- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Assessing the Toxoplasma Tachyzoite Cell Cycle Phases Using Fluorescent Ubiquitination-Based Cell Cycle Indicator
Published: Vol 16, Iss 2, Jan 20, 2026 DOI: 10.21769/BioProtoc.5588 Views: 343
Reviewed by: Marcelo S. da SilvaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2025
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Toxoplasma gondii is an apicomplexan parasite that infects a wide variety of eukaryotic hosts and causes toxoplasmosis. The cell cycle of T. gondii exhibits a distinct architecture and regulation that differ significantly from those observed in well-studied eukaryotic models. To better understand the tachyzoite cell cycle, we developed a fluorescent ubiquitination-based cell cycle indicator (FUCCI) system that enables real-time visualization and quantitative assessment of the different cell cycle phases via immunofluorescence microscopy. Quantitative immunofluorescence and live-cell imaging of the ToxoFUCCIS probe with specific cell cycle markers revealed substantial overlap between cell cycle phases S, G2, mitosis, and cytokinesis, further confirming the intricacy of the apicomplexan cell cycle.
Key features
• This protocol describes the development of the transgenic lines capable of detecting individual cell cycle phases and processes of the Toxoplasma tachyzoite cell cycle.
• Quantitative immunofluorescence analysis and real-time microscopy enable the measurement of each cell cycle phase.
• The ToxoFUCCIS probe helps to gain new insights into the highly flexible, overlapping nature of cell cycle organization in apicomplexan parasites.
Keywords: ApicomplexaGraphical overview

Background
Fluorescent ubiquitination–based cell cycle indicator (FUCCI) is a powerful technology that enables real-time visualization of the cell cycle [1–6]. FUCCI technology exploits the phase-specific proteolysis of critical cell cycle regulators. Sequential activation of major E3 ligases, APC/C and SCFSkp2, orchestrates the precise regulation of DNA replication factors. This temporal machinery controls the production of Geminin accumulation during S and G2 phases to prevent DNA re-replication. On the contrary, Cdt1 (chromatin licensing and DNA replication factor 1) is expressed in M and G1 phases and assists in licensing the origins of replication [7–9]. The original FUCCI probes invented by Sakauo-Sawana et al. in 2008 used fluorescent protein fusions of Geminin and Cdt1 and accurately detected cell cycle progression in mammalian cells in real time [2]. This technology has revolutionized cell cycle analysis. It provided a new dimension for studying cellular dynamics, enabled the assessment of drug effects on cell replication, visually distinguished proliferating and quiescent cells, and allowed the development of various stem cell populations [3,5]. Due to the lack or altered expression of the conventional FUCCI markers, the FUCCI adaptation to the unicellular apicomplexan parasite Toxoplasma gondii required technical adjustments [10,11]. To satisfy FUCCI marker requirements, a candidate protein should be abundant and localized in distinguishable cellular compartments such as the nucleus and cytoplasm, and its cell cycle expression should be controlled. According to these criteria, a DNA replication factor, TgPCNA1 (proliferating cellular nuclear antigen 1), was identified as a suitable FUCCI marker and successfully used to determine the organization of the T. gondii tachyzoite cell cycle [10]. While the ToxoFUCCIS cell line alone is sufficient to detect major cell cycle phases such as G1, S, and mitosis concurrent with cytokinesis (budding), the combination of ToxoFUCCIS with quantitative immunofluorescence microscopy analysis (qIFA) using a set of cell cycle markers facilitates the identification of the intervals within the composite cell cycle phase [10]. Previous studies validated cell cycle–dependent expression and localization of the suggested markers, including basal complex component TgBCC0 [12], MORN (membrane occupation and recognition nexus) repeat-containing protein TgMORN1 [11,13–15], and inner membrane complex protein TgIMC1 [16,17].
Materials and reagents
Biological materials
1. Human foreskin fibroblasts (HFF) (ATCC SCRC-1041)
2. Toxoplasma gondii RH TIR1 strain (genotype: RHΔhxgprtΔku80; TUB1:TIR1-3xFLAG, SAG1:CAT) [18]
3. Toxoplasma gondii RH Tati Δku80 strain [19]
Reagents
1. Dulbecco’s modified Eagle’s medium (DMEM) (Millipore Sigma, catalog number: D6429)
2. Fetal bovine serum (FBS) (Millipore Sigma, catalog number: F0926)
3. Antibiotic antimycotic solution (PSA) (Millipore Sigma, catalog number: A5955)
4. Hank’s balanced salt solution (Millipore Sigma, catalog number: H9394)
5. Mycoplasma Detection PCR kit (MP biomedicals, catalog number: 93050201)
6. Dimethyl sulfoxide (DMSO) (Fisher Scientific, catalog number: BP231)
7. 0.5% Trypsin EDTA solution (Fisher Scientific, catalog number: 15400054)
8. Adenosine triphosphate (ATP) (New England Biolabs, catalog number: P0756S)
9. Reduced L-glutathione (RPI, catalog number: 70188)
10. 6× gel loading dye (New England Biolabs, catalog number: B7025)
11. dNTPs (New England Biolabs, catalog number: N0447S)
12. Ethanol (Fisher Scientific, catalog number: BP28184)
13. DNA oligomers (Integrated DNA technologies, IDT, 25 nmole)
14. DNA Extraction kit (Monarch Genomic DNA purification Kit, NEB, catalog number: T3010S)
15. Miniprep kit (Qiagen, catalog number: 27106)
16. PCR Cleanup kit (Macherey Nagel, catalog number: 740609.50)
17. Phusion DNA Polymerase (New England Biolabs, catalog number: M0535S)
18. 10× Tris-Borate-EDTA (TBE) buffer (Bio-Rad, catalog number: 1610770)
19. Orange G powder (Fisher Scientific, catalog number: AC416550100)
20. 1 kb+ DNA ladder (New England Biolabs, catalog number: N3232L)
21. Gibson Assembly master mix (New England Biolabs, catalog number: E2611S)
22. E. coli NEB5α chemically competent cells with SOC media (New England Biolabs, catalog number: C2987H)
23. Agarose (Millipore Sigma, catalog number: A9539-500)
24. LB agar (Fisher Scientific, catalog number: BP1425-500)
25. LB broth (Fisher Scientific, catalog number: BP1426-500)
26. Ampicillin sodium salt (Fisher Scientific, catalog number: BP-1760-25)
27. 7.5 M ammonium acetate solution (Millipore Sigma, catalog number: A2706)
28. 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI) (Millipore Sigma, catalog number: MBD0015)
29. Rabbit anti-myc antibodies (clone 71D10) (Cell Signalling, catalog number: 2278)
30. Rabbit anti-NeonGreen antibodies (Cell Signaling Technology, catalog number: 53061S)
31. Mouse anti-Centrin antibodies (clone 20H5) (Millipore Sigma, catalog number: 04-1624)
32. Rabbit anti-TgMORN1 serum (provided by Dr. Marc-Jan Gubbels, Boston College, MA)
33. Rabbit anti-TgIMC1 serum (provided by Dr. Gary Ward, University of Vermont, VT)
34. Sodium azide (NaN3) (Fisher Scientific, catalog number: S227I-500)
35. Proteinase K (Fisher Scientific, catalog number: EO0491)
36. Anti-rabbit, cross-adsorbed secondary antibody (Invitrogen, catalog number: A11012)
37. Anti-mouse, cross-adsorbed secondary antibody (Invitrogen, catalog number: A11032)
38. Potassium chloride (KCl) (Fisher Scientific, catalog number: P217-500)
39. Monopotassium phosphate (KH2PO4) (Fisher Scientific, catalog number: BP362-500)
40. Sodium phosphate dibasic (Na2HPO4) (Fisher Scientific, catalog number: S374-500)
41. Sodium chloride (NaCl) (Fisher Scientific, catalog number: BP358)
42. Calcium chloride (CaCl2) (ThermoFisher Scientific, catalog number: 10035)
43. Magnesium chloride (MgCl2) (Fisher Scientific, catalog number: 7791-18-6)
44. HEPES (RPI, catalog number: 7365)
45. Tween-20 (Millipore Sigma, catalog number: P1379)
46. Pyrimethamine (Millipore Sigma, catalog number: SML3579)
47. Triton X-100 (Fisher Scientific, catalog number: AAA16046AE)
48. PacI endonuclease (New England Biolabs, catalog number: R0547S)
49. NsiI endonuclease (New England Biolabs, catalog number: R0127S)
50. EcoRV endonuclease (New England Biolabs, catalog number: R3195)
51. 4% paraformaldehyde solution (PFA) (Fisher Scientific, catalog number: AAJ19943K2)
52. 10 mg/mL ethidium bromide solution (Millipore Sigma, catalog number: E1510)
Laboratory supplies
1. Microcentrifuge tubes (1.7 mL) (Fisher Scientific, catalog number: 14222-171)
2. PCR tubes (Millipore Sigma, catalog number: AXYPCR02D)
3. T-25 and T-75 culture flasks (Fisher Scientific, catalog numbers: FB012936, FB012938)
4. 96-well, 24-well, and 6-well plates (Fisher Scientific, catalog numbers: FB012931, FB012929, FB 012927)
5. 15 mL conical centrifuge tubes (VWR, catalog number: 21008-212)
6. 50 mL conical centrifuge tubes (Fisher Scientific, catalog number: 12-565-270)
7. Snap cap tubes (Fisher Scientific, catalog number: 07-000-212)
8. 10, 200, 1,000 μL pipette tips (USA Scientific, catalog numbers: 1120-3710, 1120-8710, 1122-1730)
9. 5 mL, 10 mL, 25 mL serological pipettes (Fisher Scientific, catalog number: 13-678-11)
10. 10 mL Luer lock syringes (Fisher Scientific, catalog number: 14-955-459)
11. Blunt end needles 23G (Grainger, catalog number: 5FVK7)
12. Stericup filter units 500 and 1,000 mL (Fisher Scientific, catalog numbers: S2GPU05RE, S2GPU10RE)
13. 3.0 μm pore size filter membrane (Millipore Sigma, catalog number: TSTP02500)
14. Cell scrapers (Fisher Scientific, catalog number: 07-000-553)
15. 0.22 μm sterile syringe filters (Fisher Scientific, catalog number: 50-209-3153)
16. Glass pipettes (Fisher Scientific, catalog number: 13-678-20C)
17. Nitrocellulose/filter sandwich (Bio-Rad, catalog number: 1620213)
18. Petri dishes (VWR, catalog number: 25384-342)
19. 2 mm electroporator cuvette (Fisher Scientific, catalog number: FB102)
20. MyFugeTM Minispin (Alkali Scientific, catalog number: 50-233-6060)
21. 15 mm, 22 mm glass coverslips (Fisher Scientific, catalog numbers: 501215160, 12541016)
Solutions
1. Tachyzoite medium (see Recipes)
2. Pyrimethamine selection media (see Recipes)
3. 10× phosphate-buffered saline (PBS) (see Recipes)
4. Cytomix electroporation buffer pH 7.6 [20] (see Recipes)
5. 3% IFA block buffer (see Recipes)
6. IFA permeabilization buffer (see Recipes)
7. 0.8% agarose gel (see Recipes)
Recipes
1. Tachyzoite medium
DMEM supplemented with 3% FBS and 1% PSA. Store at 4 °C.
2. Pyrimethamine selection media
Tachyzoite medium (Recipe 1) with 1 μM pyrimethamine. Store at 4 °C.
3. 10× PBS
1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, and 20 mM KH2PO4 in a final volume of 1 L. Adjust pH to 7.4. Store at room temperature.
4. Cytomix electroporation buffer pH 7.6 [20]
10 mM KPO4 (pH 7.6), 120 mM KCl, 0.15 mM CaCl2, 5 mM MgCl2, 25 mM HEPES, 2 mM EDTA. Adjust the solution to the final volume of 1 L. Filter-sterilize and store at 4 °C.
5. 3% IFA block buffer
500 mL of 1× PBS and 15 mL of FBS. Run through a Stericup filter and add 500 μL of 2 mM NaN3. Store at 4 °C.
6. IFA permeabilization buffer
900 mL of nanopure water, 100 mL of 10× PBS, 1 mL of 2 mM NaN3, and 5 mL Triton X-100.
7. 0.8% agarose gel
0.8 g of agarose in 100 mL of 1× TBE buffer. Microwave until clear, cool down, and add 5 μL of 10 mg/mL ethidium bromide solution.
Equipment
1. Autoclave (Steris, model: Amsco Lab 250)
2. Biological safety cabinet (Labconco, model: 302411000)
3. Benchtop centrifuge with 15 and 50 mL conical vial holders (Eppendorf, model: 5910R)
4. Water bath at 37 °C (Shel Lab, model: SWB15)
5. Cell culture incubator at 34 and 37 °C with 5% CO2 (Eppendorf, catalog number: 6731010015)
6. Incubator at 37 °C (Fisher Scientific, model number: FFCO300DABB)
7. Bacterial shaker at 37 °C (New Brunswick Scientific, model: I26R)
8. Sterile Pyrex borosilicate glassware for solutions and reagents (test tubes, beakers, storage vials)
9. Amaxa Nucleofector II electroporator (Lonza, catalog number: 40700053)
10. Hemacytometer (Fisher Scientific, catalog number: 2671154)
11. Inverted-phase contrast microscope (Nikon, model: TMS-F and Olympus, model: CKX53SF)
12. Zeiss Axio Observer inverted microscope equipped with filters for multichannel fluorescence imaging (green, DAPI, and red)
13. Gel imaging system (Foto dyne Incorporated, model: 1-1430)
14. Microcentrifuge (Eppendorf, catalog number: 2231001179)
15. Microwave (Black+Decker, catalog number: 810004819726)
16. Pipettes and carousel (Fisher, catalog numbers: 13684251, 3116000015)
17. Motorized pipette controller (Eppendorf, catalog number: 4430000018)
18. Protein transfer apparatus (Bio-Rad, model: Trans-Blot Turbo System)
19. SDS-PAGE electrophoresis apparatus (Bio-Rad, model: Mini-Protean Tetra Cell)
20. Thermocycler (ThermoFisher Scientific, catalog number: 4484073)
21. Refrigerator (LG, model: LRTLS2403S)
22. Freezers -20 and -80 °C (Fisher Scientific, catalog number: FBH07CFSA and Eppendorf, model: CryoCube F740hi)
23. Liquid nitrogen storage (Thermo Scientific, catalog number: 7400)
24. Nanodrop spectrophotometer (Thermo Scientific, catalog number: 13400519)
25. Vacuum aspirator (Barnant Company, catalog number: 400-3910)
26. Heat block (Fisher Scientific, catalog number: 88-860-023)
Software and datasets
1. Google Chrome (Google), Safari, or any other web browser
2. SnapGene (Version 8.0)
3. Zen 3.2 (Zeiss)
4. NEB ligation calculator (New England Biolabs) (nebiocalculator.neb.com)
Procedure
A. Creating ToxoFUCCIS strain
1. Retrieving gene information from the ToxoDB database (VEuPathDB)
a. Genomic sequences and coding regions of T. gondii genes of interest were obtained from the ToxoDB database (www.toxodb.org).
b. Find the nucleotide sequence of the coding 5’ and 3’ UTR regions of TgPCNA1 on the TGME49_247460 gene page.
c. From the drop-down menu, located on the left, select “Sequences” to access and download the predicted genomic sequence.
2. Primer design and PCR amplification
a. Use a forward and reverse primer with ligation-independent cloning (LIC) overhangs. The PCNA1 forward primer includes a 5’LIC overhang for vector annealing, fused to the TgPCNA1 5’UTR sequence. The 5’end of the reverse primer includes a LIC overhang for the vector annealing, followed by the last 27 nucleotides of TgPCNA1 ORF excluding the stop codon.
PCNA1_For: tacttccaatccaatttaatgcacgaaatccgcggcacgc
PCNA1_Rev: tcctccacttccaattttagcctcatccatcatagagtcgtccatgcc
b. Purchase primers from Integrated DNA Technologies (www.idtdna.com).
c. Use a DNA Extraction kit to isolate genomic DNA from the parental T. gondii RH TIR1 strain [21]. Culture and collect parasites as described in section D below.
d. Set up a PCR reaction consisting of 100 ng of genomic DNA, 2 μL of 10 mM dNTPs, 5 μL of 10μ M PCNA1_For primer, 5 μL of 10 μM PCNA1_Rev primer, 5 μL of DMSO, 20 μL of 5× Phusion buffer, and 1 μL of Phusion DNA polymerase, and adjust the volume to 100 μL with deionized water.
e. PCR thermocycling conditions: one cycle of denaturation at 98 °C for 1 min; 32 cycles of denaturation at 98 °C for 10 s, annealing at 62 °C for 15 s, and extension at 72 °C for 50 s; one cycle of extension at 72 °C for 5 min.
f. Confirm amplification of the 3.1 kb DNA fragment by mixing 5 μL of PCR mixture with 2 μL of Orange G dye and resolving the PCR product on a 0.8% agarose gel with a 1 kb+ DNA ladder. Purify the amplified DNA fragment using a PCR Cleanup kit.
g. Use a nanodrop to measure the DNA concentration of the purified PCR product.
3. Gibson assembly cloning and E.coli plasmid transformation
a. Vector preparation: Linearize 5 μg of pLIC-2×NeonGreen-DHFR [10] vector DNA with endonuclease PacI following the manufacturer’s recommendation. Confirm the vector linearization by agarose gel electrophoresis, purify linear DNA using a DNA Extraction kit, and measure DNA concentration using a nanodrop spectrophotometer.
b. Use the NEB ligation calculator to determine the DNA amount required for a 1:3 (vector: insert) molar ratio.
Optional: Use SnapGene software to simulate Gibson Assembly and generate the pLIC-TgPCNA1-2×NeonGreen-DHFR plasmid map.
c. Gibson assembly reaction: mix on ice 100 ng of PacI-treated pLIC-2xNeonGreen-DHFR plasmid DNA, 3× molar excess of purified TgPCNA1 DNA fragment, and 5 μL of 2× NEBuilder HiFi DNA assembly master mix. Adjust the volume to 10 μL with nuclease-free water. Incubate the reaction in a thermocycler at 50 °C for 15 min.
d. Mix 50 μL of chemically competent E.coli NEB5α bacteria with 2 μL of the annealed Gibson assembly mixture and incubate on ice for 30 min. Heat-shock bacteria for 30 s in the heat block set to 42 °C. Chill the reaction on ice for 2 min. Add 950 μL of SOC media and recover transformed cells in a bacterial shaker at 250 rpm for 60 min at 37 °C. Spread 100 μL of the transformed E.coli NEB5α cells onto LB agar plates with 100 μg/mL ampicillin. Incubate the plates overnight at 37 °C. Set up overnight liquid cultures of 6–8 individual colonies in 5 mL of LB broth with 100 μg/mL ampicillin. Grow cultures in the bacterial shaker for 16–18 h at 250 rpm at 37 °C.
e. Pellet E.coli NEB5α cells expressing pLIC-TgPCNA1-2xNeonGreen-DHFR plasmid in a tabletop centrifuge at 6,800× g for 3 min. Extract plasmid DNA using QIAGEN Prep Spin Miniprep kit. Measure DNA concentration by spectrophotometry. The expected yield range is 300–800 ng/μL.
f. Validate the correct plasmid assembly by Sanger sequencing using the following primers:
pLIC_For: gtggataaccgtattaccgcctttg
NeonGreen_Rev: accatcgttggggttaccagtac
g. Linearize 50 μg of pLIC-TgPCNA1-2xNeonGreen-DHFR plasmid with endonuclease EcoRV according to the manufacturer’s recommendation. Confirm plasmid linearization by agarose gel electrophoresis. Precipitate DNA in a 70% ethanol solution with 0.75 M ammonium acetate. Store at -20 °C until further use.
4. Parasite transfection, cloning, and selection of transgenic clones
a. Culturing parasites: Infect a T-25 flask of confluent HFF cells with 106 freshly egressing RH TIR1 parasites. Grow parasites until they lyse 75%–80% of the host monolayer for 36–48 h at 37 °C. Release parasites from the host cells by passing the scraped monolayer through a blunt-end 23G needle 3–4 times. Separate parasites from the host cell remnants using a 3.0 μm pore-size filter and pellet them in a 1.7 mL conical tube using a tabletop centrifuge at 1,800× g for 15 min at 4 °C. Aspirate the supernatant and resuspend the parasite in 300 μL of Cytomix buffer supplemented with 0.2 M ATP and 0.5 M reduced L-glutathione (complete Cytomix).
b. Pellet 50 μg of endonuclease-digested and ethanol-precipitated pLIC-TgPCNA1 2xNeonGreen-DHFR in the microcentrifuge at 15,000× g for 10 min. Aspirate and air dry the DNA pellet. Rehydrate the DNA pellet in 100 μL of parasite suspension in complete Cytomix and transfer the mixture into a 2 mm electroporation cuvette.
c. Electroporate parasites with a single 1.7–1.8 kV/176–200 μs pulse using Amaxa Nucleofector II set on program T016. Immediately transfer parasites into a tachyzoite medium in a T-75 flask with a confluent HFF monolayer and recover them for 24 h at 37 °C with 5% CO2.
d. Replace growth medium with pyrimethamine selection medium. Pass cultures 2–3 times in selection media until a stable drug-resistant population is established.
e. Check the plasmid integration efficiency by live fluorescence microscopy of the polyclonal population. Infect HFF monolayers on coverslips with 103 tachyzoites under selection. After 24 h of growth in the tachyzoite medium, fix slides with 4% PFA (5 min, room temperature), incubate in IFA permeabilization buffer (10 min, room temperature), followed by IFA blocking buffer (30 min, room temperature), and stain with DAPI (nuclear dye). Visualize parasites expressing TgPCNA1NG in a Zeiss Axio Observer inverted microscope using ET-GFP block.
f. Clone parasites by limiting dilution. Scrape HFF monolayers infected with a pyrimethamine-resistant population of tachyzoites, pass parasites through a G23 blunt-end needle, and separate from ruptured host cells using a 3 mm filter. Count parasites using a haemocytometer according to the manufacturer’s recommendations. Dilute the parasite suspension in the tachyzoite medium to a final concentration of 250/mL. Using an 8-channel pipette, perform serial 1:1 parasite dilutions in a 96-well plate to achieve a final concentration of 1–2 parasites per well. Grow cloned cultures at 37 °C with 5% CO2 for 10 days to allow plaque development.
g. Identify wells with individual plaques under an inverted light microscope (Nikon TMS-F). Expand clonal parasites by growing in 24-well and T-25 plates.
h. Confirm live TgPCNA1NG expression. Grow clonal parasites in the tachyzoite medium on glass coverslips for 18–20 h at 37 °C with 5% CO2. Fix slides with 4% PFA (5 min, room temperature), incubate in IFA permeabilization buffer (10 min, room temperature), and then incubate in the IFA blocking buffer (30 min, room temperature) and stain with DAPI (nuclear dye). Visualize parasites expressing TgPCNA1NG in a Zeiss Axio Observer inverted microscope using ET-GFP block.
i. Confirm the plasmid integration in individual clones by PCR. Prepare a crude genomic DNA sample of individual clones. Pellet parental and transgenic clonal parasites from 100 μL of parasite suspension in the growth medium in 200 mL PCR tubes using a Minispin centrifuge for 5 min at room temperature. Remove the supernatant and resuspend the parasites in 42 μL of PBS with 0.5 μL of Proteinase K solution, 2.5 μL of DMSO, and 5 μL of 5× HF-Phusion buffer. Incubate the reaction in the thermocycler at 37 °C for 1 h, followed by 50 °C for 2 h and 95 °C for 15 min.
j. Prepare three diagnostic PCR primers. A forward Primer 1 is nested in the TgPCNA1 gene. A reverse Primer 2 is complementary to TgPCNA1 3’UTR. A reverse Primer 3 is complementary to the NeonGreen sequence. The combination of Primer 1 and Primer 2 amplifies a 1.3 kb locus of the native TgPCNA1, while the combination of Primer 1 and Primer 3 will amplify a 1.1 kb locus of TgPCNA1 fused with NeonGreen. Note that published ToxoFUCCIS parasites carry TgPCNA1NG as a second copy [10].
Primer 1: GAAGCCCAAGCCTTTTGCGAAT
Primer 2: CGAAGGAAACCATCGATGCCA
Primer 3: ACCATCGTTGGGGTTACCAGTAC
k. Set up four PCR reactions. Test parental and transgenic parasite genomes using Primer 1/Primer 2 and Primer 1/Primer 3 combinations. The thermocycling conditions are as follows: one cycle of denaturation at 98 °C for 1 min; 32 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 15 s, and extension at 72 °C for 25 s; and one cycle of extension at 72 °C for 5 min. Confirm DNA amplification by mixing 5 μL of PCR mixture with 2 μL of Orange G dye and resolving PCR products on a 0.8% agarose gel with a 1 kb+ DNA ladder.
l. Confirm the TgPCNA1NG protein expression in the transgenic clones by western blot using the anti-mNeonGreen antibody as previously described [10]. Expand and cryopreserve at least three clonal populations of RH TIR1 expressing TgPCNA1NG protein for future use.
B. Endogenous C-terminal tagging of TgBCC0
1. Retrieve the TgBCC0 genomic sequence (TGME49_294860) from ToxoDB database (www.toxodb.org).
2. Primer design and PCR amplification
a. Use BCC0_For and BCC0_Rev primers to amplify the 3’end of TgBCC0 ORF. The forward primer includes a 5’LIC overhang for the vector annealing and TgBCC0-specific sequence. The 5’ end of the reverse primer includes LIC overhang for the vector annealing, followed by the last 24 nucleotides of TgBCC0 ORF, excluding the stop codon. Order the TgBCC0 tagging primers from IDT DNA (www.idtdna.com).
BCC0_For: tacttccaatccaatttaatgcaaggtgctccaggaacagagag
BCC0_Rev: tcctccacttccaattttagcgtaaaacgtcacttgtctccccag
b. Amplify and purify a 2.4 kb TgBCC0 genomic fragment as described in step A2 using proper primer annealing temperature and PCR extension.
3. Cloning via Gibson assembly and transformation in E.coli
a. Follow the steps described in step A3 and clone the TgBCC0 gene fragment into the pLIC-SM-3xmyc-CAT vector [10].
4. Parasite transfection, cloning, and selection of clones
a. Create an alternative ToxoFUCCI model in RH Tati Δku80 strain using the protocol described in section A [10,19].
b. Electroporate 50 mg of NsiI-linearized and ethanol-precipitated pLIC-TgBCC0-SM-3xmyc-CAT plasmid in the RH Tati Δku80 ToxoFUCCI tachyzoites following the procedure described in step A4. Select a chloramphenicol-resistant parasite population. Obtain and verify individual RH Tati Δku80 ToxoFUCCI clones expressing TgBCC0myc by IFA, PCR, and western blot.
C. Measurement of tachyzoite cell cycle phases
1. Use ToxoFUCCIS strain alone or expressing cell cycle markers or co-stained with antibodies against cell cycle markers to identify the individual cell cycle phases. In the absence of a reliable synchronization technique, the durations of the cell cycle phases can be determined using a combination of quantitative live and immunofluorescent microscopy (Figure 1A). The live TgPCNA1NG fluorescence detects ToxoFUCCIS parasites actively replicating DNA [10].
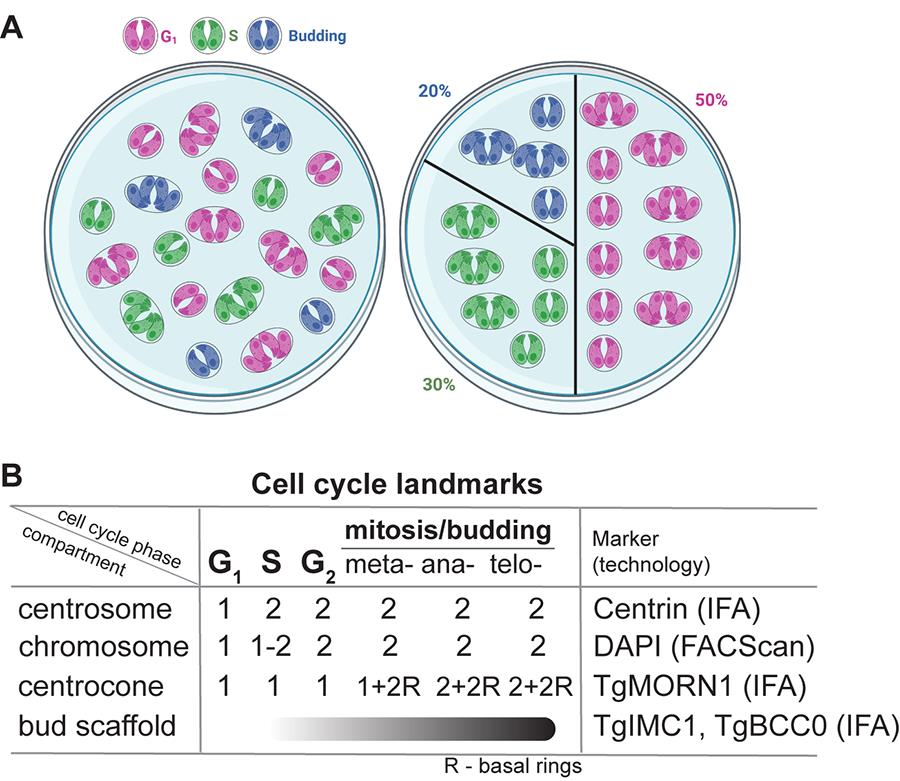
Figure 1. Quantitative immunofluorescence microscopy (qIFA) approach to examine tachyzoite cell cycle. (A) A cartoon representation of the asynchronously growing tachyzoite vacuoles randomly distributed in the microscopic field on the left and organized according to the cell cycle phases on the right. The color represents a vacuole in a specific cell cycle stage. (B) A matrix designed to distinguish cell cycle phases based on the presence and the number of various mitotic structures (compartments). R: basal (MORN) ring.
2. Supplement ToxoFUCCIS live fluorescence with anti-Centrin antibody staining to quantify centrosomes [10,22,23].
3. Use anti-MORN1 serum to visualize spindle morphology and basal rings [10–12,14,24].
4. Use anti-TgIMC1 antibodies to identify tachyzoites undergoing cytokinesis (budding) [10,17,25].
Note: Tachyzoites maintain relative cell cycle synchrony within the vacuole. Yet, parasite populations are asynchronous, and the fraction of vacuoles in each cell cycle stage reflects the time parasites spend in each stage (Figure 1A). For example, the tachyzoite cell cycle is composed of ~50% G1, 30% S, and 20% of vacuoles undergoing overlapping mitosis (M) and budding (C, cytokinesis) [10,26,27]. Therefore, a random 100 vacuoles will represent a combination of ~50 vacuoles of parasites with a single centrosome (G1), ~50 vacuoles of parasites with duplicated centrosomes (S + G2 + M/C), of which ~15–20 vacuoles of parasites will have variable size buds (M/C), and ~10 vacuoles will have parasites with basal rings attached to a single or a duplicated centrocone (meta- and anaphases of mitosis) (Figure 1B).
5. Quantitative immunofluorescence
a. Scrape infected HFF monolayers, pass three times through a G23 blunt-end needle, and separate freshly egressing parasites using a 3 mm filter (ToxoFUCCIS strain or derivatives). Infect coverslips with confluent HFF monolayers with 103 filtered parasites. Important: Incubate for 15–20 min at 37 °C with 5% CO2 and wash infected monolayers 2–3 times with Hank’s buffer to remove non-invaded parasites. Grow parasites at 34 °C with 5% CO2 for 18 h. It allows parasites to complete two replication cycles. The resulting population primarily consists of cells at the optimal stage for cell cycle evaluation: vacuoles of four synchronously dividing parasites [10].
b. Perform IFA. Fix slides with 4% PFA (5 min, room temperature), incubate in IFA permeabilization buffer (10 min, room temperature), incubate in the IFA blocking buffer (30 min, room temperature), and stain with appropriate antibodies and DAPI (nuclear dye). Visualize TgPCNA1NG in green, markers in red, and DAPI in blue channels using a Zeiss Axio Observer inverted microscope.
c. Important: Choose microscopic fields without bias. Count all parasites within each vacuole according to parameters defined in the experiment. Perform three biological replicates of each experiment. Use three people to do counts.
6. Cell cycle phase measurement
a. G1 phase: Quantify 500 random ToxoFUCCIS parasites and determine how many of them exhibit diffused nuclear TgPCNANG staining and a single centrosome visualized with anti-Centrin antibodies (Figure 2, G1 panel).
b. S phase: Quantify 500 random ToxoFUCCIS parasites and determine how many of them exhibit aggregated nuclear TgPCNANG staining that corresponds to the active replication foci (Figure 2, S-phase panel).
c. S/C (budding) interval: Use a RH Tati Δku80 ToxoFUCCI line expressing one of the earliest markers of cytokinesis TgBCC0myc [10]. Stain parasites with anti-myc antibodies. Quantify 500 parasites with aggregated nuclear TgPCNANG (active DNA replication) and determine the fraction that also expresses TgBCC0myc (Figure 2, S/C panel).
d. S/M/C interval: Stain ToxoFUCCIS parasites with anti-TgMORN1 antibodies. The TgMORN1 localizes to the spindle compartment centrocone and basal complex, representing the parasite cytoskeleton. Quantify 500 parasites with aggregated nuclear TgPCNANG (active DNA replication) and determine the fraction of the parasites that have a single centrocone with two MORN1 rings (basal complexes of the future buds), likely representing a prophase of the mitosis (Figure 2, S/M/C panel).
e. M/C interval: Stain ToxoFUCCIS parasites with anti-TgIMC1 antibodies. Quantify 500 parasites with diffused nuclear and bright conoidal TgPCNANG building TgIMC1 positive internal buds (Figure 2, M/C panel).
i. To distinguish metaphase and anaphase, stain ToxoFUCCIS parasites with anti-TgMORN1 antibodies. Metaphase parasites have diffused nuclear and bright conoidal TgPCNANG and a single centrocone associated with two MORN1 rings. Anaphase parasites have diffused nuclear and bright conoidal TgPCNANG and a duplicated centrocone, each associated with a MORN1 ring.
ii. To distinguish telophase, stain ToxoFUCCIS parasites with anti-TgIMC1 antibodies. Telophase parasites have two nuclei (use DAPI stain), bright conoidal TgPCNANG, and two nearly mature internal buds within the recognizable mother cytoskeleton.
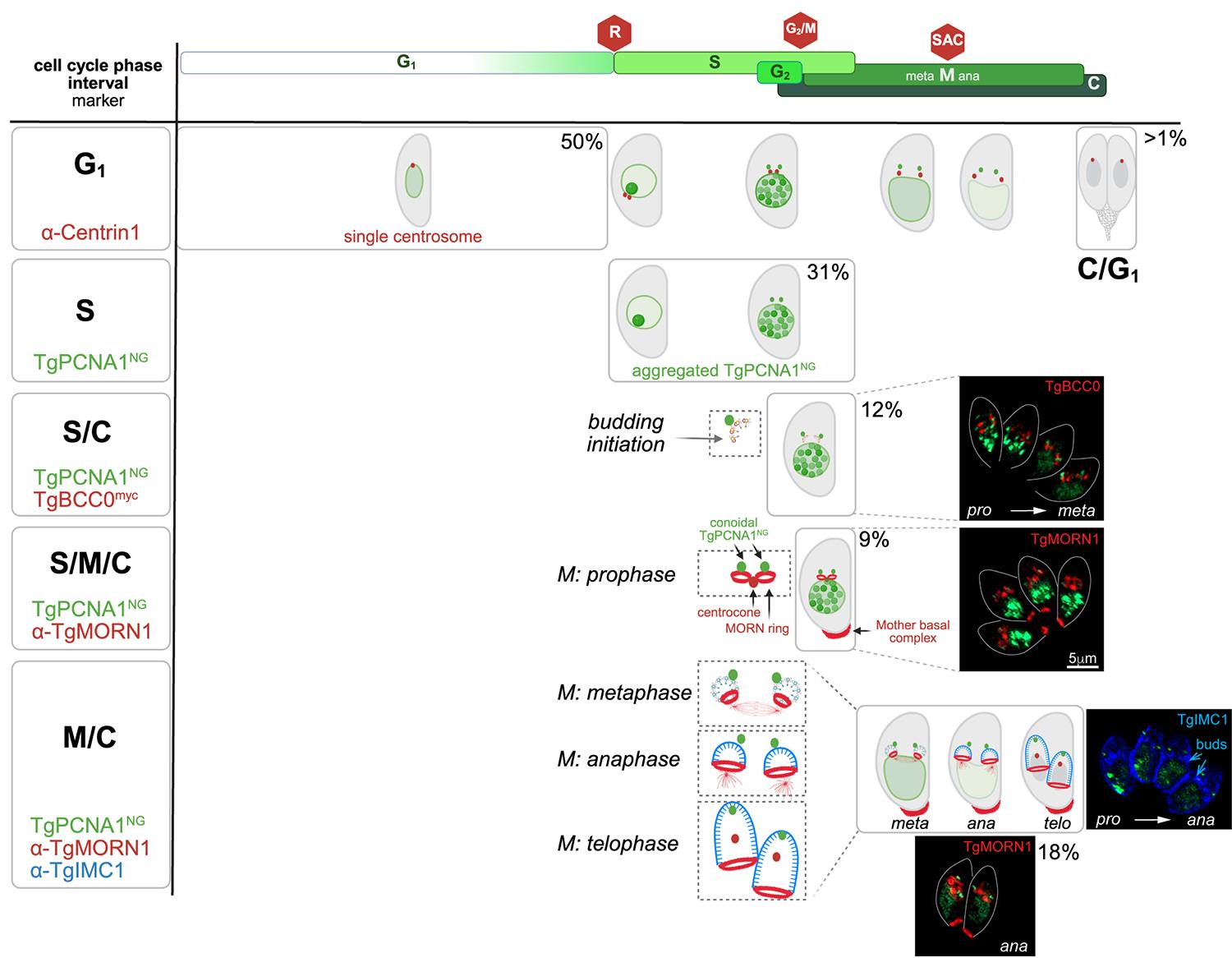
Figure 2. Measurement of the tachyzoite cell cycle phases using ToxoFUCCIS strain. The upper panel shows T. gondii cell cycle organization. Cell cycle phases and their relative durations are indicated. The red stop signs mark three major checkpoints. The left vertical panel lists cell cycle phases or intervals and markers used to determine a phase duration or an overlap of the indicated phases. The main window shows cartoons of the tachyzoites undergoing cell cycle progression and depicts the phase of interest. The corresponding live/immunofluorescence microscopy images of the selected intervals are shown on the right. The percentage is the time that the tachyzoite spends in the indicated cell cycle frame. As a reference, the first panel shows all major morphological transitions of the tachyzoite, including the transition to the next division cycle (C/G1).
Data analysis
We used a T-test in our quantifications to determine whether the difference between the means of two groups was statistically significant. For example, we quantified data from three independent experiments to compare the proportions of replicating and non-replicating parasites. Statistical differences between groups were assessed using an unpaired two-tailed t-test in Excel. The formula used to calculate the t-value is as follows:
where X1 and X2 are means of the two samples (replicating and non-replicating), s12 and s22 are the variances of the two samples, and n1 and n2 are the sample sizes (three biological replicates, n = 3). A value <0.05 was considered statistically significant.
Validation of protocol
This protocol was used to build the ToxoFUCCIS reporter system expressing the green fluorescent DNA replication factor TgPCNA1, as described in Batra et al. [10]. This transgenic line and its derivatives were instrumental in establishing the cell cycle organization of replicating T. gondii tachyzoites. Live-cell and immunofluorescent microscopy validated the ToxoFUCCIS probe. In addition, flow cytometry analysis confirmed the reproducibility of the ToxoFUCCIS signal distributed across the asynchronous population. Furthermore, the strain was capable of detecting cell cycle checkpoints, including TgCrk6-mediated spindle assembly checkpoint and TgCrk4-regulated G2/M transition [11,14].
General notes and troubleshooting
General notes
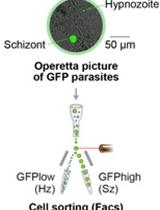
1. Attempts to attach NeonGreen to endogenous TgPCNA1 were unsuccessful, likely because the TgPCNA1NG cannot form the functional trimer ring on the replicating DNA. Therefore, TgPCNA1NG was introduced as a second copy, which did not alter tachyzoite survival or cell cycle organization (see Graphical abstract) [10].
2. The ToxoFUCCIS strain may lose TgPCNA1NG expression after 50 passages; therefore, earlier passages should be preserved and used in experiments.
3. The S-phase starts with the emergence of a single TgPCNA1NG aggregate, followed by amplification and spread of aggregates throughout the nucleus, and ends with a few or a single aggregate [10]. Therefore, the S-phase duration should account for a variable number of TgPCNA1NG aggregates. Note that TgPCNA1NG accumulates in conoids of the daughter buds after 60% of S phase is completed [10]. The conoidal accumulation remains post–S phase and should not be mistaken for DNA replication foci. Please use co-staining with DAPI to distinguish nuclear and cytoplasmic TgPCNA1NG aggregates.
4. The ToxoFUCCIS approach can be used to study the effects of specific proteins on cell cycle progression by creating an auxin-induced degradation model of the gene of interest in the ToxoFUCCIS background. For example, we validated Toxoplasma checkpoint kinases by creating and examining the ToxoFUCCIS strains co-expressing TgCrk6AID-HA or TgCrk4 AID-HA [10].
Acknowledgments
Author contributions: Conceptualization, E.S.S., M.B.; Investigation, M.B.; Writing—Original Draft, M.B, E.S.S.; Writing—Review & Editing, E.S.S., M.B.; Funding acquisition, E.S.S.; Supervision, E.S.S. We thank Dr. Daria A. Naumova (Mercor LLC) for editing the final draft. This project was supported by grants from the National Institute of Health to E.S.S. (R01AI175640, R03AI155834, R21AI178797). Some figures were created using BioRender and Adobe Illustrator 2025: Graphical abstract (biorender.com/illustrations/6900e6283b5ca980ff291810), Figure 1 (biorender.com/illustrations/67141bdff50aaa0a23a9ffc4), and Figure 2 (biorender.com/illustrations/6902489fd22ecc3885c09bf4).
This protocol was used in Batra et al. [10].
Competing interests
The authors declare that they have no competing interests.
References
- Ando, R., Sakaue-Sawano, A., Shoda, K. and Miyawaki, A. (2023). Two coral fluorescent proteins of distinct colors for sharp visualization of cell-cycle progression. Cell Struct Funct. 48(2): 135–144. https://doi.org/10.1247/csf.23028
- Sakaue-Sawano, A., Kurokawa, H., Morimura, T., Hanyu, A., Hama, H., Osawa, H., Kashiwagi, S., Fukami, K., Miyata, T., Miyoshi, H., et al. (2008). Visualizing Spatiotemporal Dynamics of Multicellular Cell-Cycle Progression. Cell. 132(3): 487–498. https://doi.org/10.1016/j.cell.2007.12.033
- Yano, S. and Hoffman, R. M. (2018). Real-Time Determination of the Cell-Cycle Position of Individual Cells within Live Tumors Using FUCCI Cell-Cycle Imaging. Cells. 7(10): 168. https://doi.org/10.3390/cells7100168
- Yin, K., Ueda, M., Takagi, H., Kajihara, T., Sugamata Aki, S., Nobusawa, T., Umeda‐Hara, C. and Umeda, M. (2014). A dual‐color marker system for in vivo visualization of cell cycle progression in Arabidopsis. Plant J. 80(3): 541–552. https://doi.org/10.1111/tpj.12652
- Zielke, N. and Edgar, B. A. (2015). FUCCI sensors: powerful new tools for analysis of cell proliferation. WIREs Dev Biol. 4(5): 469–487. https://doi.org/10.1002/wdev.189
- Zielke, N., Korzelius, J., van Straaten, M., Bender, K., Schuhknecht, G. F., Dutta, D., Xiang, J. and Edgar, B. A. (2014). Fly-FUCCI: A Versatile Tool for Studying Cell Proliferation in Complex Tissues. Cell Rep. 7(2): 588–598. https://doi.org/10.1016/j.celrep.2014.03.020
- Arias, E. E. and Walter, J. C. (2007). Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 21(5): 497–518. https://doi.org/10.1101/gad.1508907
- Li, X., Zhao, Q., Liao, R., Sun, P. and Wu, X. (2003). The SCFSkp2 Ubiquitin Ligase Complex Interacts with the Human Replication Licensing Factor Cdt1 and Regulates Cdt1 Degradation. J Biol Chem. 278(33): 30854–30858. https://doi.org/10.1074/jbc.c300251200
- Sugiyama, M., Sakaue-Sawano, A., Iimura, T., Fukami, K., Kitaguchi, T., Kawakami, K., Okamoto, H., Higashijima, S. i. and Miyawaki, A. (2009). Illuminating cell-cycle progression in the developing zebrafish embryo. Proc Natl Acad Sci USA. 106(49): 20812–20817. https://doi.org/10.1073/pnas.0906464106
- Batra, M., Marsilia, C., Awshah, D., Hawkins, L. M., Wang, C., Chaput, D., Naumova, D. A. and Suvorova, E. S. (2025). Deciphering cell cycle organization of Toxoplasma endodyogeny. mBio. 16(8): e01119–25. https://doi.org/10.1128/mbio.01119-25
- Hawkins, L. M., Wang, C., Chaput, D., Batra, M., Marsilia, C., Awshah, D. and Suvorova, E. S. (2024). The Crk4-Cyc4 complex regulates G2/M transition in Toxoplasma gondii. EMBO J. 43(11): 2094–2126. https://doi.org/10.1038/s44318-024-00095-4
- Engelberg, K., Bechtel, T., Michaud, C., Weerapana, E. and Gubbels, M. J. (2022). Proteomic characterization of the Toxoplasma gondii cytokinesis machinery portrays an expanded hierarchy of its assembly and function. Nat Commun. 13(1): e1038/s41467–022–32151–0. https://doi.org/10.1038/s41467-022-32151-0
- Gubbels, M. J., Vaishnava, S., Boot, N., Dubremetz, J. F. and Striepen, B. (2006). A MORN-repeat protein is a dynamic component of theToxoplasma gondiicell division apparatus. J Cell Sci. 119(11): 2236–2245. https://doi.org/10.1242/jcs.02949
- Hawkins, L. M., Naumov, A. V., Batra, M., Wang, C., Chaput, D. and Suvorova, E. S. (2022). Novel CRK-Cyclin Complex Controls Spindle Assembly Checkpoint in Toxoplasma Endodyogeny. mBio. 13(1): e03561–21. https://doi.org/10.1128/mbio.03561-21
- Heaslip, A. T., Dzierszinski, F., Stein, B. and Hu, K. (2010). TgMORN1 Is a Key Organizer for the Basal Complex of Toxoplasma gondii. PLoS Pathog. 6(2): e1000754. https://doi.org/10.1371/journal.ppat.1000754
- Hu, K., Mann, T., Striepen, B., Beckers, C. J. M., Roos, D. S. and Murray, J. M. (2002). Daughter Cell Assembly in the Protozoan ParasiteToxoplasma gondii. Mol Biol Cell. 13(2): 593–606. https://doi.org/10.1091/mbc.01-06-0309
- Uy, J. N., Lou, Q., Zhou, Z. H. and Bradley, P. J. (2025). Toxoplasma IMC1 is a central component of the subpellicular network and plays critical roles in parasite morphology, replication, and infectivity. bioRxiv. e657516. https://doi.org/10.1101/2025.06.03.657516
- Brown, K., Long, S. and Sibley, L. (2018). Conditional Knockdown of Proteins Using Auxin-inducible Degron (AID) Fusions in Toxoplasma gondii. Bio Protoc. 8(4): e2728. https://doi.org/10.21769/bioprotoc.2728
- Sheiner, L., Demerly, J. L., Poulsen, N., Beatty, W. L., Lucas, O., Behnke, M. S., White, M. W. and Striepen, B. (2011). A Systematic Screen to Discover and Analyze Apicoplast Proteins Identifies a Conserved and Essential Protein Import Factor. PLoS Pathog. 7(12): e1002392. https://doi.org/10.1371/journal.ppat.1002392
- Soldati, D. and Boothroyd, J. C. (1993). Transient Transfection and Expression in the Obligate Intracellular Parasite Toxoplasma gondii. Science. 260(5106): 349–352. https://doi.org/10.1126/science.8469986
- Long, S., Brown, K. M., Drewry, L. L., Anthony, B., Phan, I. Q. H. and Sibley, L. D. (2017). Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog. 13(5): e1006379. https://doi.org/10.1371/journal.ppat.1006379
- Alvarez, C. A. and Suvorova, E. S. (2017). Checkpoints of apicomplexan cell division identified in Toxoplasma gondii. PLoS Pathog. 13(7): e1006483. https://doi.org/10.1371/journal.ppat.1006483
- Chen, C. T. and Gubbels, M. J. (2013). The Toxoplasma gondii centrosome is the platform for internal daughter budding as revealed by a Nek1 kinase mutant. J Cell Sci. 126(Pt 15): e123364. https://doi.org/10.1242/jcs.123364
- Engelberg, K., Bauwens, C., Ferguson, D. J. P. and Gubbels, M. J. (2025). Co-dependent formation of the Toxoplasma gondii subpellicular microtubules and inner membrane skeleton. mBio. 16(9): e01389–25. https://doi.org/10.1128/mbio.01389-25
- Dubey, R., Harrison, B., Dangoudoubiyam, S., Bandini, G., Cheng, K., Kosber, A., Agop-Nersesian, C., Howe, D. K., Samuelson, J., Ferguson, D. J. P., et al. (2017). Differential Roles for Inner Membrane Complex Proteins across Toxoplasma gondii and Sarcocystis neurona Development. mSphere. 2(5): e00409–17. https://doi.org/10.1128/msphere.00409-17
- Behnke, M. S., Wootton, J. C., Lehmann, M. M., Radke, J. B., Lucas, O., Nawas, J., Sibley, L. D. and White, M. W. (2010). Coordinated Progression through Two Subtranscriptomes Underlies the Tachyzoite Cycle of Toxoplasma gondii. PLoS One. 5(8): e12354. https://doi.org/10.1371/journal.pone.0012354
- Radke, J. (2001). Defining the cell cycle for the tachyzoite stage of Toxoplasma gondii. Mol Biochem Parasitol. 115(2): 165–175. https://doi.org/10.1016/s0166-6851(01)00284-5
Article Information
Publication history
Received: Nov 7, 2025
Accepted: Dec 15, 2025
Available online: Jan 8, 2026
Published: Jan 20, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Batra, M. and Suvorova, E. S. (2026). Assessing the Toxoplasma Tachyzoite Cell Cycle Phases Using Fluorescent Ubiquitination-Based Cell Cycle Indicator. Bio-protocol 16(2): e5588. DOI: 10.21769/BioProtoc.5588.
Category
Microbiology > Microbial cell biology > Cell imaging
Microbiology > in vivo model > Protozoan
Cell Biology > Cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.