- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Highly Efficient siRNA Transfection Method in Primary Cultured Cortical Neurons
Published: Vol 16, Iss 2, Jan 20, 2026 DOI: 10.21769/BioProtoc.5567 Views: 297
Reviewed by: Olga KopachAnonymous reviewer(s)

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Transfecting neurons remains technically challenging due to their sensitivity. Conventional methods, such as Lipofectamine 2000 or Lipofectamine RNAiMAX, often result in significant cytotoxicity, which limits their utility. Although lentiviral transfection offers high efficiency, it is hindered by high costs and complex procedures. This experiment employs a small interfering RNA (siRNA)-specific transfection reagent from the Kermey company. This reagent is a novel nanoparticle-based lipid material designed for the efficient delivery of oligonucleotides, including siRNA, into a wide range of cell types. Its efficacy in achieving high transfection efficiency in neurons, however, has not yet been established. After several days of in vitro neuronal culture, researchers can perform a simple transfection procedure using this reagent to achieve robust transfection efficiency. Notably, the protocol does not require medium replacement 6–8 h post-transfection, streamlining the workflow and minimizing cellular stress.
Key features
• Based on Kermey’s siRNA-specific transfection reagent, we present a method for efficient in vitro transfection of siRNA into primary cultured mouse cortical neurons.
• No observable adverse effects are detected in the transfected neurons during the entire experiment.
• This method enables consistent and efficient knockdown of the target protein.
• Phosphoglycerate dehydrogenase (PHGDH) siRNA and siNC (negative control) siRNA can be transfected into neuronal cells after 72 h of in vitro culture.
Keywords: NeuronGraphical overview
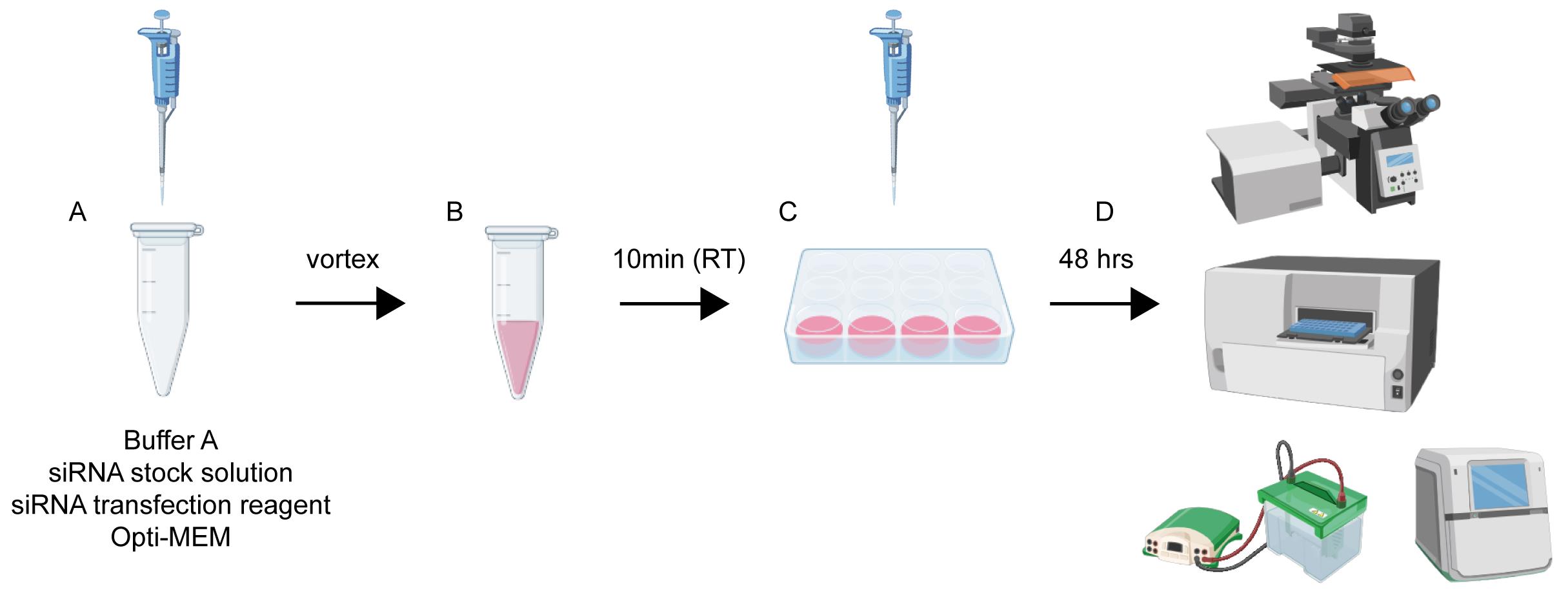
Workflow and timeline for neuronal transfection. (A) Reagents are sequentially added to a microcentrifuge tube in volumes appropriate for the specific culture plate. (B) The mixture from step A is vortexed thoroughly and incubated at room temperature for 10 min to allow complex formation. (C) The transfection mixture is then added dropwise and evenly to the neuronal culture medium. (D) Forty-eight hours post-transfection, cells are ready for downstream assays, including western blotting, immunofluorescence staining, and cell viability analysis.
Background
Investigating neuronal survival and function is essential for understanding the structure, development, and function of the nervous system. It is also critical for addressing neurological disorders such as neurodegenerative diseases, stroke, and neuropsychiatric conditions [1,2]. Effective gene-targeting strategies are vital to elucidate gene functions, clarify causal relationships and underlying molecular signaling pathways, and develop novel therapeutic approaches to modulate neuronal death and function.
Currently, small interfering RNA (siRNA)-mediated gene knockdown is widely used in various cell types and cell lines in vitro and even in vivo through specialized delivery techniques [3]. However, delivering siRNA into primary neuronal cells remains a significant technical challenge and a costly endeavor [4]. Primary neurons, due to their post-mitotic nature, are inherently more difficult to transfect than proliferating cells and are highly susceptible to cell death [5]. Presently, viral vector-mediated siRNA delivery is commonly employed for neurons and offers reliable efficacy along with long-term stability. However, this approach involves complex procedures, high costs, and potential cytotoxicity [6,7]. In contrast, conventional transfection methods, such as Lipofectamine 2000 or RNAiMAX, frequently induce neuronal toxicity, compromising cell viability and potentially confounding experimental outcomes. Moreover, these reagents typically yield low transfection efficiency in primary neurons [7]. Additionally, nucleofection is only applicable to neurons on the day of plating, when they are in suspension and have not yet extended neurites [8]. Given these limitations, there is a pressing need to develop safer, more efficient, and neuron-compatible transfection strategies that enable robust siRNA delivery in primary cultured neurons without compromising their physiological integrity.
To address these limitations, we developed an optimized transfection protocol for mouse primary neurons using Kermey’s siRNA-specific transfection reagent. This newly developed and commercialized product is a novel nano-lipid material designed for efficient oligonucleotide delivery into various cell types. Comparative analyses with conventional reagents, such as Lipofectamine 2000 and RNAiMAX, demonstrate that this new method achieves superior siRNA delivery efficiency with minimal cytotoxicity, robust target gene knockdown, and excellent preservation of neuronal viability and physiological characteristics. These advantages are likely attributable to the unique design of the nano-lipid material, which minimizes immune activation and membrane-related cellular stress. This advancement provides researchers with a reliable, cost-effective tool for conducting mechanistic studies in primary neuronal systems.
Materials and reagents
Biological materials
1. C57BL/6L mouse [Ji’nan Pengyue Laboratory Animal Breeding Co., Ltd. (China), origin: Ji’nan Shandong, China]
Reagents
1. Poly-D-lysine (Sigma, catalog number: P6407-10X5MG)
2. NeurobasalTM medium (1×) (Gibco, catalog number: 21103049)
3. B-27 supplement (50×) (Gibco, catalog number: 17504044)
4. siRNA transfection reagent (Kermey, catalog number: MLR0201)
5. LipofectamineTM RNAiMAX transfection reagent (Thermo Fisher Scientific, catalog number: 13778150)
6. GlutaMAXTM (100×) (Gibco, catalog number: 35050061)
7. Dulbecco’s modified Eagle medium (DMEM) basic (1×) (Gibco, catalog number: 11995065)
8. Penicillin-streptomycin liquid (100×) (Solarbio, catalog number: P1400)
9. Phosphate-buffered solution (PBS), 0.01 M (powder, pH 7.2–7.4) (Solarbio, catalog number: P1010)
10. Opti-MEM (1×) (Gibco, catalog number: 31985047)
11. Fluorescent dye-labeled siRNA negative control (GenePharma)
12. Standard negative control (siNC) (GenePharma, sequence: UUCUCCGAACGUGUCACGUtt)
13. Custom siRNA targeting PHGDH (GenePharma, sequence: UCGGCAGAAUUGGAAGAGAtt)
14. Triton X-100 (Solarbio, catalog number: T8200)
15. Albumin (bovine fraction V) (BSA) (MeilunBio, CAS: 9048-46-8)
16. DAPI (Solarbio, catalog number: S2110)
17. Paraformaldehyde (PFA), 4% (Solarbio, catalog number: P1110)
18. Primary antibody used in this study: PHGDH (ProteinTech, catalog number: 14719-1-AP)
19. Second antibody used in this study: HRP-conjugated Affinipure goat anti-rabbit IgG (H+L) (SA00001-2) (ProteinTech)
20. CCK8 Assay kit (MeilunBio, CAS: MA0218)
21. Fetal bovine serum (FBS) (Sigma, catalog number: F0193-500ML)
Solutions
1. Triton X-100, 0.5% (see Recipes)
2. BSA, 5% (see Recipes)
3. Neurobasal medium (see Recipes)
4. Trypsin, 0.025% (see Recipes)
5. Complete medium (see Recipes)
6. PDL 10× (see Recipes)
7. PDL 1× (see Recipes)
Recipes
1. Triton X-100, 0.5%
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Triton X-100, 100% | 0.5% (v/v) | 250 μL |
| PBS | n/a | 49.75 mL |
| Total | n/a | 50 mL |
2. BSA, 5%
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| BSA | 1/50 | 5 g |
| PBS | n/a | 100 mL |
| Total | n/a | 100 mL |
3. Neurobasal medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| NeurobasalTM medium | 48/50 | 48 mL |
| B-27 Supplement | 1/50 | 1 mL |
| GlutaMAXTM | 1/100 | 0.5 mL |
| Penicillin-streptomycin, liquid | 1/100 | 0.5 mL |
| Total | n/a | 50 mL |
4. Trypsin, 0.025%
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Trypsin, 0.25% | 1/10 | 5 mL |
| DMEM | 9/10 | 45 mL |
| Total | n/a | 50 mL |
5. Complete medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DMEM | 44/50 | 44 mL |
| FBS | 1/10 | 5 mL |
| GlutaMAXTM | 1/100 | 0.5 mL |
| Penicillin-streptomycin, liquid | 1/100 | 0.5 mL |
| Total | n/a | 50 mL |
6. PDL 10×
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Poly-D-lysine | n/a | 5 mg |
| PBS | n/a | 50 mL |
| Total | n/a | 50 mL |
7. PDL 1×
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| PDL, 10× | 1/10 | 1 mL |
| PBS | n/a | 9 mL |
| Total | n/a | 10 mL |
Laboratory supplies
1. 24-well tissue culture plate (JET, catalog number: TCP011124)
2. 24-well round coverslip (WHB, catalog number: WHB-24-CS)
3. 50 mL centrifuge tube (LABSELECT, catalog number: CT-012-50)
4. 15 mL centrifuge tube (LABSELECT, catalog number: CT-002-15A)
5. 1.5 mL centrifuge tube (LABSELECT, catalog number: MCT-001-150)
6. 0.22 μm PES syringe filter (Biosharp, catalog number: BS-PES-22)
7. 50 mL syringe (Beyotime, catalog number: FS850-20pcs)
8. 200 μL RNase-free pipette (Biosharp, catalog number: BS-200-TRS)
9. 10 μL RNase-free pipette (Biosharp, catalog number: BS-RT-10)
Equipment
1. Dissection microscope (RWD, model: 77001S)
2. Dissection tools: forceps (RWD, model: FC4001R-11) and iris scissors (RWD, model: S12003-09)
3. Biosafety cabinet (Esco, model: AC2-4S1)
4. CO2 incubator (Thermo Scientific, model: Forma Steri-Cycle i160 CR)
5. SpectraMax ABS (Molecular Devices, USA)
6. Low-speed desktop centrifuge (BAIOU, model: TDL-320)
7. Amersham ImageQuantTM 800 GxP (Cytiva, model: 29653452)
8. Confocal microscope (Nikon, model: Nikon C2)
Software and datasets
1. Image J [NIH, Java 1.8.0_345 (64-bit)]
Procedure
A. Preparation of cortical neuron cultures
1. One day in advance, coat the required well plates or culture dishes with 1× PDL, ensuring the bottom of each well is completely covered. The following day, aspirate the PDL and wash the wells three times with sterile PBS. Allow the plates to dry in an incubator before use.
2. Euthanize a pregnant C57BL/6 mouse at embryonic day 15–18 (E15–E18) using CO2 asphyxiation. Open the abdominal cavity, carefully remove the fetuses, and immediately transfer them into ice-cold PBS supplemented with 1% penicillin-streptomycin.
3. Carefully remove the fetal membranes with fine scissors and separate the heads at the neck region. Under a dissecting microscope, peel away the skin from the heads, then open the skulls along the midline to expose the brains. Gently remove the meninges, isolate the cortical tissue, and transfer it into pre-chilled DMEM. Perform all procedures on ice to maintain tissue viability.
4. Aspirate the DMEM and digest the tissue with 0.025% trypsin-EDTA. Gently triturate the tissue using a pipette to break it into smaller fragments. Incubate at 37 °C for 15–20 min, gently agitating the mixture three times during incubation to ensure uniform digestion.
5. Terminate the digestion by adding complete medium. Filter the cell suspension once, then centrifuge at 322× g for 5 min.
6. After centrifugation, carefully discard the supernatant and resuspend the pellet in fresh, prewarmed neurobasal medium. Filter the resuspended cell suspension again through a filter mesh. For cell counting, load 10 μL of the filtered suspension into a clean hemocytometer. Count the cells under a microscope, calculate the average count, and multiply this value by the total volume of the cell suspension in milliliters to determine the total cell number.
7. Seed the cells according to the experimental requirements (in this protocol, plate 1 × 106 cells per well in a 12-well plate).
8. Replace half of the medium with fresh, prewarmed neurobasal medium every 3 days and routinely monitor the neuronal health and morphology.
B. Primary neuron transfection with siRNA transfection reagent from Kermey
1. Perform transfection when neurons reach 4–5 days in vitro (DIV4–5), ensuring a minimum culture period of 3 days prior to transfection. This protocol is designed for a 24-well plate format (refer to Table 1 for scaling adjustments to other plate formats). Prepare transfection complexes using a 20 μM stock solution of siRNA.
Note: Table 1 specifies reagent volumes for different culture plate formats.
Table 1. Transfection complex preparation
| Plates | siRNA stock solution | Buffer A | siRNA transfection reagent |
|---|---|---|---|
| 24-well | 0.5 μL | 7 μL | 2 μL |
| 12-well | 1 μL | 14 μL | 4 μL |
| 6-well | 2 μL | 28 μL | 8 μL |
2. To prepare the siRNA-transfection complex for a single well of a 24-well plate, begin by adding 7 μL of buffer A into a sterile microcentrifuge tube. Next, add 0.5 μL of siRNA solution (from the 20 μM stock) and mix thoroughly by vortexing or pipetting. Then, add 2 μL of siRNA transfection reagent and mix completely. Incubate the mixture at room temperature for 10 min. Subsequently, add 45 μL of Opti-MEM medium and mix by pipetting up and down approximately five times to yield the final transfection complex, which is now ready for immediate use (Figure 1).
3. For transfection, gently add the prepared complex dropwise to the neuronal culture in the 24-well plate. Gently rock the plate back and forth 3–5 times to ensure uniform distribution of the solution.
4. After transfection, incubate the cells at 37 °C in a 5% CO2 atmosphere. For optimal assessment of knockdown efficiency, analyze protein levels 48–72 h post-transfection. In this protocol, neuronal protein samples were collected at a 48-h time point for subsequent analysis.
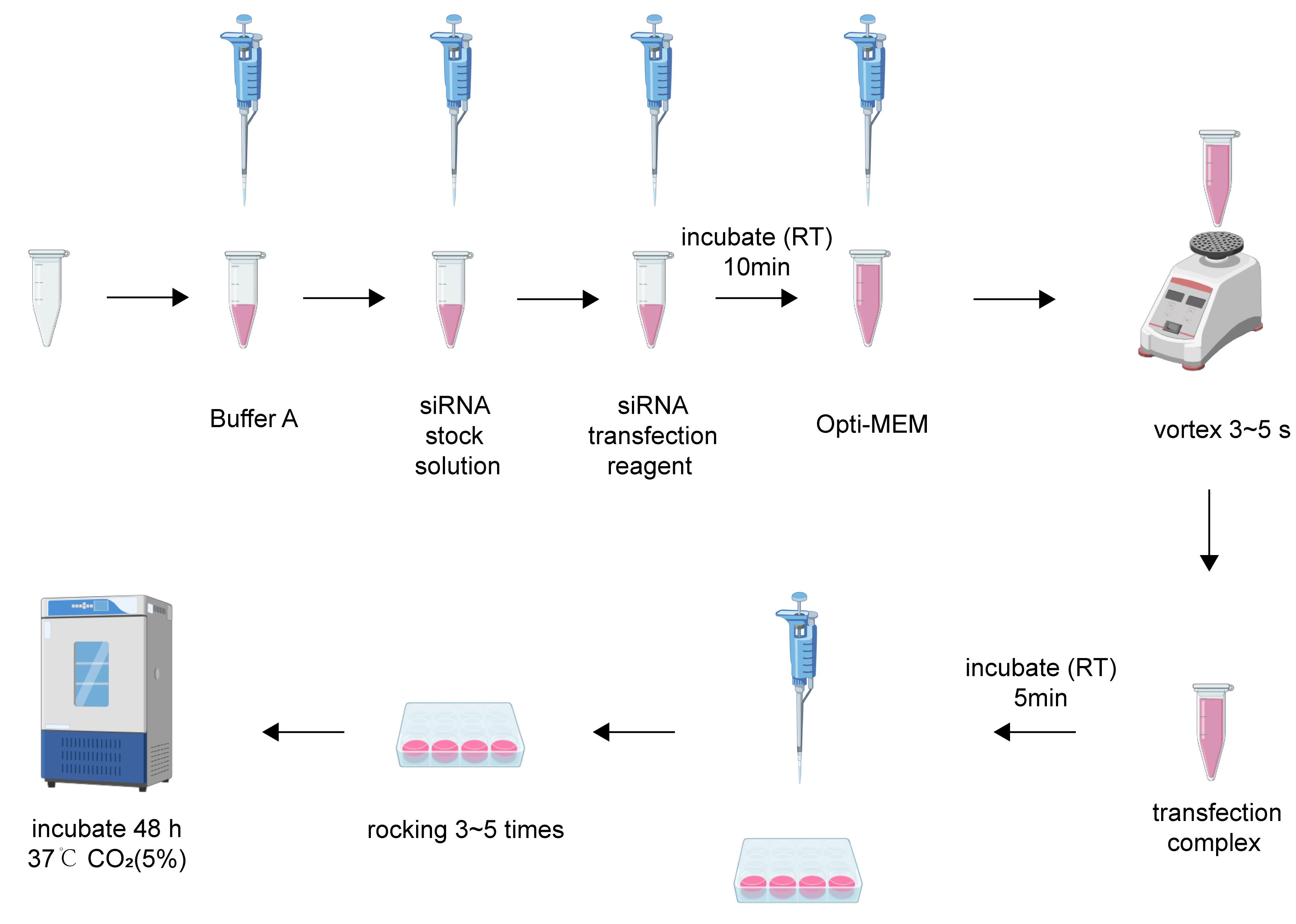
Figure 1. Steps of siRNA transfection
C. Primary neuron transfection with LipofectamineTM RNAiMAX transfection reagent (Thermo Fisher Scientific)
1. Perform transfection when neurons have been in culture for 4–5 days in vitro (DIV4–5).
2. Dilute Lipofectamine® RNAiMAX reagent in Opti-MEM medium (Table 2).
3. Dilute siRNA-standard negative control (siNC) or siPHGDH in Opti-MEM.
4. Combine the diluted siRNA with the diluted Lipofectamine® RNAiMAX reagent at a 1:1 ratio.
5. Incubate the mixture for 5 min at room temperature.
6. Add the siRNA–lipid complex dropwise to the neuronal culture.
7. After 6–8 h, replace the medium containing the transfection mixture with prewarmed neurobasal medium.
Note: Table 2 specifies reagent volumes for different culture plate formats.
Table 2. Transfection complex preparation of LipofectamineTM RNAiMAX transfection reagent
| Plates | siRNA stock solution | LipofectamineTM RNAiMAX transfection reagent | Opti-MEM medium |
|---|---|---|---|
| 24-well | 1.5 μL | 1.5 μL | 25 μL |
| 12-well | 3 μL | 3 μL | 50 μL |
| 6-well | 6 μL | 6 μL | 100 μL |
D. Analyze neuronal viability and transfection efficiency
1. Cell viability analysis: Analyze neuronal morphology by microscopy 48 h post-transfection.
Note: Neurons transfected with Lipofectamine® RNAiMAX reagent exhibited extensive cell death, whereas neurons transfected with the siRNA transfection reagent from Kermey displayed normal morphology. To further assess cell viability, we performed a CCK-8 assay. Primary cortical neurons seeded in 48-well plates were subjected to transfection. After 48 h, CCK-8 reagent was added to each well, followed by incubation for an additional 4–6 h. We measured the absorbance (OD) at 450 nm using a SpectraMax ABS microplate reader to evaluate cell viability. As shown in Figure 2, cell viability in the Lipofectamine RNAiMAX group was reduced to approximately 50% compared to the control group, whereas only a slight decrease was observed with the Kermey siRNA transfection reagent.
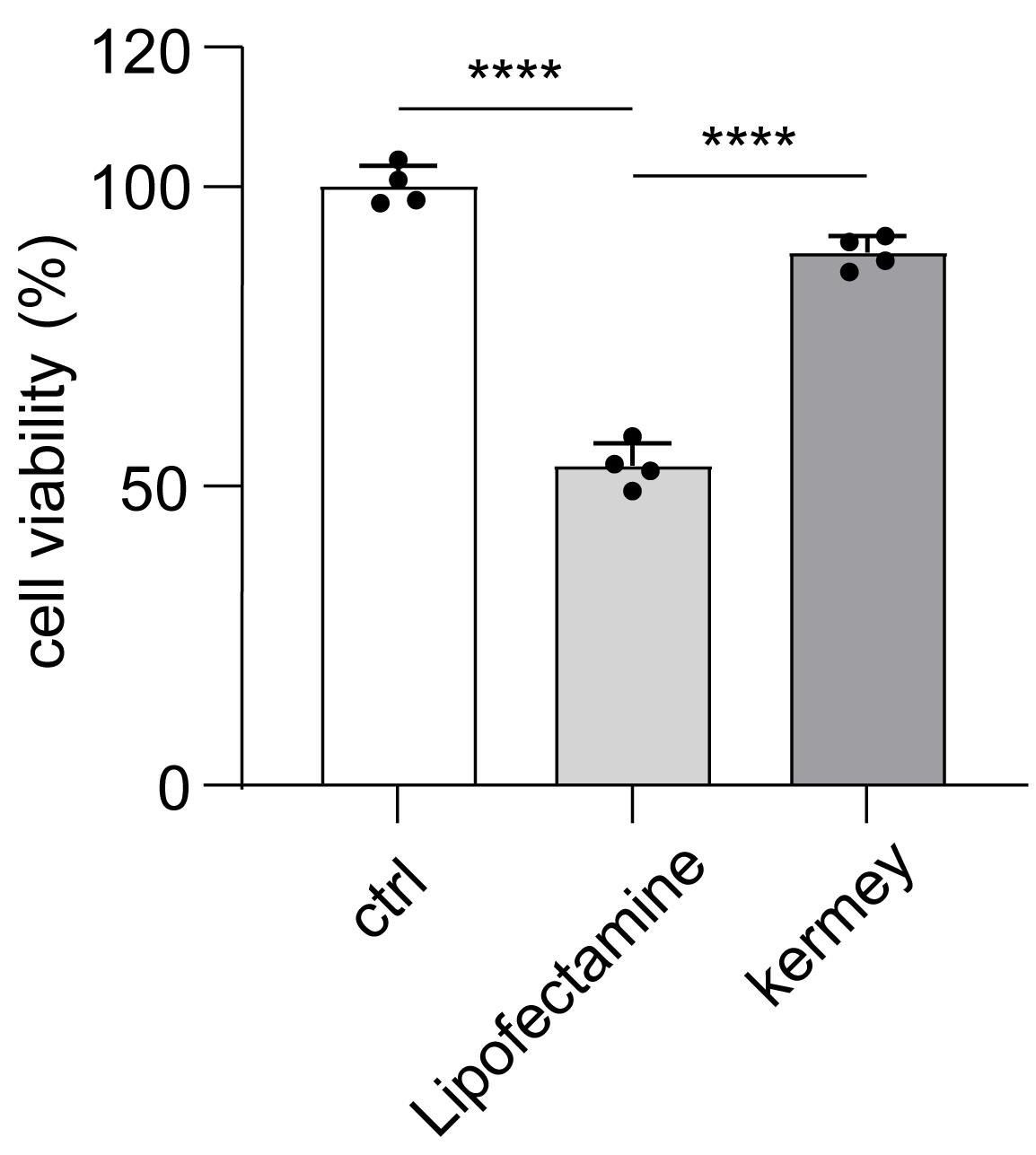
Figure 2. Cell viability analysis of neurons after transfection with siNC using Kermey transfection reagent for 48 h. n = 4. The data are means ± SD, for all panels: *P < 0.05, **P < 0.01, ***P < 0.001, n.s. no significance by one-way ANOVA analysis.
2. siRNA transfection efficiency analysis
a. Forty-eight hours after transfecting neurons with fluorescently labeled siRNA using the Kermey reagent, remove the cell-covered coverslips from the incubator and rinse them three times with PBS (1×), allowing 5 min per wash.
b. Next, fix the samples with 4% PFA for 10 min, followed by three additional 5-min washes in PBS.
c. Permeabilize the cells with 0.5% Triton X-100 for 10 min and wash three times with PBS, each for 5 min.
d. Afterward, block nonspecific binding sites by incubating the samples in 5% BSA for 1 h. Carefully lift the coverslips from the wells using a bent 1 mL syringe needle, and invert them cell-side down onto a glass slide precoated with a drop of DAPI-containing mounting medium. Seal the edges of the coverslip with fingernail polish and allow the slide to dry completely at room temperature for 5–10 min.
e. Finally, image the samples using a Nikon C2 confocal microscope following standard operating protocols.
Note: Most neurons were successfully transfected with negative control siRNA (Figure 3B), as indicated by the evenly distributed green fluorescence throughout the cytoplasm, demonstrating the effective delivery of siRNA using the Kermey transfection reagent.
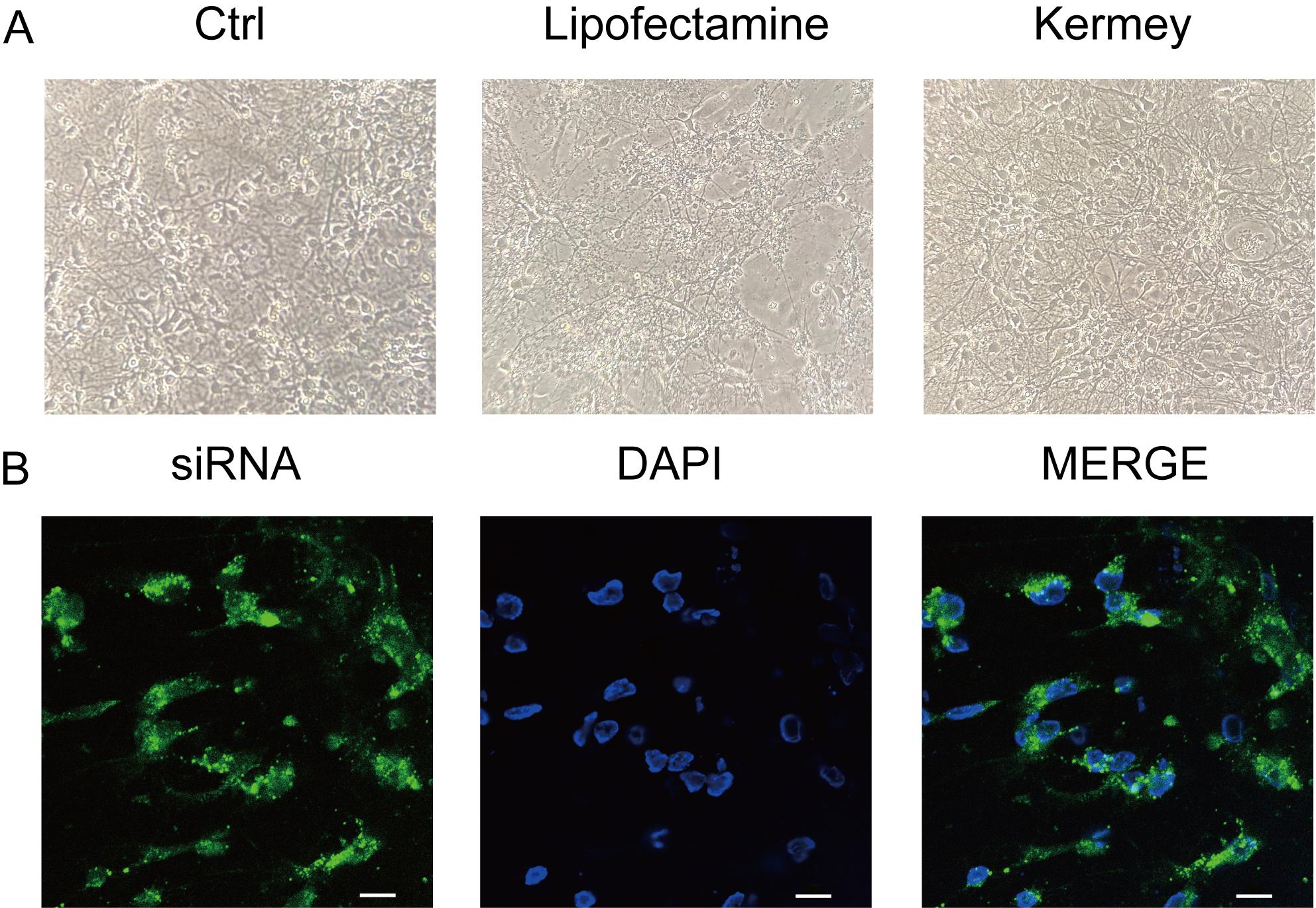
Figure 3. Morphology of primary cortical neurons and siRNA transfection efficiency. (A) Cortical neurons 48 h after transfection with siNC using Kermey transfection reagent or Lipofectamine® RNAiMAX reagent. (B) Confocal microscopy images showing cortical neurons transfected with fluorescent dye-labeled siRNA using Kermey transfection reagent. The siRNA is localized in the cytoplasm (green), while nuclei are stained with DAPI. Scale bars represent 10 μm. The data are representative of three independent experiments.
3. Analysis of gene targeting efficiency
To analyze gene knockdown efficiency, primary neurons were transfected with a control siRNA (siNC) and a validated siRNA targeting PHGDH (siPHGDH) [9], as described in section B. Furthermore, 3-phosphoglycerate dehydrogenase (PHGDH) is notable as a key enzyme that functions as the primary rate-limiting enzyme in the serine biosynthesis pathway, facilitating the conversion of 3-phosphoglycerate to 3-phosphohydroxypyruvate. After 48 h, we performed the immunoblot analysis. Total proteins were extracted from cells with lysis buffer containing protease (1% P6730) and phosphatase inhibitors (1% P1260). The proteins were separated by SDS-PAGE and transferred to PVDF membranes. The membranes were blocked with 5% skim milk in TBST for 2 h at room temperature, followed by overnight incubation with primary antibodies and a subsequent 1-h incubation with secondary antibodies. Then, membranes were incubated with primary antibodies overnight and secondary antibodies for 1 h. Membranes were exposed to a chemiluminescence detection system. The protein abundance of PHGDH was significantly reduced in neurons transfected with siPHGDH, compared with neurons transfected with siNC (Figure 4).

Figure 4. siRNA transfection reagent from Kermey can effectively knock down the expression of target proteins in neurons. Representative immunoblot analysis of PHGDH expression in primary cultured neurons after transfection with siNC and siPHGDH by using the transfection reagent from Kermey. The data are representative of three independent experiments.
Data analysis
Note: Data are processed using Microsoft Office Excel and analyzed using GraphPad Prism software.
1. Cell viability is determined by measuring the OD at 450 nm. Percent over control was calculated as a measure of cell viability. Calculation is as follows:
Cell survival rate = [(As-Ab)/(Ac-Ab)] × 100%
As: Absorbance of the experimental well (containing cell culture medium, CCK-8, and the test drug).
Ac: Absorbance of the control well (containing cell culture medium, CCK-8, without the test drug).
Ab: Absorbance of the blank well (containing culture medium without cells and the test drug, with CCK-8).
2. We analyzed the western blot results using ImageJ by calculating the ratio of PHGDH band intensity to the corresponding actin band intensity for both the experimental and control groups.
3. One-way ANOVA analysis was used to analyze the difference between the control, lipofectamine, and Kermey groups.
4. All experiments were performed three times unless otherwise indicated.
Validation of protocol
To validate the protocol, we evaluated neuronal morphology, siRNA transfection efficiency, and target gene expression following gene knockdown in neurons. Notably, neurons transfected using Lipofectamine® RNAiMAX exhibited extensive cell death, whereas those transfected with the siRNA transfection reagent from Kermey maintained normal morphological features (Figure 3A). Furthermore, confocal microscopy revealed efficient transfection, with the majority of neurons successfully delivering the negative control siRNA (Figure 3B). Additionally, western blot analysis confirmed a significant reduction in PHGDH expression in neurons transfected with siPHGDH, compared to those treated with siNC (Figure 4).
General notes and troubleshooting
General notes
1. During the 24–72 h post-transfection period, complete medium replacement is unnecessary when using Kermey transfect reagent. However, partial medium supplementation should be performed as needed to compensate for evaporation, particularly at the edges of the culture vessel.
2. This protocol has been validated exclusively in primary neuron cultures. The transfection efficiency and cellular viability parameters have not been tested in other neural cell types, including but not limited to astrocytes, microglia, oligodendrocytes, or neural progenitor cells.
3. The earliest successful transfection was achieved at DIV3. For earlier timepoints, protocol optimization may be necessary.
4. This procedure is compatible with media heated to 37 °C and subsequently equilibrated to room temperature.
5. When initiating transfections, it is recommended to assess transfection efficiency. Concurrently, select cells that exhibit strong transfection signals and appear morphologically healthy.
6. Use RNase-free pipette tips during transfection to prevent RNA degradation.
Troubleshooting
Problem 1: The siRNA-mediated knockdown effect was not significant.
Possible cause: Variations in cell density and physiological states across cultures might compromise transfection efficacy, as high-density or confluent regions may require higher siRNA concentrations than those specified in our current protocol to achieve effective gene silencing.
Solution: When working with high-density cultures, prepare the siRNA-transfection mixture at an increased concentration (e.g., 1.5× or 2× concentration) while preserving the optimal reagent ratios. However, if cells exhibit poor health prior to transfection, it is strongly recommended to first optimize the cell culture conditions, such as adjusting plating density, refining medium composition, and ensuring strict control of incubation parameters (37 °C, 5% CO2 with humidity regulation) to improve overall cellular fitness and transfection responsiveness.
Problem 2: Neurons exhibit low viability.
Possible cause: Low cell density may impair siRNA uptake, while excessive siRNA concentrations can induce cytotoxicity, leading to reduced neuronal survival.
Solution: Increase cell number or decrease siRNA concentration.
Acknowledgments
W.X. designed and performed most experiments, analyzed data, and prepared the manuscript; L.Y. helped with neuronal culture; and Y.C. and Z.Z. conceptualized the research, directed the study, and prepared the manuscript. All authors revised the manuscript. This work was supported by the Science and Technology Support Plan for Youth Innovation of Colleges and Universities of Shandong Province of China (2022KJ146), National Natural Science Foundation of China (32470984), and Natural Science Foundation of Shandong Province (ZR2024MH302).
Competing interests
The authors declare no conflicts of interest.
Ethical considerations
Male 9~10 weeks C57BL/6 mice were obtained from Ji’nan Pengyue Laboratory Animal Breeding Co., Ltd. (China). The mice were housed under specific pathogen-free conditions with a constant temperature and humidity on a 12 h light/dark cycle. Food and water were given ad libitum.
All animal experiments were conducted in compliance with National Institutes of Health guidelines and were approved by the institutional animal care and use committee of Qingdao University (QYFY WZLL 30494). All the animal experiments conform to the ARRIVE guidelines.
References
- Pfisterer, U. and Khodosevich, K. (2017). Neuronal survival in the brain: neuron type-specific mechanisms. Cell Death Dis. 8(3): e2643–e2643. https://doi.org/10.1038/cddis.2017.64
- Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M. and Brown, G. C. (2018). Neuronal Cell Death. Physiol Rev. 98(2): 813–880. https://doi.org/10.1152/physrev.00011.2017
- Dana, H., Chalbatani, G. M., Mahmoodzadeh, H., Gharagouzlo, E., Karimloo, R., Rezaiean, O., Moradzadeh, A., et al. (2017). Molecular Mechanisms and Biological Functions of siRNA. Int J Biomed Sci. 13(2): 48–57. https://doi.org/10.59566/ijbs.2017.13048
- Tseng, S., Chen, P. and Hwang, E. (2025). Primary Neuronal Culture and Transient Transfection. Bio Protoc. 15(1363): e5169. https://doi.org/10.21769/bioprotoc.5169
- Marwick, K. F. M. and Hardingham, G. E. (2017). Transfection in Primary Cultured Neuronal Cells. Methods Mol Biol. 2799: 137–144. https://doi.org/10.1007/978-1-4939-7321-7_6
- Xia, H., Mao, Q., Paulson, H. L. and Davidson, B. L. (2002). siRNA-mediated gene silencing in vitro and in vivo. Nat Biotechnol. 20(10): 1006–1010. https://doi.org/10.1038/nbt739
- Karra, D. and Dahm, R. (2010). Transfection Techniques for Neuronal Cells: Table 1. J Neurosci. 30(18): 6171–6177. https://doi.org/10.1523/jneurosci.0183-10.2010
- Zeitelhofer, M., Vessey, J. P., Xie, Y., Tübing, F., Thomas, S., Kiebler, M. and Dahm, R. (2007). High-efficiency transfection of mammalian neurons via nucleofection. Nat Protoc. 2(7): 1692–1704. https://doi.org/10.1038/nprot.2007.226
- Lv, M., Duan, Z., Tan, J., Liu, J., Wang, Q., Wang, C., Zhang, Z., Sun, X., Liu, R., Cui, Y., et al. (2025). PHGDH-mediated serine synthesis in astrocytes supports neuroinflammation by sustaining NADH level to promote histone acetylation. Cell Death Dis. 16(1): e1038/s41419–025–07732–8. https://doi.org/10.1038/s41419-025-07732-8
Article Information
Publication history
Received: Oct 9, 2025
Accepted: Dec 10, 2025
Available online: Dec 26, 2025
Published: Jan 20, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Wang, X., Li, Y., Sun, X., Cui, Y. and Zhang, Z. (2026). A Highly Efficient siRNA Transfection Method in Primary Cultured Cortical Neurons. Bio-protocol 16(2): e5567. DOI: 10.21769/BioProtoc.5567.
Category
Neuroscience > Cellular mechanisms
Cell Biology > Cell-based analysis > Gene expression
Molecular Biology > RNA > RNA interference
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.