- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A One-Step Method for Efficient Purification of Functional Cas9 Protein
(*contributed equally to this work) Published: Vol 16, Iss 3, Feb 5, 2026 DOI: 10.21769/BioProtoc.5594 Views: 157
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2025

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The CRISPR/Cas9 system is a cornerstone technology in genome editing. Delivery of pre-assembled Cas9 ribonucleoprotein (RNP) complexes exhibits distinct advantages, including reduced off-target effects and lower immunogenicity. Conventional methods for purifying Cas9 protein typically involve multi-step chromatography and the cleavage of fusion tag, which are time-consuming and result in diminished yields. In this study, we present a simplified, one-step purification strategy for functional Streptococcus pyogenes Cas9 (SpCas9) using the ubiquitin (Ub) fusion system in Escherichia coli. The N-terminal Ub fusion not only improves protein solubility but also facilitates high-yield production of the His-Ub-Cas9 fusion protein. Importantly, the Ub tag does not require proteolytic removal during purification, allowing direct one-step purification of the fusion protein via nickel-affinity chromatography. The purified His-Ub-Cas9 retains robust DNA cleavage activity in vivo, as validated in zebrafish embryos. This protocol greatly simplifies the production of functional Cas9 protein, facilitating its broad application in genome editing.
Key features
• The Ub fusion system enables single-step purification of Cas9 in E. coli using Ni-NTA chromatography, eliminating the protease cleavage step.
• This method yields over 8 mg/L of high purity (>95%), functional Cas9 protein, suitable for direct use in RNP complex assembly.
• The purified His-Ub-Cas9 maintains high genome editing activity in vivo, as demonstrated in zebrafish embryos.
Keywords: SpCas9Background
The CRISPR-Cas9 system has revolutionized genome editing by enabling precise, programmable modifications of DNA sequences across diverse biological systems. Owing to its simplicity, efficiency, and versatility, the CRISPR-Cas9 system has become an essential tool for basic scientific research, biotechnology applications, and the advancement of therapeutic strategies [1]. Although plasmid- and mRNA-based delivery methods are commonly used, Cas9 ribonucleoprotein (RNP) delivery offers distinct advantages, including higher editing precision, lower off-target effects, and elimination of genomic integration risks [2–4]. Consequently, the RNP-based CRISPR/Cas9 system has been widely adopted across a variety of organisms, enabling applications including resistance marker-free knockouts in fungi, high-efficiency precise editing in plant protoplasts, and the generation of stable mutant animal models such as zebrafish for functional analysis in vivo [5–7]. However, the production of high-purity Cas9 protein remains challenging due to its large size (158 kDa) and the complexities of conventional purification workflows [8].
Conventional protocols often involve multiple purification steps, such as affinity, ion-exchange, and size-exclusion chromatography, as well as protease cleavage to remove fusion tags [9,10]. These multi-step processes not only increase time and cost but also lead to significant protein loss [11]. To overcome these limitations, we developed a single-step purification method using a ubiquitin fusion strategy. The Ub tag significantly enhances soluble expression of the fusion protein in E. coli [12]. Furthermore, it can be cleaved by endogenous deubiquitylases in eukaryotic cells [13], eliminating the need for in vitro tag removal and thereby avoiding operational complexity and low recovery yields. The Ub fusion approach simplifies the purification while maintaining Cas9’s structural and functional integrity. Comparative analysis confirms that the purified Ub-Cas9 exhibits nuclease activity comparable to that of commercial Cas9 in vivo.
This protocol describes the construction of His-Ub-Cas9 expression plasmid, its expression in E. coli BL21(DE3), and one-step purification using Ni-NTA affinity chromatography. We also provide detailed procedures for in vivo functional validation in zebrafish embryos. The entire process can be completed within one week and yields over 8 mg/L of Cas9 protein with >95% purity and robust in vivo editing activity.
In summary, we present a streamlined, efficient, and cost-effective method for obtaining high-purity functional Cas9 protein, which will facilitate its broader application in genome editing and gene therapy research.
Materials and reagents
Biological materials
1. pET-28b-Cas9-His plasmid (Addgene, catalog number: 47327)
2. pHUE plasmid [12]
3. pCMV-Tag2B (Agilent Technologies, catalog number: 211172)
4. E. coli BL21 (DE3) cells (NEB, catalog number: C2527H)
5. E. coli DH5α cells (NEB, catalog number: C2988J)
Reagents
1. Phanta Max Super-Fidelity DNA Polymerase (Vazyme, catalog number: P505)
2. TIANgel Purification kit (TIANGEN, catalog number: DP219-03)
3. Restriction enzyme BamH I, 10× K buffer (TAKARA, catalog number: 1010S)
4. Restriction enzyme Hind III (TAKARA, catalog number: 1060S)
5. Exnase II, 5× CE II buffer (Vazyme, catalog number: C112)
6. 2× Hieff® PCR Master Mix (with dye) (Yeasen, catalog number: 10101)
7. TIANprep Mini Plasmid kit (TIANGEN, catalog number: DP103-03)
8. Tryptone (Beyotime, catalog number: ST799)
9. Yeast extract (Beyotime, catalog number: ST969)
10. Sodium chloride (NaCl) (MCKLIN, catalog number: S805275)
11. Agar powder (Solarbio, catalog number: A8190)
12. Ampicillin (Beyotime, catalog number: ST008)
13. 50× TAE buffer (Beyotime, catalog number: ST716)
14. Agarose (Yeasen, catalog number: 10208ES60)
15. Super Red DNA gel stain (Biosharp, catalog number: BS354B)
16. 10× DNA loading buffer (TAKARA, catalog number: SD0012)
17. DL 2000 DNA marker (TAKARA, catalog number: 3427A)
18. Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Yeasen, catalog number: 10902ES08)
19. M5 protease inhibitor cocktail, EDTA-free (100× DMSO) (Mei5bio, catalog number: MF182-plus-0)
20. Triton X-100 (Biosharp, catalog number: BS084)
21. Glycerol (MACKLIN, catalog number: G810575)
22. Imidazole (Sigma-Aldrich, catalog number: I2399)
23. 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES) (Sigma, catalog number: H3375)
24. Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (MACKLIN, catalog number: T917415)
25. Potassium hydroxide (KOH) (XILONG, catalog number: 10202101)
26. Potassium chloride (KCl) (XILONG, catalog number: 10200501)
27. Coomassie Blue R-250 (AMRESCO, catalog number: C0472)
28. Methanol (XILONG, catalog number: 12801001)
29. Acetic acid (XILONG, catalog number: 12705701)
30. Coomassie Blue G-250 (Beyotime, catalog number: ST030)
31. Ethanol (XILONG, catalog number: 12803401)
32. Phosphoric acid (XILONG, catalog number: 12704101)
33. Glycine (Solarbio, catalog number: G8200)
34. Tris base (Solarbio, catalog number: T80)
35. Sodium dodecyl sulfate (SDS) (Solarbio, catalog number: S8010)
36. 1 M Tris-HCl (Solarbio, catalog number: T1020)
37. 1.5 M Tris-HCl (Solarbio, catalog number: T1010)
38. 30% acrylamide-bisacrylamide (Biosharp, catalog number: BL513b)
39. Ammonium persulfate (APS) (MACKLIN, catalog number: 801037)
40. N,N,N’,N’-Tetramethylethylenediamine (TEMED) (Solarbio, catalog number: T8090)
41. Isopropyl alcohol (XILONG, catalog number: 12802501)
42. SDS loading buffer (5×) (Beyotime, catalog number: P0015)
43. M5 HiClear prestained protein ladder (10–180 kDa) (Mei5bio, catalog number: MF212)
44. Sodium phosphate dibasic (Na2HPO4) (XILONG, catalog number: 10105901)
45. Disodium phosphate dodecahydrate (Na2HPO4·12H2O) (XILONG, catalog number: 1010590104204)
46. Potassium dihydrogen phosphate (KH2PO4) (XILONG, catalog number: 10204401)
47. Calcium chloride (CaCl2) (GHTECH, catalog number: 111-2003)
48. Sodium bicarbonate (NaHCO3) (XILONG, catalog number: 10104201)
49. Cas9-N-NLS nuclease (Genscript, catalog number: Z03388)
50. Sodium hydroxide (NaOH) (XILONG, catalog number: 10103101)
51. T7 Endonuclease I (Vazyme, catalog number: EN303)
52. Liquid nitrogen (local distributor)
Solutions
1. Luria-Bertani (LB) medium (see Recipes)
2. 100 mg/mL ampicillin (see Recipes)
3. LB medium with ampicillin (see Recipes)
4. LB agar plate with ampicillin (see Recipes)
5. 1 M IPTG (see Recipes)
6. Lysis buffer (see Recipes)
7. Elution buffer (see Recipes)
8. Storage buffer (see Recipes)
9. 10% APS (see Recipes)
10. 5× Tris-glycine buffer (see Recipes)
11. 10 mg/mL BSA (see Recipes)
12. Coomassie blue staining solution (see Recipes)
13. Destaining solution (see Recipes)
14. 1× PBS (see Recipes)
15. 10× PBS (see Recipes)
16. 5× Bradford reagent (see Recipes)
17. Embryo medium (see Recipes)
18. 50 mM NaOH (see Recipes)
19. 100 mM Tris-HCl (pH 8.0) (see Recipes)
Recipes
1. Luria-Bertani (LB) medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Tryptone | 1% (w/v) | 10 g |
| Yeast extract | 0.5% (w/v) | 5 g |
| NaCl | 1% (w/v) | 10 g |
| ddH2O | n/a | ~980 mL |
| Total | n/a | 1 L |
Autoclave at 121 °C for 20 min. Store at room temperature.
2. 100 mg/mL ampicillin
| Reagent | Final concentration | Amount |
|---|---|---|
| Ampicillin | 100 mg/mL | 1 g |
| ddH2O | n/a | 10 mL |
Sterilize the solution with a 0.22 μm syringe filter. Store at -20 °C.
3. LB medium with ampicillin
| Reagent | Final concentration | Amount |
|---|---|---|
| LB medium | 1× | 50 mL |
| 100 mg/mL ampicillin | 100 μg/mL | 50 μL |
Store at 4 °C.
4. LB agar plate with ampicillin
| Reagent | Final concentration | Amount |
|---|---|---|
| LB broth | 25 g/L | 25 g |
| Agar | 15 g/L | 15 g |
| 100 mg/mL ampicillin | 100 μg/mL | 1 mL |
| ddH2O | n/a | ~980 mL |
| Total | n/a | 1 L |
a. Dissolve LB broth in ~980 mL of ddH2O. Transfer the medium to a 1-L glass bottle.
b. Add 15 g of agar, drop in a stir bar, and autoclave at 121 °C for 20 min.
c. Stir the medium and allow to cool to 50–60 °C.
d. Add 1 mL of 100 mg/mL ampicillin solution. Stir well.
e. Transfer 20 mL of the solution to 10-cm Petri dishes using a serological pipette.
f. Allow the agar plates to cool and dry in a laminar flow hood for 30 min.
g. Store the plates at 4 °C in a plastic bag.
5. 1 M IPTG
| Reagent | Final concentration | Amount |
|---|---|---|
| IPTG | 1 M | 2.4 g |
| ddH2O | n/a | 10 mL |
a. Weigh 2.4 g of IPTG powder and place it into a 15 mL tube.
b. Add 8 mL of ddH2O. Mix thoroughly until dissolved, then bring the volume up to 10 mL with ddH2O.
c. Sterilize the solution by filtration through a 0.22 μm syringe filter.
d. Aliquot into small volumes (1 mL per aliquot) and store at -20 °C.
6. Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Tris-HCl, pH 7.5 | 20 mM | 4 mL |
| NaCl | 500 mM | 5.844 g |
| Glycerol | 5% (v/v) | 10 mL |
| Imidazole | 40 mM | 0.544 g |
| ddH2O | n/a | ~170 mL |
| Total | n/a | 200 mL |
a. Weigh 5.844 g of NaCl and 0.544 g of imidazole and transfer to a clean 250 mL beaker.
b. Add about 170 mL of ddH2O and stir using a magnetic stir bar until completely dissolved.
c. Add 4 mL of 1 M Tris-HCl (pH 7.5) and 10 mL of glycerol to the solution and mix completely.
d. Adjust the pH to 7.5 with 1 M HCl or 1 M NaOH, then bring the final volume to 200 mL with ddH2O.
e. Filter the buffer through a 0.22 μm syringe filter and store at 4 °C.
7. Elution buffer
Elution buffer 1: Lysis buffer with 80 mM imidazole
Elution buffer 2: Lysis buffer with 500 mM imidazole
Filter the buffer through a 0.22 μm syringe filter. Store at 4 °C.
8. Storage buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| HEPES | 20 mM | 0.952 g |
| KCl | 150 mM | 2.235 g |
| Glycerol | 10% (v/v) | 20 mL |
| 500 mM TCEP | 1 mM | 400 μL |
| ddH2O | n/a | ~160 mL |
| Total | n/a | 200 mL |
a. Dissolve 0.143 g of TCEP in 1 mL of ddH2O to prepare a 500 mM stock solution. Store at 4 °C for several weeks.
b. Prepare the buffer by combining the components above.
c. Include 400 μL of the 500 mM TCEP stock to a final concentration of 1 mM.
d. Adjust the pH to 7.5 with 1 M KOH.
e. Filter the buffer through a 0.22 μm syringe filter. Store at 4 °C for up to one week.
9. 10% APS
| Reagent | Final concentration | Amount |
|---|---|---|
| APS | 10% (w/v) | 1 g |
| ddH2O | n/a | 10 mL |
Store at 4 °C for up to one week.
10. 5× Tris-glycine buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris | 0.125 M | 15.1 g |
| Glycine | 1.25 M | 94 g |
| SDS | 0.5% (w/v) | 5.0 g |
| ddH2O | n/a | ~980 mL |
| Total | n/a | 1 L |
11. 10 mg/mL BSA
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 10 mg/mL | 0.1 g |
| ddH2O | n/a | 10 mL |
12. Coomassie blue staining solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Coomassie blue R-250 | 0.05% (w/v) | 0.5 g |
| Methanol | 50% (v/v) | 500 mL |
| Acetic acid | 10% (v/v) | 100 mL |
| ddH2O | n/a | ~380 mL |
| Total | n/a | 1 L |
Filter through filter paper after preparation and store at room temperature.
13. Destaining solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Methanol | 50% (v/v) | 500 mL |
| Acetic acid | 10% (v/v) | 100 mL |
| ddH2O | n/a | ~380 mL |
| Total | n/a | 1 L |
14. 1× PBS
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 137 mM | 8 g |
| KCl | 2.7 mM | 0.2 g |
| Na2HPO4 | 10 mM | 1.42 g |
| KH2PO4 | 2 mM | 0.27 g |
| ddH2O | n/a | ~980 mL |
| Total | n/a | 1 L |
a. Adjust the pH to 7.4 by adding HCl, then bring the final volume to 1 L with ddH2O.
b. Autoclave at 121 °C for 20 min and store at room temperature.
15. 10× PBS
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 1.37 M | 80 g |
| KCl | 27 mM | 2 g |
| Na2HPO4·12H2O | 80 mM | 29 g |
| KH2PO4 | 20 mM | 2.7 g |
| ddH2O | n/a | ~980 mL |
| Total | n/a | 1 L |
Adjust the pH to 7.4 by adding HCl, then bring the final volume to 1 L with ddH2O.
16. 5× Bradford reagent
| Reagent | Final concentration | Amount |
|---|---|---|
| Coomassie blue G-250 | 0.05% (w/v) | 50 mg |
| 95% ethanol | 23.75% (v/v) | 25 mL |
| 85% H3PO4 | 42.5% (v/v) | 50 mL |
| ddH2O | n/a | ~20 mL |
| Total | n/a | 100 mL |
a. Dissolve 50 mg of Coomassie Brilliant Blue G-250 in 25 mL of 95% ethanol with stirring.
b. Add 50 mL of 85% H3PO4.
c. Bring the volume to 100 mL with ddH2O.
d. Filter through filter paper and store at 4 °C.
17. Embryo medium
| Reagent | Final concentration | Amount |
|---|---|---|
| NaCl | 60 mM | 3.5 g |
| KCl | 0.67 mM | 0.05 g |
| NaHCO3 | 2.4 mM | 0.2 g |
| CaCl2 | 0.9 mM | 0.1 g |
| ddH2O | n/a | ~980 mL |
| Total | n/a | 1 L |
Store at room temperature.
18. 50 mM NaOH
| Reagent | Final concentration | Amount |
|---|---|---|
| NaOH | 50 mM | 0.1 g |
| ddH2O | n/a | 50 mL |
19. 100 mM Tris-HCl (pH 8.0)
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris base | 100 mM | 0.606 g |
| ddH2O | n/a | 50 mL |
a. Dissolve Tris base in 40 mL of ddH2O.
b. Stir the solution and adjust the pH to 8.0 with HCl.
c. Adjust the final volume to 50 mL with ddH2O.
Laboratory supplies
1. Pipette tips: 20, 200, 1,000 μL (Biosharp, catalog numbers: T-001-10, T-001-200, T-001-1000)
2. Pipette (Eppendorf, catalog numbers: 4924000029, 4924000053, 4924000088)
3. 200 μL PCR tube (LABSELECT, catalog number: PST-0208-FT-C)
4. 1.5 and 2 mL microcentrifuge tube (Biofil, catalog numbers: CFT000015, CFT000020)
5. 15 and 50 mL conical tube (Biosharp, catalog numbers: CT-002-15, CT-002-50)
6. 250 mL and 1 L Erlenmeyer flask (Biosharp, catalog numbers: 57202, 57402)
7. 250 mL and 1 L beaker (Biosharp, catalog numbers: BS-LFB-250, BS-LFB-1000)
8. 0.22 μm syringe filter (Millipore, catalog number: SLGVR33RS)
9. 20 mL syringe (Beyotime, catalog number: FS820-40pcs)
10. 10 cm Petri dish (Yeasen, catalog number: 84009ES50)
11. Disposable plastic cuvette (Yeasen, catalog number: 84201ES20)
12. Ni-NTA resin (GE Healthcare, catalog number: 17-5318-02)
13. PD-10 desalting column (GE Healthcare, catalog number: 17-0851-01)
14. 30 kDa MWCO (Amicon Ultra, catalog number: UFC901024)
15. Glass capillary tube (WPI, catalog number: 1B100F-4)
16. Microloader pipette tip (Eppendorf, catalog number: 5242 956.003)
17. Surgical scalpel (Beyotime, catalog number: FS205)
18. PARAFILM® M (Merck, catalog number: P7793)
Equipment
1. Thermal cycler (Eppendorf, catalog number: 6337000078)
2. Water bath (Tongjun, model: HH-11-2)
3. Centrifuge (Eppendorf, model: 5418R/5804R)
4. Incubator (YETUO, model: XMA-600)
5. Shaker incubator (NOKI, model: TS-200B)
6. 28 °C incubator (LICHEN, model: LC-HN-25S)
7. Analytical balance (Sartorius, model: GL124-1SCN)
8. UV spectrophotometer (SHIMADZU, model: UV1900i)
9. SevenEasy pH meter (METTLER TOLEDO, model: S20K)
10. Diaphragm vacuum pump (Xiangmingjie, model: XMJ-15T)
11. Nanodrop one (Thermo Fisher, catalog number: 840-317400)
12. DNA electrophoresis system (JUNYI, model: JT300C)
13. Mini-PROTEAN Tetra Vertical Electrophoresis Cell (Bio-Rad, catalog number: 165-8000)
14. Bio-Rad PowerPac HV power supply (Bio-Rad, catalog number: 164-5050)
15. DNA agarose gel imaging equipment (Tanon, model: 2500R)
16. Ultrasonic cell breaker (Yetuo, model: YT-JY96-IIN)
17. Flaming/brown micropipette puller (SUTTER, model: P-97)
18. Microinjector (Eppendorf, catalog number: 5176)
19. Stereomicroscope (CNOPTEC, model: SZ810)
20. Magnetic hotplate stirrer (SCILOGEX, catalog number: 80301321159999)
21. Vortex mixer (YEASEN, catalog number: 80172ES01)
22. Milli-Q® EQ 7000 water purification system (Millipore, catalog number: ZEQ7000T0C)
23. Autoclave (Boxun, model: YXQ-100SII)
24. Fume hood (Esco, model: Frontier Acela)
25. Drying oven (Jinghongsh, model: DHG-9070A)
26. Metal bath (Yooning, model: GH-100)
Software and datasets
1. SnapGene, for molecular cloning design and DNA sequence analysis (version 6.0.2)
2. GraphPad Prism 9, for statistics and data visualization (version 9.3.1)
3. ImageJ, for image processing (version 1.53a)
Procedure
The one-step purification protocol to obtain highly purified Ub-Cas9 by metal affinity chromatography was designed as shown in Figure 1.
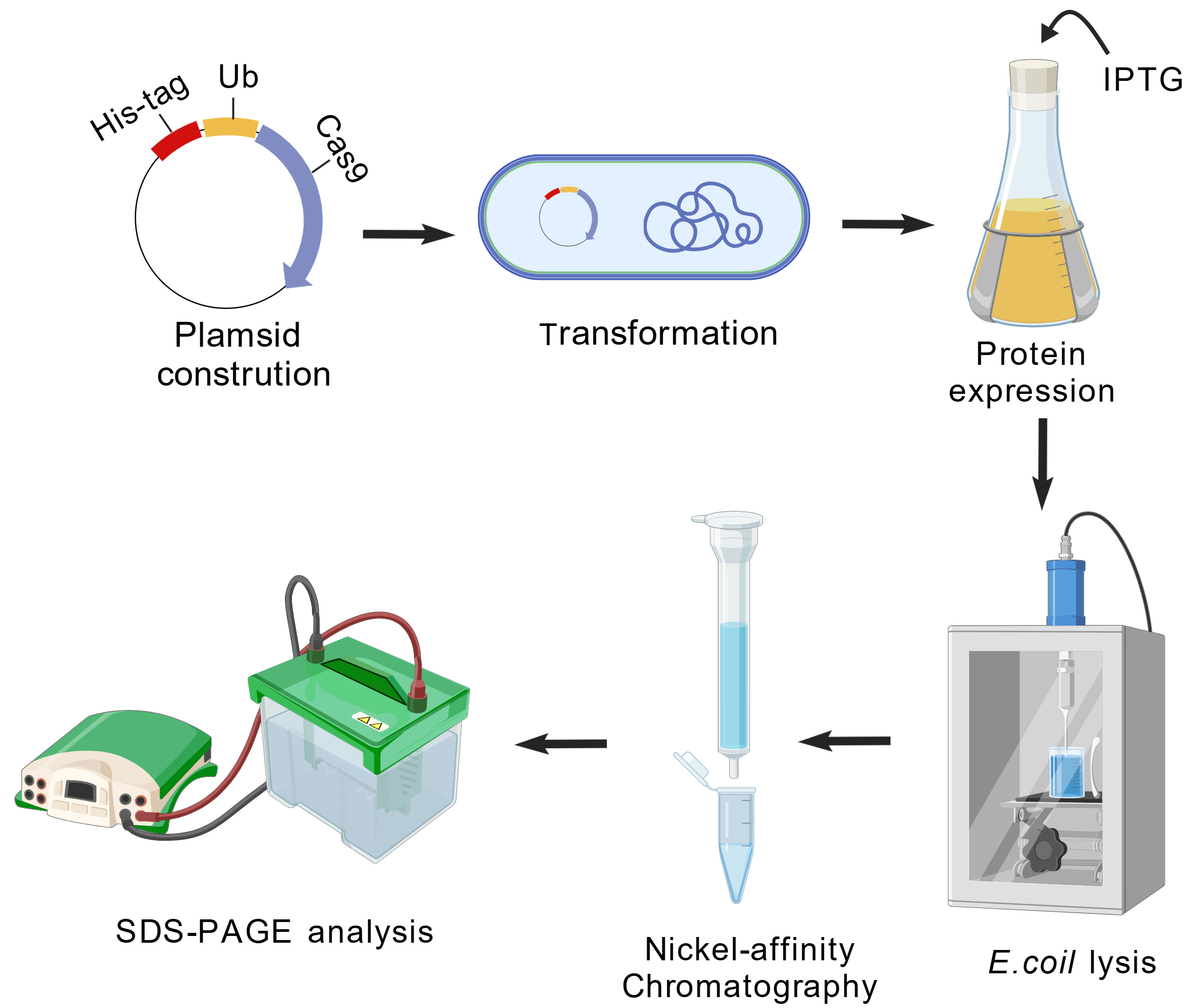
Figure 1. Schematic workflow of recombinant protein expression and purification in E. coli. Created with BioGDP.com [14].
A. Plasmid construction for His-Ub-Cas9 expression
1. Amplification of the Cas9 gene
a. Design PCR primers:
In our work, primers were designed for the recombinational cloning of the Cas9 gene into the linearized pHUE vector. Each primer incorporates a 5′ homology arm complementary to the vector ends (shown in uppercase) and a 3′ sequence that targets the Cas9 coding sequence in the pET-28b-Cas9-His template (Addgene #47327) (shown in lowercase):
Forward primer
5′-GGTGTTGCGCCTCCGCGGTGGAatggacaagaagtactccattgggc-3′
Reverse primer
5′-GTTAGCAGCCGGATCTAAGCTTAATCgactttccgtttttttttcgggc-3′
b. Prepare the following components in a 200 μL PCR tube (Table 1):
Table 1. PCR reaction components
| Component | Volume (μL) |
|---|---|
| Template DNA (Addgene #47327) | × (approximately 30 ng) |
| Forward primer (10 μM) | 1 |
| Reverse primer (10 μM) | 1 |
| Phanta Max Super-Fidelity DNA Polymerase | 0.4 |
| dNTP mix (10 mM each) | 0.4 |
| 2×Phanta Max buffer | 10 |
| ddH2O | up to 20 μL |
c. The PCR is performed in a thermal cycler using the following program (Table 2):
Table 2. PCR amplification program
| Step | Temperature | Time | Cycle number |
| 1 | 95 °C | 3 min | 1 |
| 2 | 95 °C | 15 s | 30 |
| 65 °C | 15 s | ||
| 72 °C | 30 s | ||
| 3 | 72 °C | 5 min | 1 |
| 4 | 4 °C | Hold | 1 |
d. Mix 20 μL of the PCR product with 2 μL of 10× DNA loading buffer.
e. Load the mixture onto a 1% agarose gel.
f. Perform electrophoresis in TAE buffer at 120 V for 30 min.
g. Excise the band corresponding to the Cas9 DNA fragment at the expected size of 4329 bp.
h. Purify the DNA fragment using a TIANgel Purification kit according to the manufacturer’s protocol.
2. Linearization of the pHUE expression vector
a. The pHUE expression vector, which contains an N-terminal 10× His tag followed by a ubiquitin (Ub) sequence, is linearized by double digestion with the restriction enzymes BamH I and Hind III.
b. Prepare the following components in a 200 μL tube (Table 3):
Table 3. Double digestion reaction components
| Component | Volume (μL) |
|---|---|
| pHUE plasmid | × (approximately 1 μg) |
| 10× K buffer | 2 |
| BamH I | 0.75 |
| Hind III | 0.75 |
| ddH2O | up to 20 μL |
c. Mix the reaction components thoroughly by pipetting.
d. Incubate the reaction at 37 °C for 2 h in a thermal cycler or water bath.
e. After digestion, mix 20 μL of the digestion product with 2 μL of 10× DNA loading buffer.
f. Load the mixture onto a 1% agarose gel and perform electrophoresis in TAE buffer at 120 V for 30 min.
g. Excise the gel fragment containing the linearized pHUE vector.
h. Purify the linearized vector using a TIANgel Purification kit according to the manufacturer’s protocol.
3. Homologous recombination
a. Prepare the following components in a 200 μL tube (Table 4):
Table 4. Homologous recombination reaction components
| Component | Volume (μL) |
|---|---|
| 5×CE II buffer | 1.5 |
| Purified Cas9 PCR fragment | 3 (approximately 180 ng) |
| Linearized pHUE vector | 2.5 (approximately 110 ng) |
| Exnase II | 0.5 |
Note: Please refer to the manufacturer’s instructions for the optimal amounts of the linearized vector and insert fragment.
b. Incubate the reaction at 37 °C for 30 min in a thermal cycler or water bath.
c. Incubate the reaction in ice for 5 min to finish the recombination. The recombinant plasmid can proceed immediately to the next step. Alternatively, store it at -20 °C.
4. Transformation of E. coli
a. Thaw the chemically competent E. coli DH5α cells on ice.
b. Add 7.5 μL of the recombination reaction product to 100 μL of E. coli DH5α competent cells.
c. Mix well by tapping and incubate the mixture on ice for 30 min.
d. Heat-shock the cells at 42 °C for 90 s in a water bath.
e. Immediately return the tube to ice for 2 min.
f. Add 1 mL of sterile LB medium into the tube.
g. Recover the cells at 37 °C for 60 min with shaking at 220 rpm.
h. Centrifuge the culture at 6,000× g for 5 min to collect the cells. Resuspend the pellet in 200 μL of LB medium with antibiotic, then spread 100 μL onto an LB agar plate containing 100 μg/mL ampicillin.
i. Incubate the plate overnight at 37 °C.
5. Verification of positive clones
a. The next day, pick 8 single colonies for screening.
b. Perform colony PCR using Cas9-specific primers to identify positive clones:
i. Inoculate each picked colony into 200 μL of LB medium with ampicillin in a PCR tube and incubate at 37 °C with shaking for 2–4 h.
ii. Use 2 μL of the resulting culture as template in a 20 μL PCR reaction prepared as follows (Table 5):
Table 5. Colony PCR reaction components
| Component | Volume (μL) |
|---|---|
| Colony culture | 2 |
| Forward primer (10 μM) | 1 |
| Reverse primer (10 μM) | 1 |
| 2×Hieff® PCR Master Mix | 10 |
| ddH2O | up to 20 μL |
Note: The Cas9-specific primers used are as follows:
Forward primer: 5′-ctctctttgagcttgaaaacggcc-3′
Reverse primer: 5′-caccgccggagccacccccagatc-3′
Alternatively, vector-specific primers flanking the insertion site may also be used for clone confirmation.
iii. Run the PCR using the following program (Table 6):
Table 6. Colony PCR amplification program
| Step | Temperature | Time | Cycle number |
| 1 | 95 °C | 5 min | 1 |
| 2 | 95 °C | 30 s | 32 |
| 55 °C | 30 s | ||
| 72 °C | 1 min | ||
| 3 | 72 °C | 5 min | 1 |
| 4 | 4 °C | Hold | 1 |
iv. Analyze 10 μL of each PCR product by electrophoresis on a 1% agarose gel. A positive clone yields a specific band of approximately 600 bp.
c. Inoculate positive clones into 5 mL of LB medium containing 100 μg/mL ampicillin.
d. Incubate the culture overnight at 37 °C with shaking at 220 rpm.
e. Extract plasmid from the overnight cultures using a TIANprep Mini Plasmid kit.
f. Verify the plasmid sequence by DNA sequencing with appropriate primers. The successfully constructed expression plasmid, designated pHUE-His-Ub-Cas9, is depicted in Figure S1, and its complete coding sequence is provided in Dataset S1.
B. Expression of His-Ub-Cas9 in E. coli
1. Small-scale expression test
Prior to large-scale culture, it is advisable to conduct a small-scale expression test to confirm protein expression and refine induction conditions. The procedure is as follows:
a. Transform the pHUE-His-Ub-Cas9 plasmid into E. coli BL21(DE3) using the method outlined in step A4.
Note: E. coli BL21(DE3) is ideal for inducible expression because it provides tight, IPTG-inducible control via the T7 RNA polymerase and lacks key proteases to minimize recombinant protein degradation.
b. Inoculate a single colony from the plate into 5 mL of LB medium with ampicillin and incubate overnight at 37 °C with shaking at 220 rpm.
c. The next day, dilute the overnight culture 1:100 into three separate 10 mL aliquots of LB medium with ampicillin, each dispensed into a 50 mL flask.
d. Incubate all flasks at 37 °C with shaking until optical density at 600 nm (OD600) reaches 0.6–0.8.
e. Once OD600 reaches 0.6–0.8, collect 2 mL of culture from one flask by centrifugation at 12,000× g for 5 min. This sample serves as the uninduced control.
f. Add IPTG to the remaining two cultures to a final concentration of 0.4 mM to initiate recombinant protein expression.
g. Transfer the two induced cultures to separate temperature-controlled shakers. Incubate one at 16 °C and the other at 25 °C, with continuous shaking for 20 h.
h. After the 20-h induction, harvest 2 mL of each induced culture by centrifugation at 12,000× g for 5 min. Resuspend all cell pellets in 100 μL of 1× SDS-PAGE loading buffer, boil for 10 min, and analyze 10 μL of each sample by SDS-PAGE followed by Coomassie Blue staining.
i. Successful expression is indicated by a clearly intensified band at the expected molecular weight (~180 kDa for His-Ub-Cas9) in the induced sample compared to the uninduced control. In our work, comparison of the two induction temperatures revealed that induction at 25 °C yielded a more prominent target band, indicating higher soluble protein production. Therefore, the 25 °C condition was adopted for all subsequent large-scale cultures (Figure 2).
2. Large-scale culture preparation for induction
a. Following a successful small-scale expression test, a corresponding 10% glycerol stock of the E. coli BL21(DE3)/pHUE-His-Ub-Cas9 strain was used to seed the large-scale culture. Specifically, 500 μL of this frozen stock was used to inoculate 10 mL of LB medium with ampicillin in a sterile 50 mL Erlenmeyer flask.
b. Incubate the culture overnight at 37 °C with shaking at 220 rpm.
c. The following day, transfer 5 mL of the starter culture into 500 mL of fresh LB medium with ampicillin in a 1-L Erlenmeyer flask.
d. Incubate the main culture at 37 °C with shaking at 220 rpm.
e. Monitor the OD600 every 30 min by sampling 2 mL of the culture and measuring it in a spectrophotometer. Use sterile LB medium with ampicillin as the blank.
f. Continue incubation until the OD600 reaches 0.6–0.8, which typically takes 2–3 h.
Note: Ensure all steps are performed under sterile conditions to prevent contamination.
3. Induction of protein expression and cell harvesting
a. When the OD600 reaches 0.6–0.8, add IPTG to the culture at a final concentration of 0.4 mM. For a 500 mL culture, add 200 μL of 1 M IPTG stock solution.
b. Transfer the culture to a 25 °C shaking incubator and incubate for 20 h at 220 rpm to induce protein expression.
c. After induction, transfer the culture to centrifuge bottles. Harvest the cells by centrifugation at 6,000× g for 10 min at 4 °C.
d. Carefully discard the supernatant. Flash-freeze the cell pellets in liquid nitrogen and store them at -80 °C for subsequent use.
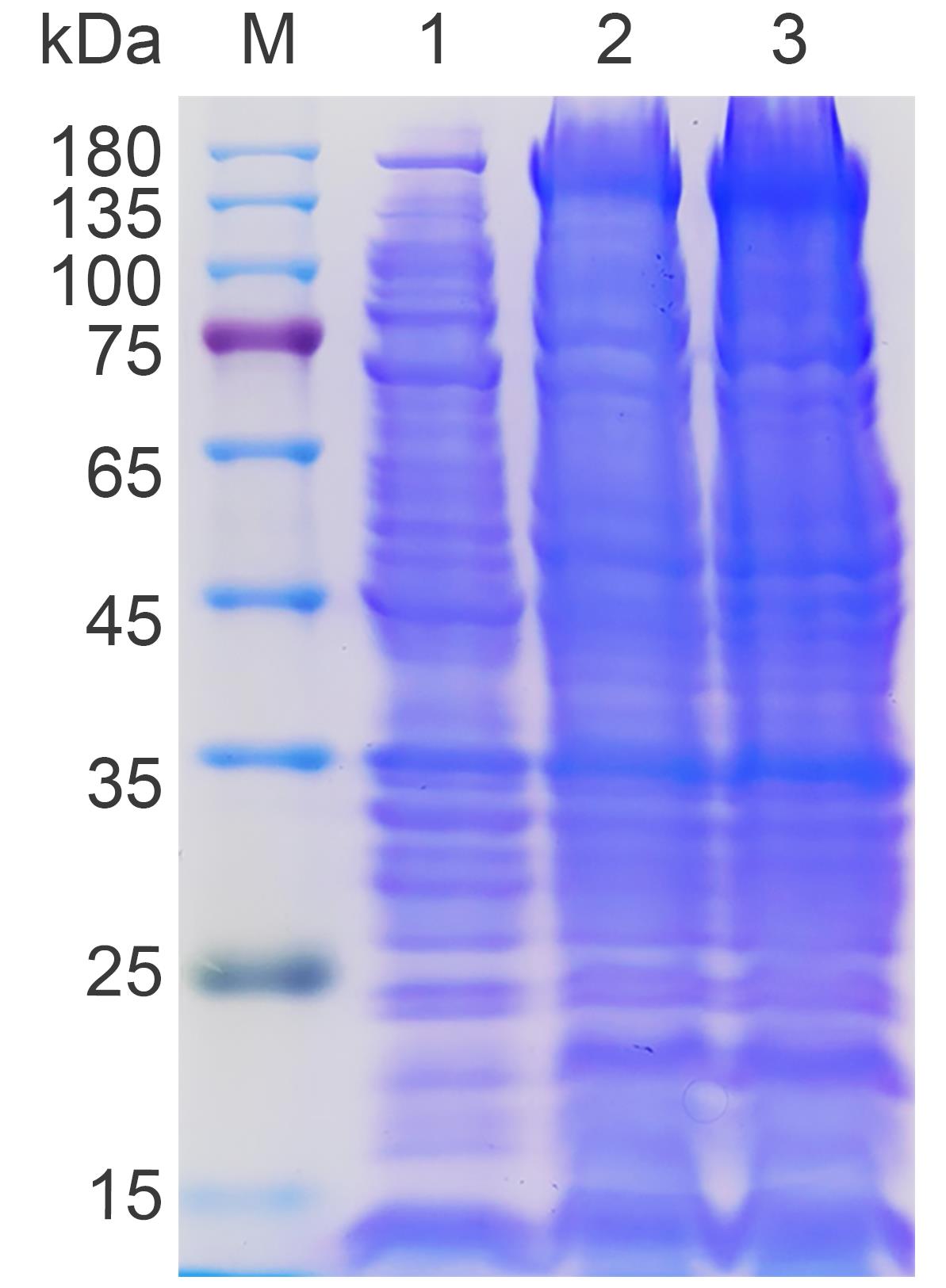
Figure 2. Small-scale expression test of His-Ub-Cas9 protein. Lane 1: Uninduced control. Lane 2: 16 °C induction. Lane 3: 25 °C induction. The arrow marks the His-Ub-Cas9 protein (~180 kDa). The results demonstrate that induction at 25 °C yields the highest amount of target protein.
C. Single-step purification of His-Ub-Cas9 by nickel-affinity chromatography
1. Lysis E. coli by sonication
a. The day before the experiment, transfer the frozen cell pellet from -80 °C to a 4 °C refrigerator to thaw overnight.
b. Resuspend the thawed pellet in 19 mL of lysis buffer.
c. Thaw the protease inhibitor cocktail at room temperature and add it to the resuspended cells at a 1:100 ratio.
d. Keep the cell suspension in an ice bath throughout the subsequent steps.
e. Set the sonicator to the following parameters: 3 s pulse on, 8 s pulse off, for a total duration of 10 min.
f. Fully immerse the sonication probe and perform lysis while ensuring the sample tube is maintained in an ice bath.
g. Add Triton X-100 to the lysate to a final concentration of 1% (v/v).
h. Incubate the mixture in a shaking ice bath for 30 min.
i. Transfer the lysate to pre-chilled centrifuge tubes.
j. Centrifuge at 13,000× g for 10 min at 4 °C.
k. Collect the supernatant and filter it through a 0.22 μm syringe filter.
2. Nickel-affinity chromatography purification
a. Pack a chromatography column with 2 mL of Ni-NTA agarose resin.
b. Equilibrate the column with 10 mL of lysis buffer by gravity flow.
c. Reserve an 80 μL sample of the filtered lysate as the “load” sample for later analysis. Load the remaining lysate onto the column at the slowest flow rate, collecting the flowthrough in a clean tube.
d. Wash the column with 15 mL of lysis buffer to remove nonspecifically bound proteins and collect the wash fractions.
e. For stepwise elution, first elute with 3 mL of elution buffer 1 (lysis buffer containing 80 mM imidazole), collecting three 1 mL fractions (designated as E1–E3).
f. Then, elute with 12 mL of elution buffer 2 (lysis buffer containing 500 mM imidazole), collecting twelve 1 mL fractions (designated as E4–E15) (Figure 3).
Note: All procedures involving protein samples should be performed on ice to maintain protein stability. Furthermore, for higher throughput, the purification can also be performed using prepacked Ni-NTA columns on UPLC systems, which enable precise flow control, gradient elution, and in line UV monitoring.
3. Analysis of purification fractions by SDS-PAGE and Coomassie Blue staining
3.1. Preparation of protein samples
a. Transfer an 8 μL sample from each fraction to separate tubes.
b. Add 2 μL of 5× SDS-PAGE loading buffer to each sample.
c. Heat the mixture at 100 °C for 10 min for SDS-PAGE analysis.
3.2. Preparation of 10% SDS-PAGE gel
a. Gel casting system assembly:
i. Thoroughly clean and dry two glass plates and 1.0 mm spacers.
ii. Assemble plates with spacers and secure in the casting frame.
iii. Perform a brief leak test by filling the assembly with deionized water.
b. Preparation of 10% separating gel
i. For two gels, combine the following components sequentially in a 50 mL tube (Table 7):
Table 7. Components for casting a 10% separating gel (for two gels)
| Component | Volume (mL) |
|---|---|
| H2O | 4.0 |
| 30% acrylamide/Bis solution | 3.3 |
| 1.5 M Tris-HCl, pH 8.8 | 2.5 |
| 10% SDS | 0.1 |
| 10% APS | 0.1 |
| TEMED | 0.004 |
ii. After adding TEMED, mix the solution immediately by swirling.
iii. Pour the gel solution into the assembled plates to approximately two-thirds of their total height.
iv. Carefully overlay the gel solution with a layer of isopropyl alcohol to eliminate bubbles and ensure a flat gel surface.
v. Allow the gel to polymerize at room temperature for 30 min.
c. Preparation of stacking gel
i. After polymerization, pour off and discard the isopropyl alcohol layer.
ii. Prepare the stacking gel mixture for two gels in a 50 mL tube with the following components (Table 8):
Table 8. Components for casting the stacking gel (for two gels)
| Component | Volume (mL) |
|---|---|
| H2O | 2.7 |
| 30% acrylamide/Bis solution | 0.67 |
| 1.0 M Tris-HCl, pH 6.8 | 0.5 |
| 10% SDS | 0.04 |
| 10% APS | 0.04 |
| TEMED | 0.004 |
iii. Mix quickly and pour the solution onto the separating gel.
iv. Insert a plastic comb carefully to avoid bubbles.
v. Allow the gel to polymerize at room temperature for 30 min.
3.3. Electrophoresis
a. Mount the gel securely in the electrophoresis chamber.
b. Prepare 1× Tris-glycine running buffer by diluting the 5× Tris-glycine running buffer with deionized water (e.g., mix 200 mL of 5× stock with 800 mL of water to obtain 1 L of 1× buffer), then fill the chamber with 1× buffer.
c. Carefully remove the comb.
d. Load 10 μL of each prepared protein sample and 5 μL of prestained protein ladder into individual wells.
e. To prepare a standard curve, dilute the 10 mg/mL BSA stock solution to 1 mg/mL by adding 70 μL of ddH2O and 20 μL of 5× SDS loading buffer to 10 μL of the BSA stock. Load 1, 2, 3, 4, and 5 μL of this 1 mg/mL BSA solution into the five empty wells. This represents a range of 1–5 μg of protein (designated as B1–B5) (Figure 3).
Note: This BSA series creates a standard curve for semi-quantitative analysis of protein concentration based on band intensity after Coomassie blue staining.
f. Run the gel at a constant voltage of 80 V until the bromophenol blue enters the separating gel. Then, increase the voltage to 120 V and continue running until the dye front reaches the bottom of the gel.
3.4. Coomassie Blue staining and destaining
a. Staining procedure:
i. Carefully separate the glass plates and transfer the gel to a suitable container.
ii. Add sufficient Coomassie Blue R-250 staining solution to completely submerge the gel.
iii. To accelerate staining, heat the gel in staining solution using a microwave for 15–30 s. Repeat this heating once, then incubate the gel on a shaker for 10 min.
b. Destaining procedure:
i. Pour off the staining solution (which can be saved and reused).
ii. Add sufficient destaining solution to cover the gel.
iii. Similarly, microwave the gel for 15–30 s twice to accelerate destaining, then place it on a rocker.
iv. Change destaining solution every 30–60 min until the gel background becomes clear.
3.5. Imaging and analysis
Transfer the destained gel onto a white light transilluminator and capture images (Figure 3). Analyze the protein band patterns across all collected fractions (Table 9).
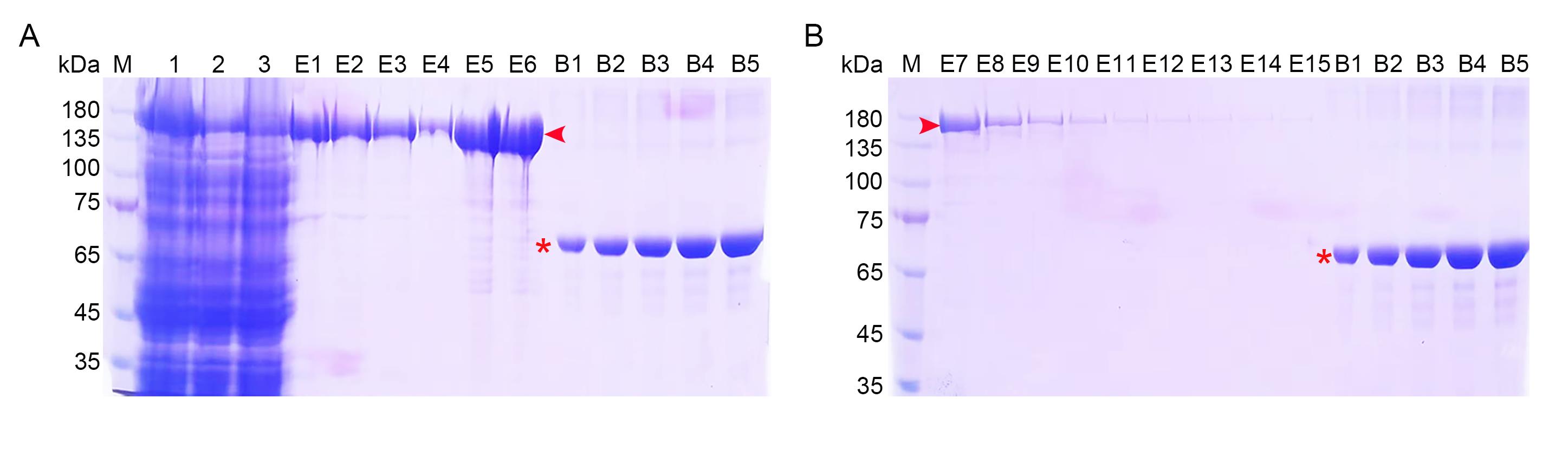
Figure 3. Expression and purification of the Cas9 protein. SDS-PAGE analysis of the purification of Ub-Cas9. (A) Early-elution fractions. Lane 1, E. coli lysate; 2, flowthrough; 3, wash solution; E1–E3, eluted fractions with 80 mM imidazole; E4–E6, eluted fractions with 500 mM imidazole. (B) Late-elution fractions with 500 mM imidazole (E7–E15). For both panels, lanes B1–B5 contain 1–5 μg of BSA (asterisks) as a quantitative standard. The Ub-Cas9 protein is indicated by red arrowheads. M, protein standards.
Table 9. Protein yield of purified Ub-Cas9 from 500 mL of bacterial culture
| Sample name | Concentration (mg/mL) | Volume (mL) | Total protein (mg) |
| Metal affinity enrichment | |||
| Ni-NTA elution 1 | 0.558 | 1 | 0.558 |
| Ni-NTA elution 2 | 0.486 | 1 | 0.486 |
| Ni-NTA elution 3 | 0.375 | 1 | 0.375 |
| Ni-NTA elution 4 | 0.129 | 1 | 0.129 |
| Ni-NTA elution 5 | 1.140 | 1 | 1.140 |
| Ni-NTA elution 6 | 1.122 | 1 | 1.122 |
| Ni-NTA elution 7 | 0.203 | 1 | 0.203 |
| Ni-NTA total | 4.013 | ||
| Purity | 95.85% | ||
D. Buffer exchange, concentration, and storage of purified His-Ub-Cas9
1. Desalting procedure
a. Wash the PD-10 desalting column with 10× PBS four times, filling the column completely each time.
b. Equilibrate the column with storage buffer four times.
c. Identify fractions containing purified Ub-Cas9 based on SDS-PAGE analysis. Typically, fractions with >90% purity (as judged by SDS-PAGE) and a protein concentration >0.1 mg/mL are pooled. Add a maximum of 2.5 mL of the protein sample to the column and discard the flowthrough.
d. Elute the protein with 3.5 mL of storage buffer and collect the entire eluate in a clean tube.
Note: If the pooled sample volume exceeds 2.5 mL, use multiple PD-10 columns in parallel or perform sequentially. If the sample volume is less than 2.5 mL, adjust the volume to 2.5 mL by adding storage buffer before loading.
e. The purity of the desalted protein is verified by SDS-PAGE (Figure S2). In our work, it showed that high purity (>95%) was maintained following buffer exchange.
f. Wash the column with 10× PBS four times, followed by four washes with 1× PBS.
g. Store the column filled with 1× PBS at 4 °C.
2. Protein concentration
a. Load the desalted protein into an Amicon Ultra centrifugal filter device (30 kDa MWCO).
b. Concentrate the protein by centrifugation at 3,000× g for 30–45 min at 4 °C.
c. Discard the flowthrough and add the remaining protein solution to the concentrator.
d. Repeat centrifugation until the final volume reaches 0.5–1 mL.
e. Gently pipette the concentrated protein into a clean microcentrifuge tube.
Note: To monitor potential protein loss, the flowthrough can be tested using a Bradford assay. Blue color indicates protein leakage, which may be due to membrane damage or overloading. This concentration step is optional and can be omitted based on the user’s requirements. Please note that this process may cause low sample recovery due to adsorptive losses, over-concentration, or passage of the sample through the membrane.
3. Flash-freezing and storage
a. Aliquot the protein into 10 μL portions in sterile microcentrifuge tubes.
b. Snap-freeze the aliquots by immersing them in liquid nitrogen for 30 s.
c. Transfer and store the frozen aliquots at -80 °C for long-term preservation.
d. Clearly label the protein name, concentration, and date.
E. In vivo genome editing in zebrafish embryos via Cas9 RNP microinjection
Note: The procedure for evaluating the genome editing efficiency of the purified Ub-Cas9 is outlined in Figure 4.
1. Preparation of microinjection needles
a. Horizontally place a glass capillary tube in the micropipette puller clamps.
b. Align the center of the capillary with the heating filament.
c. Set the pulling parameters as follows: HEAT 650, PULL 60, VEL 100, TIME 140, P 500.
d. Run the pulling program to produce two injection needles.
e. Carefully remove the needles and store in a storage box.
f. Needles trimming:
i. Position the needle tip under a stereomicroscope.
ii. Using a surgical scalpel, cleanly and quickly trim the tip at a 45° angle.
iii. Examine the trimmed tip under the microscope to confirm the size and quality of the opening.
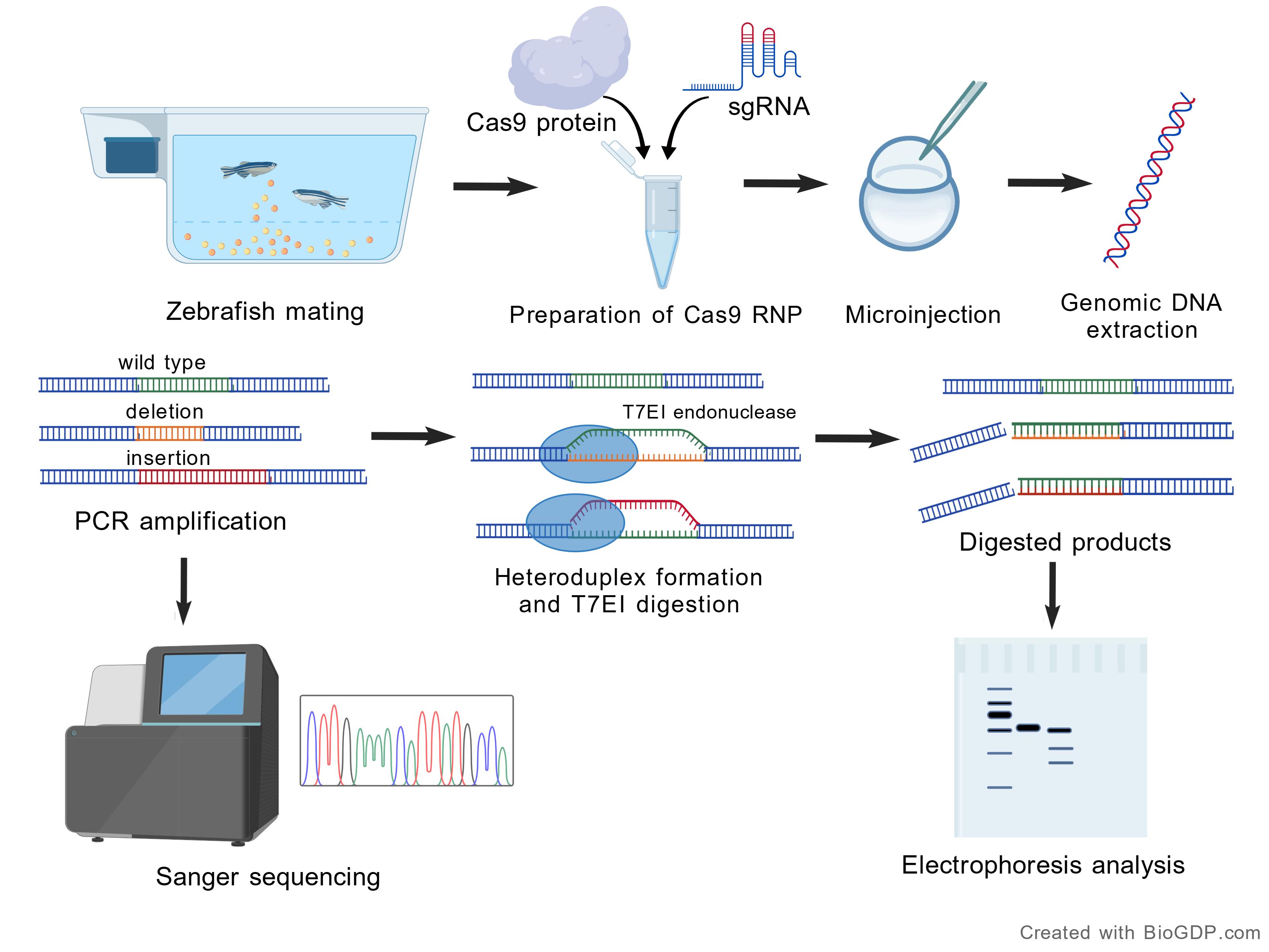
Figure 4. Schematic workflow for in vivo genome editing in zebrafish embryos via Cas9 RNP microinjection. Created with BioGDP.com [14].
2. Zebrafish mating and embryo collection
a. The evening before embryo injection, set up breeding tanks with dividers.
b. Fill tanks with system water and place healthy adult male and female zebrafish in separate compartments of tanks.
c. The next morning, remove the dividers to initiate mating.
d. Collect embryos within 0.5–1 h after spawning and transfer them to Petri dishes containing embryo medium.
e. Under a stereomicroscope, select fertilized embryos and discard unfertilized or abnormal embryos.
3. Microinjection of Cas9 RNP into zebrafish embryos
3.1. Preparation of Cas9 RNP complex
a. Thaw His-Ub-Cas9 protein, commercial Cas9 protein, and target sgRNA on ice.
b. Prepare the RNP complexes in nuclease-free tubes as follows (Table 10):
Table 10. RNP complex preparation for microinjection
| Complex | Component | Volume (μL) | Final concentration |
| RNP 1 | His-Ub-Cas9 | 1 | 300 ng/μL |
| sgRNA | 1 | 200 ng/μL | |
| RNP 2 | Commercial Cas9 | 1 | 300 ng/μL |
| sgRNA | 1 | 200 ng/μL |
c. Mix components gently and keep the prepared RNP complexes on ice until injection.
Note: Always add Cas9 protein to sgRNA. The reverse order may cause protein aggregation and reduce editing efficiency.
3.2. Microinjection procedure:
a. Align 50–100 one-cell stage embryos in an injection mold.
b. Backfill a prepared microneedle with 2 μL of RNP solution using a microloader pipette tip.
c. Mount the needle onto the microinjector and adjust pressure to form consistent droplets.
d. Under a stereomicroscope at 2× magnification, inject 1 nL of the RNP solution into the yolk of each embryo.
e. Transfer injected embryos to fresh embryo medium and incubate at 28 °C. Meanwhile, the uninjected WT embryos are used as the negative control group.
3.3. Genomic DNA extraction from embryos
a. At 24 h post-fertilization (hpf), transfer embryos into PCR strip tubes (two embryos per tube).
b. Add 10 μL of 50 mM NaOH to each tube to lyse the embryos.
c. Incubate the embryos at 95 °C for 20 min in a thermal cycler.
d. Cool the tubes to room temperature and neutralize the reaction with 10 μL of 100 mM Tris-HCl (pH 8.0).
e. Vortex the samples and centrifuge briefly. Use 4 μL of the supernatant as the PCR template. The remaining genomic DNA can be stored at -20 °C for several weeks.
3.4. PCR amplification of target region
a. Prepare a 20 μL PCR reaction as shown below (Table 11):
Table 11. PCR reaction components
| Component | Volume (μL) |
|---|---|
| Zebrafish genomic DNA | 4 |
| Forward primer (10 μM) | 1 |
| Reverse primer (10 μM) | 1 |
| 2× Hieff® PCR Master Mix | 10 |
| ddH2O | up to 20 μL |
Note: The sequence of PCR amplification of zebrafish afap1l1b spanning the sgRNA target is shown in Dataset S2. Primers are designed to flank the sgRNA target site, generating an amplicon of 300–500 bp for the T7 endonuclease I (T7EI) assay. The sequence of the primers is as follows:
Forward primer: 5′-CAAAGCTGGTACCATTTCCA-3′
Reverse primer: 5′-GTAGGCCAAAACCACTAGTG-3′
b. Run the following PCR program (Table 12):
Table 12. PCR amplification program
| Step | Temperature | Time | Cycle number |
| 1 | 95 °C | 5 min | 1 |
| 2 | 95 °C | 30 s | 32 |
| 55 °C | 30 s | ||
| 72 °C | 1 min | ||
| 3 | 72 °C | 5 min | 1 |
| 4 | 4 °C | Hold | 1 |
c. Confirm successful amplification by analyzing 5 μL of PCR product via electrophoresis on a 1.5% agarose gel.
d. Denature and reanneal the PCR products to allow heteroduplex formation using the following program (Table 13):
Table 13. Thermal cycling program for heteroduplex formation
| Temperature | Time | Ramp |
| 95 °C | 10 min | |
| 95 °C | 1 min | 2 |
| 85 °C | 1 min | 0.3 |
| 75 °C | 1 min | 0.3 |
| 65 °C | 1 min | 0.3 |
| 55 °C | 1 min | 0.3 |
| 45 °C | 1 min | 0.3 |
| 35 °C | 1 min | 0.3 |
| 25 °C | 1 min | 0.3 |
| 25 °C | 1 min |
3.5. T7 endonuclease I (T7EI) assay
a. Digest reannealed PCR products in a 10 μL reaction (Table 14):
Table 14. T7 endonuclease I (T7EI) digestion reaction components
| Component | Volume (μL) |
|---|---|
| Reannealed PCR product | 6 |
| T7 endonuclease I | 0.1 |
| 10× T7EI reaction buffer | 1 |
| ddH2O | to 10 μL |
b. Incubate the reaction at 37 °C for 30 min to allow for complete digestion of heteroduplex DNA.
Note: T7EI serves as a detection tool for Cas9-induced mutations. It identifies DNA mismatches by cleaving the heteroduplex structures formed when PCR products from the edited genomic region are reannealed.
3.6. Editing efficiency analysis
a. Analyze T7EI digestion products by electrophoresis on a 2% agarose gel.
b. Identify cleaved bands indicating successful genome editing (Figure 5). In a standard T7EI assay, the intact PCR product (e.g., ~449 bp in this study) is cleaved into two smaller fragments upon digestion of the heteroduplex DNA formed between wildtype and mutant alleles. Specifically, the cleaved bands observed at approximately 274 and 175 bp (Figure 5) correspond to these digestion products, confirming the presence of targeted mutations.
c. Quantify editing efficiency as the percentage of successfully edited samples within a pool (Table 15). A sample is considered to be positive if cleaved bands (274 and 175 bp) are visibly detected in the T7EI assay. Editing efficiency is calculated as follows: (Number of positive samples) / (Total number of samples in the pool) × 100%. For example, if cleaved bands were observed in 6 out of 7 samples within a pool, the editing efficiency for that pool is 85.71%.
Note: The variation in the intensity and pattern of cleaved bands across different pools may arise from minor differences in microinjection volume or RNP activity between embryos, as well as variability in genomic DNA extraction and PCR amplification efficiency.
3.7. Sequence verification
a. Purify the remaining PCR products from step E.3.4 using a TIANgel Purification kit.
b. Submit the purified product for Sanger sequencing.
c. Analyze sequencing results for mutations at the target site (Figure 6).
Note: The general methodology for generating and evaluating CRISPR-Cas9-induced mutations in zebrafish embryos followed established protocols [15].
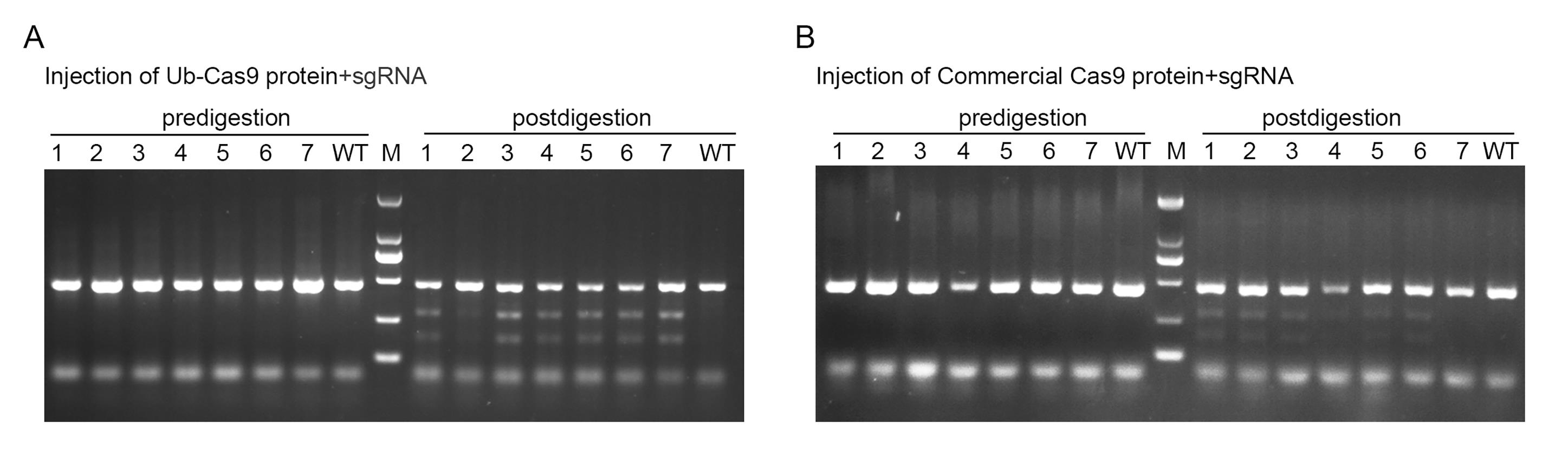
Figure 5. Functional validation of purified Ub-Cas9 protein by in vivo endonuclease assay. (A) Embryos injected with RNP complexes containing purified Ub-Cas9. (B) Embryos injected with RNP complexes containing commercial Cas9. For both panels, “predigestion” lanes show the PCR product amplified from the target site, and “postdigestion” lanes show the products after T7EI treatment. Cleaved bands confirm successful genome editing.
Table 15. Comparison of nuclease activity between Ub-Cas9 and commercial Cas9 in vivo
| Sample group | In vivo cleavage efficiency (%) | ||
|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 3 | |
| Ub-Cas9 | 85.71 | 100 | 71.43 |
| Commercial Cas9 | 71.43 | 85.71 | 85.71 |
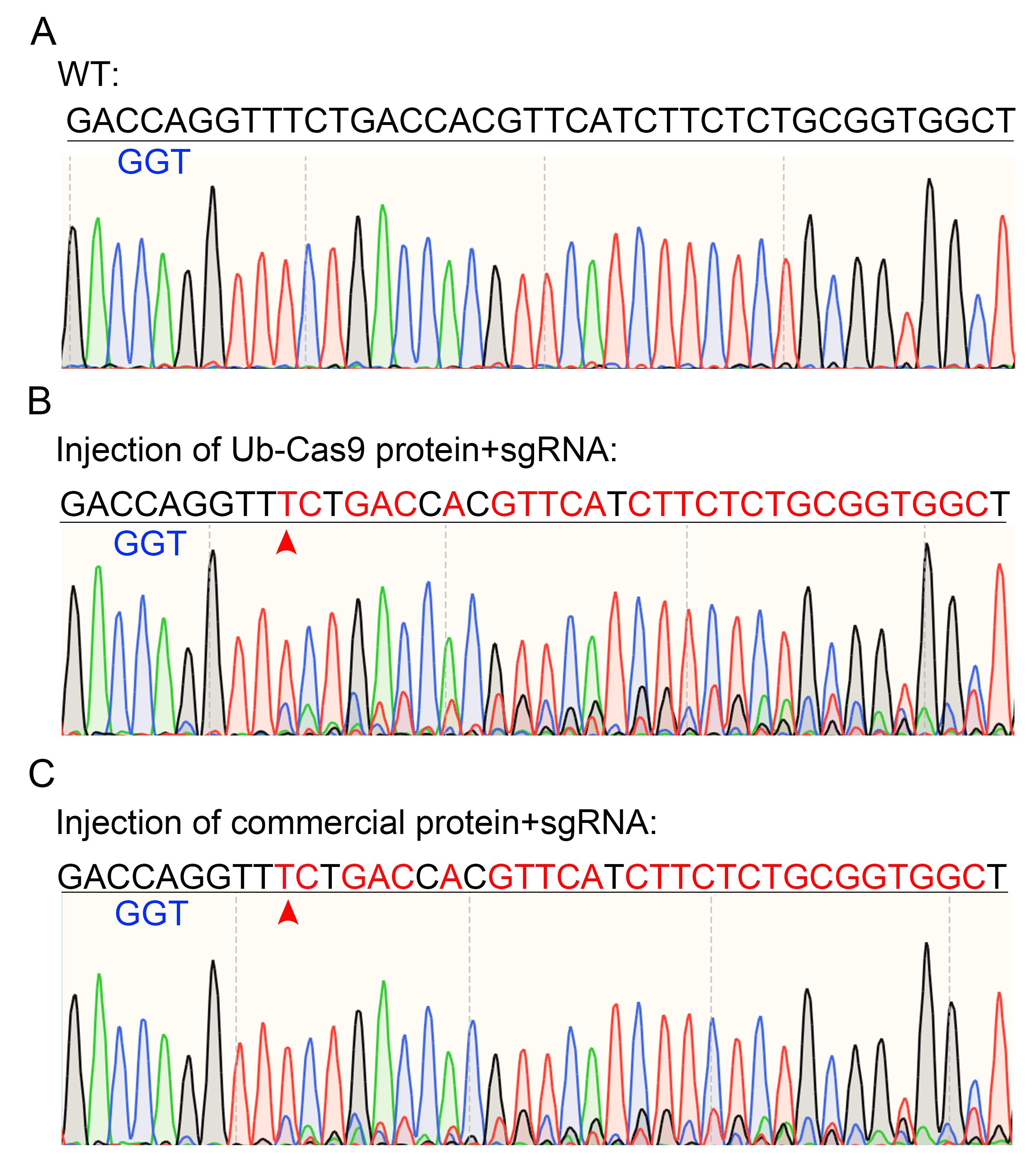
Figure 6. Sequencing results of PCR products from Figure 5. (A) Uninjected control. (B) Cleaved sample by injection of Ub-Cas9 and sgRNA. (C) Cleaved sample by injection of commercial Cas9 and sgRNA. The blue nucleotides indicate the PAM sequence of sgRNA. The red nucleotides highlight the mismatched bases. An arrowhead marks the cleavage site in the target sequence.
Data analysis
The concentration of Ub-Cas9 was quantified by gel densitometry using ImageJ software against a BSA standard curve. The total protein yield was calculated from eluted fractions obtained from a 500 mL LB culture and is reported as the yield per liter of culture. Protein purity was assessed by analyzing the relative band intensity of Ub-Cas9 within its lane using ImageJ.
Sequencing of PCR amplicons from embryos injected with commercial Cas9 or purified His-Ub-Cas9 RNPs confirmed successful targeted genome editing. Chromatograms from both groups displayed overlapping peaks downstream of the protospacer-adjacent motif (PAM) site, a pattern characteristic of heterogeneous indel mutations resulting from non-homologous end joining (NHEJ) repair (Figure 6). No such peak overlapping was observed in uninjected control samples.
Validation of protocol
This protocol has been effectively validated through both direct experimental evidence and its application in published research. The one-step purification method consistently yields over 8 mg/L of high-purity (>95%) His-Ub-Cas9 protein, as demonstrated by SDS-PAGE analysis (Figure 3, Table 9). The functional integrity of the purified protein was confirmed by its robust genome-editing activity in zebrafish embryos, achieving high efficiency comparable to commercial Cas9 in T7EI assays across multiple replicates (Figures 5–6, Table 15). Furthermore, this protocol (or parts of it) has been used and validated in the following research article:
Duan et al. [13]. Single-step purification of functional Cas9 protein via the ubiquitin expression system. International journal of biological macromolecules (Figures 2D, 4A, and 5).
Limitations
Although the one-step Ub-Cas9 purification strategy offers significant advantages in simplicity and high yield, several limitations warrant consideration. First, cleavage of the ubiquitin tag relies on endogenous deubiquitinating enzymes (DUBs) activity, which may vary across different cell types and species, potentially affecting editing efficiency. Second, while RNP delivery reduces off-target risks, the biocompatibility and potential off-target effects of Ub-Cas9 require systematic evaluation prior to therapeutic application. Future studies should employ sensitive off-target screening methods (e.g., GUIDE-seq) to comprehensively assess its specificity and safety profile.
Supplementary information
The following supporting information can be downloaded here:
1. Dataset S1. Gene sequence encoding His-Ub-Cas9.
2. Dataset S2. DNA sequence of the zebrafish afap1l1b PCR amplification spanning the sgRNA target site.
3. Figure S1. Plasmid map of pHUE-His-Ub-Cas9.
4. Figure S2. SDS-PAGE analysis of His-Ub-Cas9 after desalting.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32100659 to M.A.), Guangdong Basic and Applied Basic Research Foundation (2023A1515012586 to M.A., 2025A1515012750 to Z.Z.), and SUMC Start-up Funding (510858072 to Z.Z.). This protein purification protocol was adapted from our previously established method [13].
Competing interests
The authors declare no conflicts of interest.
References
- Doudna, J. A. and Charpentier, E. (2014). The new frontier of genome engineering with CRISPR-Cas9. Science. 346(6213): e1258096. https://doi.org/10.1126/science.1258096
- Ramakrishna, S., Kwaku Dad, A. B., Beloor, J., Gopalappa, R., Lee, S. K. and Kim, H. (2014). Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res. 24(6): 1020–1027. https://doi.org/10.1101/gr.171264.113
- Chen, K., Stahl, E. C., Kang, M. H., Xu, B., Allen, R., Trinidad, M. and Doudna, J. A. (2024). Engineering self-deliverable ribonucleoproteins for genome editing in the brain. Nat Commun. 15(1): e1038/s41467–024–45998–2. https://doi.org/10.1038/s41467-024-45998-2
- Jacobs, J. Z., Ciccaglione, K. M., Tournier, V. and Zaratiegui, M. (2014). Implementation of the CRISPR-Cas9 system in fission yeast. Nat Commun. 5(1): e1038/ncomms6344. https://doi.org/10.1038/ncomms6344
- Hu, R., Li, G., Hu, P., Niu, H., Li, W., Jiang, S., Guan, G., Xu, Q., Liu, M., Chen, L., et al. (2024). bmp10 maintains cardiac function by regulating iron homeostasis. J Genet Genomics. 51(12): 1459–1473. https://doi.org/10.1016/j.jgg.2024.10.003
- Werner, J., Zuo, W. and Doehlemann, G. (2024). CRISPR/Cas9 Ribonucleoprotein-Mediated Mutagenesis in Sporisorium reilianum. Bio Protoc. 14(1343): e4978. https://doi.org/10.21769/bioprotoc.4978
- Jiang, W., Bush, J. and Sheen, J. (2021). A Versatile and Efficient Plant Protoplast Platform for Genome Editing by Cas9 RNPs. Front Genome Ed. 3: e719190. https://doi.org/10.3389/fgeed.2021.719190
- Carmignotto, G. P. and Azzoni, A. R. (2019). On the expression of recombinant Cas9 protein in E. coli BL21(DE3) and BL21(DE3) Rosetta strains. J Biotechnol. 306: 62–70. https://doi.org/10.1016/j.jbiotec.2019.09.012
- Rajagopalan, N., Kagale, S., Bhowmik, P. and Song, H. (2018). A Two-Step Method for Obtaining Highly Pure Cas9 Nuclease for Genome Editing, Biophysical, and Structural Studies. Methods Protoc. 1(2): 17. https://doi.org/10.3390/mps1020017
- Teng, A. C., Tavassoli, M., Shrestha, S., Marks, R. M., McFadden, M. J., Evagelou, S. L., Lindsay, K., Vandenbelt, A., Li, W., Ivakine, E., et al. (2023). An efficient and cost-effective purification protocol for Staphylococcus aureus Cas9 nuclease. STAR Protoc. 4(1): 101933. https://doi.org/10.1016/j.xpro.2022.101933
- Evmenov, K., Pustogarov, N., Panteleev, D., Safin, A. and Alkalaeva, E. (2024). An Efficient Expression and Purification Protocol for SpCas9 Nuclease and Evaluation of Different Delivery Methods of Ribonucleoprotein. Int J Mol Sci. 25(3): 1622. https://doi.org/10.3390/ijms25031622
- Catanzariti, A., Soboleva, T. A., Jans, D. A., Board, P. G. and Baker, R. T. (2004). An efficient system for high‐level expression and easy purification of authentic recombinant proteins. Protein Sci. 13(5): 1331–1339. https://doi.org/10.1110/ps.04618904
- Duan, X., Zhou, Z., Zeng, X., Chen, J. and Mao, A. (2025). Single-step purification of functional Cas9 protein via the ubiquitin expression system. Int J Biol Macromol. 328: 147590. https://doi.org/10.1016/j.ijbiomac.2025.147590
- Jiang, S., Li, H., Zhang, L., Mu, W., Zhang, Y., Chen, T., Wu, J., Tang, H., Zheng, S., Liu, Y., et al. (2024). Generic Diagramming Platform (GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 53: D1670–D1676. https://doi.org/10.1093/nar/gkae973
- Luo, J., Lu, C., Wang, M. and Yang, X. (2023). Protocol for generating mutant zebrafish using CRISPR-Cas9 followed by quantitative evaluation of vascular formation. STAR Protoc. 4(4): 102753. https://doi.org/10.1016/j.xpro.2023.102753
Article Information
Publication history
Received: Nov 19, 2025
Accepted: Dec 29, 2025
Available online: Jan 13, 2026
Published: Feb 5, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Duan, X., Zhou, Z. and Mao, A. (2026). A One-Step Method for Efficient Purification of Functional Cas9 Protein. Bio-protocol 16(3): e5594. DOI: 10.21769/BioProtoc.5594.
Category
Biochemistry > Protein > Isolation and purification
Developmental Biology > Genome editing > Targeted integration
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.