on the concentration results from Qubit and the proportion of 430–780 bp fragments (target fragment) in the total library as determined by Qsep analysis, calculate the relative concentration of 430–780 bp fragments for each sample. For example, if the concentration of sample A1 is a ng/μL, and the proportion of 430–780 bp fragments is b%, then the relative concentration of 430–780 bp fragments is calculated as (a × b) ng/μL (Table 3).
2. Based on the relative concentration of 430–780 bp fragments in each sample, multiple samples should be pooled such that the total relative concentration of each sample is the same across all pools. Additionally, samples to be pooled together must be distinguished by a unique index. If there are libraries like Figure 3 in the pre-mixed library pool, it is not recommended to proceed with pooling, as such libraries have a lower fragment proportion, which can reduce the total DNA amount in the pool and cause difficulties for subsequent enzymatic digestion and sorting. The closer the concentration and fragment distribution between different libraries, the better. If data differences are not significant, the libraries can be directly mixed based on their volumes. In addition, when the total amount of the target fragment is less than two-thirds of the total amount required for pooled targets, this sample should not be used for pooling. Otherwise, it will increase the difficulty of enzyme digestion and sorting and also lead to uneven data (Table 3).
Table 3. Mixed database example
| ID | B73-1 | B73-2 | B73-3 | B73-4 | B73-5 | B73-6 | B73-7 | B73-8 |
| Library concentration (ng/μL) | 21.6 | 22.8 | 25.4 | 23.8 | 26.2 | 25.8 | 24.6 | 22.8 |
| Target fragment percentage (%) | 27.1 | 30.3 | 32.7 | 29.6 | 24.8 | 28.2 | 31.1 | 34.1 |
| Target fragment concentration (ng/μL) | 5.9 | 6.9 | 8.3 | 7.0 | 6.5 | 7.3 | 7.7 | 7.8 |
| Pooling volume (μL) | 10.3 | 8.7 | 7.2 | 8.5 | 9.2 | 8.2 | 7.8 | 7.7 |
| Target fragment total mass (ng) | 60 | 60 | 60 | 60 | 60 | 60 | 60 | 60 |
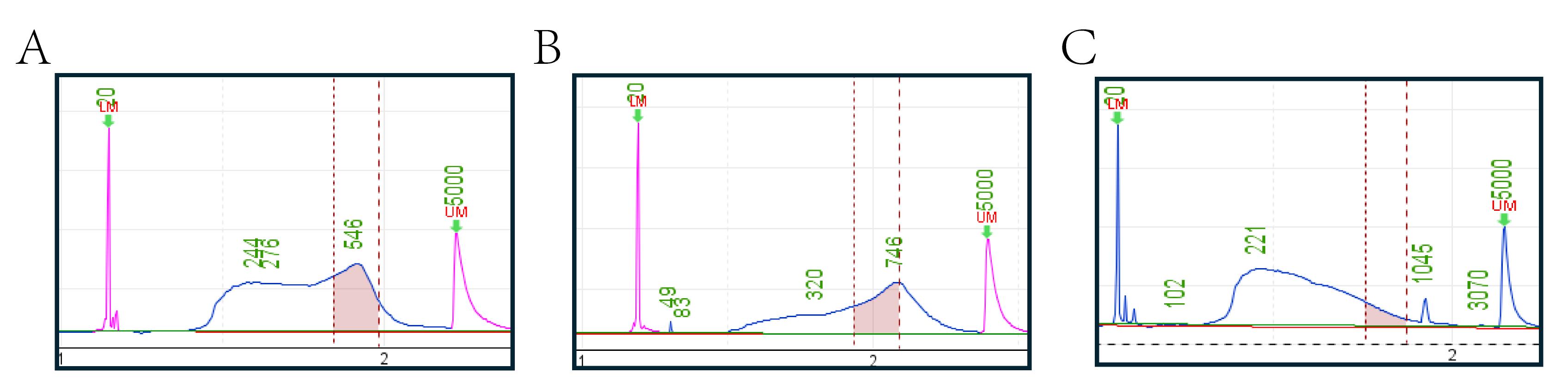
Figure 3. Quality control of the final libraries by Qsep100. A good library requires concentrated fragment distribution, with the main peak concentrated between 430 and 780 bp or nearby, to ensure that most fragments meet sequencing requirements. Good library fragments should not be concentrated in short fragments, as excessively short fragments are unsuitable for sequencing. Additionally, size selection will remove small fragments, ultimately resulting in library concentrations that fail to meet sequencing requirements. (A) Example of a good library, characterized by a fragment distribution that is predominantly concentrated within the 430–780 bp (between the first and second red vertical line) range. (B) The pooled library fragment distribution should optimally remain within the 430–780 bp range. The green text in the figure indicates the fragment sizes, while the pink peaks represent the 20 bp (first) marker and 5,000 bp (second) marker. (C) An example of a low-quality library, displaying both a fragment length distribution skewed toward overly short sizes and inadequate enrichment of the target regions. Since panel C is derived from earlier experimental results, the color of the marker peaks is not red due to version issues with the instrument system.
G. Enzymatic digestion
1. Prepare a digestion mix reaction for the pooled library provided in Table 4. MseI, MspI, and AluI were all obtained from New England Biolabs (NEB).
Note: To compensate for the substantial decrease of valid fragments in the library after digestion, multiple reactions (3–4 tubes are suitable) can be carried out in parallel. Mix the reaction thoroughly on a vortex mixer and then centrifuge briefly in a mini centrifuge to collect the mixture at the bottom of the tube.
Table 4. Library digestion system preparation
| Reagent | Volume for one reaction (μL) |
|---|---|
| Pooled library | A (1,000 ng) |
| Nuclease-free water | 42 − A |
| 10x NEB CutSmart buffer | 5 |
| MseI (10 U/μL) | 1 |
| MspI (20 U/μL) | 1 |
| AluI (10 U/μL) | 1 |
| Total | 50 |
2. Incubate the digestion reaction in a thermocycler overnight at 37 °C with the lid temperature set to 42 °C.
H. Bead clean-up (0.8×)
After the digestion step, VAHTS DNA clean beads are used to clean up the reaction products. To improve resuspension of the beads later on, warm up nuclease-free water in a metal block heater at 55 °C.
Note: Prewarming the nuclease-free water to 55 °C aids bead resuspension, particularly if they become over-dried. VAHTS DNA clean beads are usually stored in a -4 °C freezer and need to be taken out in advance and equilibrated to room temperature before use.
1. Add 40 μL of beads (0.8× ratio) to each sample. Mix the reaction thoroughly on a vortex mixer and then centrifuge briefly in a mini centrifuge to collect the mixture at the bottom of the tube.
Note: To avoid the recovery of the many short, unsequenceable fragments generated by digestion, the bead volume is lowered.
2. Incubate the mixture for 10 min at room temperature.
3. Place the 0.2 mL tubes on a 0.2 mL 8-strip tube magnet for 5–10 min. At this stage, DNA is bound to the beads until the solution becomes clear.
4. Aspirate and discard the supernatant without disturbing the bead pellet.
5. Wash the beads by adding 100 μL of 80% freshly prepared ethanol. Incubate for 1 min and remove ethanol without disturbing the beads.
6. Repeat step H5 for a second wash.
7. Carefully aspirate the remaining ethanol with a 10 μL tip.
8. Dry the beads for about 5–10 min until the beads appear dull.
Note: Over-drying (which leads to cracking) should be avoided, as it will decrease the elution efficiency of the beads.
9. Immediately remove the tube from the magnetic stand and add 20 μL of nuclease-free water preheated to 55 °C. Thoroughly resuspend the beads in the water by flicking the tube. Briefly centrifuge the tube in a microcentrifuge to collect the solution at the bottom. Incubate for 5 min at room temperature (not on the rack).
Note: Avoid excessive centrifugation, as it may pellet the beads and reduce the elution efficiency.
10. Place the tube on the magnetic stand for 5–10 min until the solution becomes clear.
11. Pipette the supernatant carefully and transfer it to a fresh tube.
I. PCR of iRAD library
If the total library yield is less than 2,000 ng, perform a library amplification PCR first. Otherwise, proceed directly to the next step of library fractionation (section J).
1. Prepare a PCR mix reaction for iRAD library following the instructions provided in Table 5. PPM, TAB, and TAE are provided with TruePrep® DNA Library Prep kit V2 for Illumina.
Note: Mix the reaction thoroughly on a vortex mixer and then centrifuge briefly in a mini centrifuge to collect the mixture at the bottom of the tube.
Table 5. iRAD library amplification system preparation
| Reagent | Volume for one reaction (μL) |
|---|---|
| iRAD library | A (100 ng) |
| Nuclease-free water | 37 − A |
| PPM | 6 |
| TAB | 10 |
| TAE | 1 |
2. Place the PCR tube into the thermocycler and run under the conditions indicated in Table 6.
Table 6. PCR amplification program
| Step | Temperature (°C) | Time | Cycle (×) |
| 1: Displacement | 72 | 3 min | 1 |
| 2: Denaturation | 98 | 30 s | 1 |
| 3.1: Denaturation | 98 | 15 s | 5 |
| 3.2: Annealing | 60 | 30 s | |
| 3.3: Extension | 72 | 3 min | |
| 4: Extend | 72 | 5 min | 1 |
| 5: Hold | 4 | Hold | 1 |
3. Purify the PCR product using beads, as described in section E.
J. Library sorting
Library quantification is performed using a Qubit fluorometer with the 1× dsDNA HS Assay kit and a Qsep100 system equipped with an S2 cartridge. The PippinHT DNA Size Selection System with 2% agarose is used in the size selection of iRAD libraries. If the library concentration is below 40 ng/μL, it needs to be concentrated utilizing a Concentrator Plus to raise the concentration above 40 ng/μL.
1. In a 0.2 mL PCR tube, dilute 1,000 ng of DNA in nuclease-free water for a final volume of 25 μL.
Note: The DNA loading volume should not exceed 2 μg per tube. If the DNA amount is insufficient, multiple parallel sorting reactions are recommended.
2. Add 5 μL of 100–600 bp marker (from 2% agarose) to the tube.
Note: Mix the reaction thoroughly on a vortex mixer and then centrifuge briefly in a mini centrifuge to collect the mixture at the bottom of the tube.
3. As per the instrument manual of PippinHT, adjust the gel cassette and start electrophoresis in marker mode following sample loading.
4. Transfer the purified library from the gel collection well to a new 1.5 mL microcentrifuge tube, with a volume of approximately 30 μL.
K. Sequencing
Paired-end sequencing (150 bp read length) of libraries is performed on an Illumina apparatus. A sequencing depth of 1–2× reference genome coverage per sample is sufficient for downstream bioinformatics analysis.
L. Data analysis
The quality of raw sequencing data was initially evaluated using FastQC (version 0.11.8) [15]. To enhance data quality, the raw reads were processed with Trimmomatic (version 0.39) [16]. FastUniq (Version 1.1) [17] was then employed to identify and eliminate duplicate reads. The cleaned, high-quality reads were aligned to a reference genome using BWA-MEM [18]. The B73_V4 reference genomes were used in this study. After alignment, the resulting BAM files underwent further processing. They were sorted using Samtools [19], and duplicate reads were marked using GATK (version 4.1.8.1) [20].
QualiMap [21] was used to analyze the distribution of genome coverage. SNPs were called using GATK’s HaplotypeCaller command with its default settings. Finally, the identified SNPs were filtered using modified parameters in GATK to remove low-quality or potentially false-positive variants. The filtering criteria included: QD < 2.0, QUAL < 30.0, SOR > 3.0, FS > 60.0, MQ < 40.0, MQRankSum < -12.5, and ReadPosRankSum < -8.0.
After analyzing, we finally obtained the genome coverage of the actual data (Figure 4), which showed good consistency with our electronic prediction results (Figure 2C).
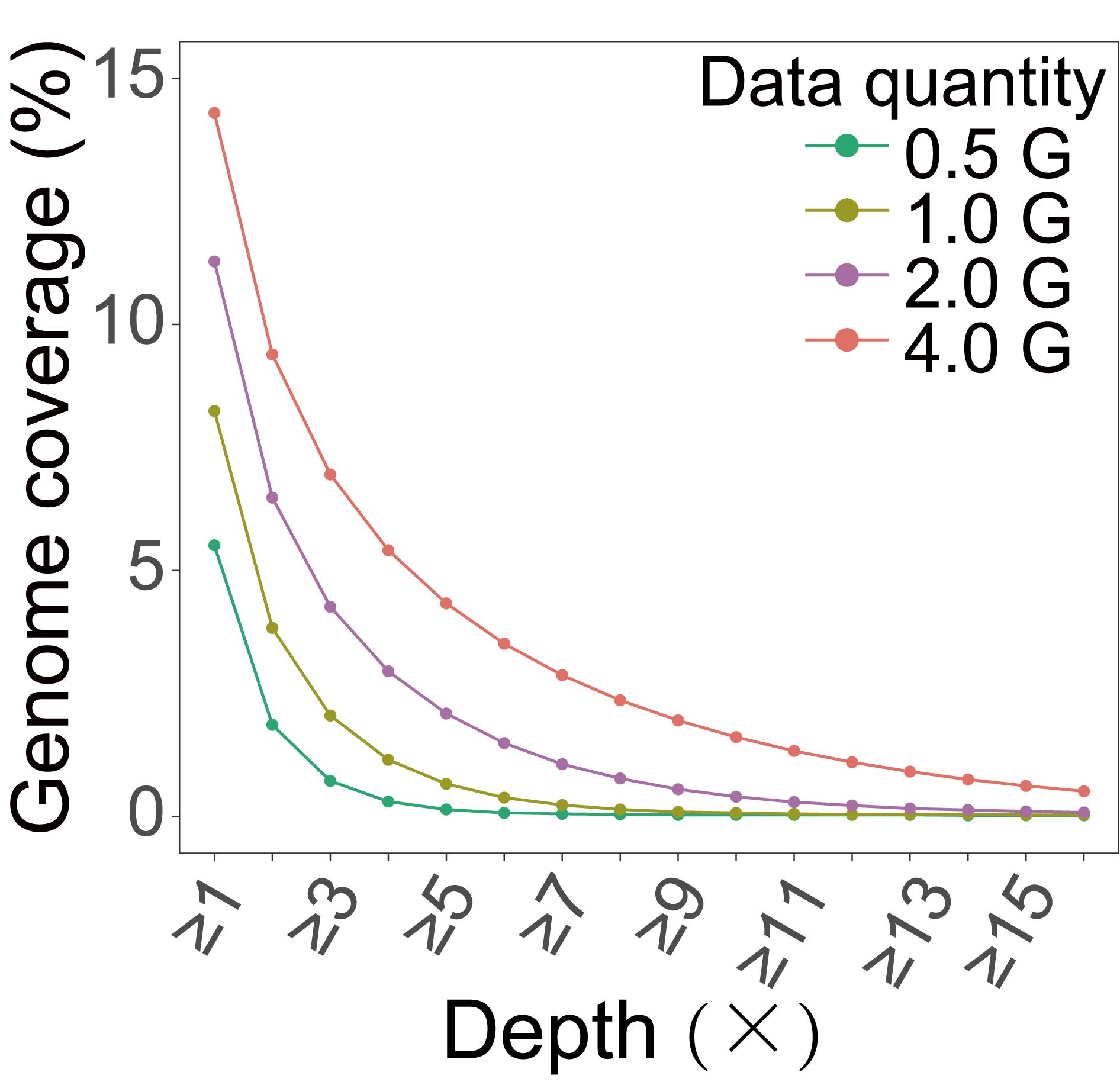
Figure 4. Genomic coverage obtained from actual data of maize. We extracted varying amounts of data from sequencing data for coverage analysis and calculated the genome coverage under different depth ranges.
We also used the IGV software to visualize the aligned data and obtained the expected results, where the distribution of reads was only visible in regions with non-dense restriction enzyme sites (Figure 5).

Figure 5. Relationship between sequencing reads and restriction enzyme (RE) recognition sites in the genome. The reads of inverse restriction site–associated DNA sequencing (iRAD-seq) are primarily enriched in non-RE recognition regions. (A) Overview of the relationship between sequencing reads and RE recognition sites across the genomic region chr1: 25,034,313-25,053,417. (B, C) Detailed depiction of fragment patterns in the subregions boxed in A.
Validation of protocol
This protocol (or parts of it) has been used and validated in the following research article(s):
Chen et al. [22]. A novel method for effectively selecting fragments not associated with restriction sites for whole-genome genotyping. BMC Biol.
We demonstrated the uniform distribution of markers across the genome and high technical reproducibility of iRAD-seq. The method was successfully applied to both evolutionary studies of natural populations and genetic analysis of breeding populations. To further contextualize its value, we compared iRAD-seq with existing methods, highlighting its advantages in workflow simplicity and cost-effectiveness. Moreover, we developed a semi-automated workflow to substantially increase its throughput.
General notes and troubleshooting
General notes
1. If the system configuration during the library preparation process can be carried out entirely on ice, it may increase the success rate of library construction.
2. After system configuration and during the magnetic bead purification steps, thorough mixing is required at each mixing step to enhance the yield and purification efficiency of the obtained product.
Troubleshooting
Problem 1: Samples with identical sequencing adapters all yielded relatively low data volumes.
Possible cause: This sequencing adapter contains a restriction enzyme site, and during enzymatic digestion, the adapter is also cleaved, resulting in failure to successfully construct the library.
Solution: Once the restriction enzyme is determined, proceed immediately with adapter screening to remove sequencing adapters containing restriction sites.
Problem 2: The final yield of the library is low.
Possible causes: The magnetic beads were taken out from the 4 °C refrigerator without equilibrating to room temperature, reaction systems were not fully mixed during binding and washing, or the elution H2O was not preheated.
Solutions: Remove the magnetic beads from the refrigerator in advance and equilibrate them to room temperature. Ensure thorough mixing during magnetic bead binding and washing steps. Prewarm the elution H2O to 55°C before use.
Problem 3: The distribution of fragments in the library is relatively small.
Possible causes: The ratio of DNA to Tn5 transposase was not properly adjusted, or the DNA quality itself is poor (the fragments were inherently short).
Solutions: Test the quality and integrity of DNA, then adjust the ratio of DNA to Tn5 transposase based on the test results, or directly determine the optimal ratio through gradient pre-experiments with DNA and Tn5 transposase.
Problem 4: After sequencing mix libraries of different species, the large-genome species fail to achieve sequencing depth commensurate with their genome size.
Possible cause: When mixing libraries, only the equal quality of target fragments is considered, without taking into account the influence of genome size.
Solution: During library pooling, libraries are mixed proportionally based on the differences in genome sizes among different species, with larger genomes requiring more mixing.
Problem 5: Sorting failed.
Possible causes: The marker has expired due to prolonged storage, the sample was not spotted, or the buffer was not added to the sample well.
Solution: Check the expiration date of the marker and use only those within their shelf life. Even if markers are within the expiration date, discard any that show signs of degradation or if problems persist, to prevent recurrence.
Acknowledgments
Y.C., Y.L., and B.Z. discussed and designed the experiments; P.C. performed the experiments; P.C., B.Z., S.Z., Z.Z., J.G., H.W., and Y.Y. performed the experiments; P.C., B.Z., Y.L., and Y.C. wrote the manuscript with contributions from all authors; H.L. and Y.X. contributed the maize population and rice line; P.C., B.Z., and H.C. participated in the data analysis and prepared the figures; Y.C. and Y.L. supervised the project. All authors have read and approved the manuscript. Funding acquisition, Y.C. This work was funded by the National Natural Science Foundation of China (32172086) and Scarce and Quality Economic Forest Engineering Technology Research Center (2022GCZX002). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests
The authors declare no conflict of interest.
References
- Chen, W., Chen, L., Zhang, X., Yang, N., Guo, J., Wang, M., Ji, S., Zhao, X., Yin, P., Cai, L., et al. (2022). Convergent selection of a WD40 protein that enhances grain yield in maize and rice. Science. 375(6587): eabg7985. https://doi.org/10.1126/science.abg7985
- Huang, X., Huang, S., Han, B. and Li, J. (2022). The integrated genomics of crop domestication and breeding. Cell. 185(15): 2828–2839. https://doi.org/10.1016/j.cell.2022.04.036
- Slovák, M., Melichárková, A., Štubňová, E. G., Kučera, J., Mandáková, T., Smyčka, J., Lavergne, S., Passalacqua, N. G., Vďačný, P., Paun, O., et al. (2022). Pervasive Introgression During Rapid Diversification of the European Mountain Genus Soldanella (L.) (Primulaceae). Syst Biol. 72(3): 491–504. https://doi.org/10.1093/sysbio/syac071
- Hirsch, C. D., Evans, J., Buell, C. R. and Hirsch, C. N. (2014). Reduced representation approaches to interrogate genome diversity in large repetitive plant genomes. Briefings Funct Genomics. 13(4): 257–267. https://doi.org/10.1093/bfgp/elt051
- Baird, N. A., Etter, P. D., Atwood, T. S., Currey, M. C., Shiver, A. L., Lewis, Z. A., Selker, E. U., Cresko, W. A. and Johnson, E. A. (2008). Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS One. 3(10): e3376. https://doi.org/10.1371/journal.pone.0003376
- Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G. and Hohenlohe, P. A. (2016). Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet. 17(2): 81–92. https://doi.org/10.1038/nrg.2015.28
- Roberts, R. J. (2003). A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 31(7): 1805–1812. https://doi.org/10.1093/nar/gkg274
- Wang, S., Meyer, E., McKay, J. K. and Matz, M. V. (2012). 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Methods. 9(8): 808–810. https://doi.org/10.1038/nmeth.2023
- Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. and Hoekstra, H. E. (2012). Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS One. 7(5): e37135. https://doi.org/10.1371/journal.pone.0037135
- Toonen, R. J., Puritz, J. B., Forsman, Z. H., Whitney, J. L., Fernandez-Silva, I., Andrews, K. R. and Bird, C. E. (2013). ezRAD: a simplified method for genomic genotyping in non-model organisms. PeerJ. 1: e203. https://doi.org/10.7717/peerj.203
- Wang, S., Lv, J., Zhang, L., Dou, J., Sun, Y., Li, X., Fu, X., Dou, H., Mao, J., Hu, X., et al. (2015). MethylRAD: a simple and scalable method for genome-wide DNA methylation profiling using methylation-dependent restriction enzymes. Open Biol. 5(11): 150130. https://doi.org/10.1098/rsob.150130
- Adey, A., Morrison, H. G., Asan, ., Xun, X., Kitzman, J. O., Turner, E. H., Stackhouse, B., MacKenzie, A. P., Caruccio, N. C., Zhang, X., et al. (2010). Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 11(12): e1186/gb–2010–11–12–r119. https://doi.org/10.1186/gb-2010-11-12-r119
- Cheng, S., Miao, B., Li, T., Zhao, G. and Zhang, B. (2024). Review and Evaluate the Bioinformatics Analysis Strategies of ATAC-seq and CUT&Tag Data. Genom Proteom Bioinf. 22(3): e1093/gpbjnl/qzae054. https://doi.org/10.1093/gpbjnl/qzae054
- Zhao, S., Zhang, C., Mu, J., Zhang, H., Yao, W., Ding, X., Ding, J. and Chang, Y. (2020). All-in-one sequencing: an improved library preparation method for cost-effective and high-throughput next-generation sequencing. Plant Methods. 16(1): 74. https://doi.org/10.1186/s13007-020-00615-3
- Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. (Accessed on 4 Oct 2018, https://www.scirp.org/reference/referencespapers?referenceid=2781642)
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15): 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
- Xu, H., Luo, X., Qian, J., Pang, X., Song, J., Qian, G., Chen, J. and Chen, S. (2012). FastUniq: A Fast De Novo Duplicates Removal Tool for Paired Short Reads. PLoS One. 7(12): e52249. https://doi.org/10.1371/journal.pone.0052249
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25(14): 1754–1760. https://doi.org/10.1093/bioinformatics/btp324
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25(16): 2078–2079. https://doi.org/10.1093/bioinformatics/btp352
- DePristo, M. A., Banks, E., Poplin, R., Garimella, K. V., Maguire, J. R., Hartl, C., Philippakis, A. A., del Angel, G., Rivas, M. A., Hanna, M., et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43(5): 491–498. https://doi.org/10.1038/ng.806
- Okonechnikov, K., Conesa, A. and García-Alcalde, F. (2015). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32(2): 292–294. https://doi.org/10.1093/bioinformatics/btv566
- Chen, P., Zhang, B., Zhao, S., Zhu, Z., Yan, Y., Chen, H., Lu, H., Xiang, Y., Li, Y., Chang, Y., et al. (2025). A novel method for effectively selecting fragments not associated with restriction sites for whole-genome genotyping. BMC Biol. 23(1): e1186/s12915–025–02330–8. https://doi.org/10.1186/s12915-025-02330-8
Article Information
Publication history
Received: Nov 20, 2025
Accepted: Jan 5, 2026
Available online: Jan 18, 2026
Published: Jan 20, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Chen, P., Zhou, S., Wang, H., Gu, J., Li, Y. and Chang, Y. (2026). Inverse Restriction Site-Associated DNA Sequencing (iRAD-seq). Bio-protocol 16(2): e5599. DOI: 10.21769/BioProtoc.5599.
Category
Molecular Biology > DNA > DNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.