- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Creating Loss-of-Function Mutants of Neurospora crassa Using a Novel CRISPR/Cas9 System
Published: Vol 16, Iss 1, Jan 5, 2026 DOI: 10.21769/BioProtoc.5562 Views: 213
Reviewed by: Alba BlesaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2024

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






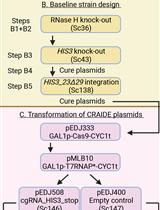

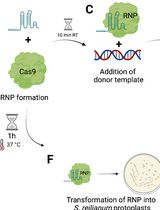
Abstract
Since its introduction, the CRISPR/Cas9 system has been used in many organisms for precise and rapid genome editing, as well as for editing multiple genes at once. This targeted mutagenesis makes it easy to analyze the function of a gene of interest (goi). The standard method for genetic manipulation of the model organism Neurospora crassa has been homologous recombination. It is well established and widely used to create knock-out or overexpression mutants. The recently developed CRISPR/Cas9 system is an addition to the toolkit for genetically manipulating N. crassa. For this protocol, a strain stably expressing the Cas9 endonuclease is required. After designing the gRNA with the online tool CHOP-CHOP, a synthetic gRNA is used to transform macroconidia via electroporation. Combining the goi-gRNA with a gRNA targeting the csr-1 gene as a selection marker allows for easy identification of colonies with mutations at the target site of the goi, since the obtained resistance to Cyclosporin A (CsA) allows for selecting editing events. The mutation type can be detected by PCR of the edited gene region followed by Sanger sequencing. This system is fast and easy to handle, offering an attractive alternative to homologous recombination, especially for targeting multiple genes simultaneously.
Key features
• This protocol allows the use of CRISPR/Cas9 in Neurospora crassa to create loss-of-function mutants.
• It can be used to create loss-of-function mutants of multiple genes in one round of transformation.
• Time-saving mutagenesis without the need for vector construction.
• In combination with csr-1 as a selection marker, the screening for successfully edited genes of interest is reduced.
Keywords: CRISPR/Cas9Graphical overview
Background
Neurospora crassa is a widely used eukaryotic model organism because of its ease of growth, genetic accessibility, and rapid life cycle. It is commonly employed in genetic research, where its genome is modified to study gene functions [1].
Traditionally, random mutagenesis methods such as UV exposure or chemicals have been used for genetic modification, which causes multiple unpredictable mutations. Identifying these mutations is time-consuming, and pinpointing the exact mutation responsible for a phenotype is difficult. Therefore, targeted mutagenesis is preferred. In N. crassa, homologous recombination (HR) has become the preferred method for creating targeted gene knockouts or overexpression lines. This involves transforming the organism with a vector containing homologous flanking sequences, the gene of interest, and/or a selection marker [2]. For higher efficiency, strains lacking nonhomologous end joining (NHEJ) are often used [3,4]. After transformation and the generation of homokaryotic transformants, validation is performed via PCR or Southern blot [3,5]. The whole process can take up to seven weeks, is labor-intensive, and is less efficient if NHEJ-deficient strains are not used [6]. The CRISPR/Cas9 system offers a faster and more precise alternative. It involves the Cas9 endonuclease and a guide RNA (gRNA) that directs editing to specific DNA sequences. When Cas9 induces a double-strand break, the cell repairs it either through NHEJ, which may introduce small insertions or deletions (indels), or homology-directed repair (HDR), which allows for the integration of new DNA sequences when a suitable donor DNA is available [7].
Although the CRISPR/Cas9 system has been adapted for various fungi [8], it has not yet been widely used in N. crassa. Another established system for N. crassa requires co-transforming multiple plasmids carrying Cas9, gRNA, and donor DNA, making it complex and labor-intensive [9]. In contrast, the system described here simplifies this process by integrating Cas9 directly into the genome and introducing naked synthetic gRNA via electroporation of macroconidia, eliminating the need to clone and co-transform Cas9/gRNA plasmids [10].
One concern with stable Cas9 expression is the potential for off-target mutations [11]. Studies in fungi suggest that limiting the duration of Cas9-gRNA complex presence can reduce this risk [12,13]. Even if Cas9 remains in the cells, controlling gRNA expression helps minimize off-target effects. To further reduce potential off-target mutations after successful mutagenesis, outcrossing the Cas9 sequence may be advisable. Co-transformation of multiple gRNAs enables simultaneous editing events at once. This ability allows the use of the csr-1 gene as a selection marker in N. crassa. It involves co-transforming a gRNA targeting csr-1 with a gRNA targeting the gene of interest (goi). If csr-1 is successfully edited and becomes non-functional, the fungus exhibits resistance to Cyclosporin A (CsA). This resistance allows selection for edited strains using CsA-containing media, simplifying the identification process. Additionally, mutations in csr-1 typically produce homokaryons [14]. Traditionally, isolating homokaryotic strains involves time-consuming steps such as crossing, microconidia isolation, or serial transfers of macroconidia. However, these steps can be bypassed by using the CRISPR/Cas9 system with csr-1 as a marker. Overall, this approach offers a time- and effort-saving, user-friendly method for generating loss-of-function mutants in N. crassa.
Materials and reagents
Biological materials
1. Neurospora crassa strain NcCas9SG [available at the Fungal Genetics Stock Center (FGSC); reference #27376]
Reagents
1. Sucrose (Roth, CAS number: 57-50-1)
2. Agar-agar (Roth, CAS number: 9002-18-0)
3. Citric acid (Roth, CAS number: 5949-29-1)
4. ZnSO4·7H2O (Merck, CAS number: 7446-20-0)
5. Fe(NH4)2(SO4)2·6H2O (Merck, CAS number: 7783-85-9)
6. CuSO4·5H2O (Merck, CAS number: 7758-99-8)
7. MnSO4·H2O (Roth, CAS number: 10034-96-5)
8. H3BO3 (Roth, CAS number: 10043-35-3)
9. Na2MoO4·2H2O (Merck, CAS number: 10102-40-6)
10. Chloroform (Roth, CAS number: 67-66-3)
11. Na3-Citrate·2H2O (Roth, CAS number: 6132-04-3)
12. KH2PO4 (Roth, CAS number: 7778-77-0)
13. NH4NO3 (Roth, CAS number: 6484-52-2)
14. MgSO4·7H2O (Merck, CAS number: 10034-99-8)
15. CaCl2·2H2O (Roth, CAS number: 10035-04-8)
16. Biotin (Roth, CAS number: 58-85-5)
17. TRIS (Roth, CAS number: 77-86-1)
18. NaCl (Roth, CAS number: 7647-14-5)
19. KCl (Roth, CAS number: 7447-40-7)
20. EtOH p.a. (Roth, CAS number: 64-15-7)
21. EDTA (Roth, CAS number: 6381-92-6)
22. Na-iodoacetate (Merck, CAS number: 305-53-3)
23. KNO3 (Roth, CAS number: 7757-79-1)
24. K2HPO4 (Roth, CAS number: 7758-11-4)
25. KOH (Roth, CAS number: 1310-58-3)
26. Cyclosporin A (biomol, catalog number: AG-CN2-0079-M100)
27. Sorbitol (Roth, CAS number: 50-70-4)
28. Tween 80 (Roth, CAS number: 9005-65-6)
29. Alt-RTM CRISPR-Cas9 tracrRNA (20 nmol) (IDT, catalog number: 1072533)
30. Duplex buffer (IDT, catalog number: 11-01-03-01)
Solutions
1. 50× Vogel’s solution (see Recipes)
2. Trace elements solution (TES) (see Recipes)
3. Vogel’s minimal medium + sucrose (VMM+S) (see Recipes)
4. Vogel’s minimal medium + sorbose-fructose-glucose + cyclosporin A (VMM+SGF+CsA) (see Recipes)
5. Sorbose-fructose-glucose solution (SGF) (see Recipes)
6. Top agar (see Recipes)
7. TPS buffer (see Recipes)
8. MKM medium (see Recipes)
9. 2× Synthetic crossing (SC) solution (see Recipes)
11. 1 M sorbitol + 0.25% Tween 80 (see Recipes)
Recipes
1. 50× Vogel’s solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Na3-Citrate·2H2O | 510 mM | 75 g |
| KH2PO4 | 1.84 M | 125 g |
| NH4NO3 | 1.25 M | 50 g |
| MgSO4·7H2O | 40.6 mM | 5 g |
| CaCl2·2H2O* | 34 mM | 2.5 g |
| TES | 1% (v/v) | 5 mL |
| Biotin | 0.1 mM | 12.5 mg |
| Chloroform** | 0.2% (v/v) | 1 mL |
| Autoclaved ddH2O | n/a | Add up to 500 mL |
| Total | n/a | 500 mL |
*Before adding, dissolve in 50 mL of ddH2O.
**Chloroform is added as a preservative and is optional.
Store Vogel’s solution in the dark at room temperature (RT).
2. Trace element solution (TES)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Citric acid | 5% (w/v) | 5 g |
| ZnSO4·7H2O | 5% (w/v) | 5 g |
| Fe(NH4)2(SO4)2·6H2O | 1% (w/v) | 1 g |
| CuSO4·5H2O | 0.25% (w/v) | 250 mg |
| MnSO4·H2O | 0.05% (w/v) | 50 mg |
| H3BO3 | 0.05% (w/v) | 50 mg |
| Na2MoO4·2H2O | 0.05% (w/v) | 50 mg |
| Chloroform* | 0.2% (v/v) | 0.2 mL |
| Autoclaved ddH2O | n/a | Add up to 100 mL H2O |
| Total | n/a | 100 mL |
*Chloroform is added as a preservative and is optional.
Store TES at 4 °C.
3. Vogel’s minimal medium + sucrose (VMM+S)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 50× Vogel’s solution | 1× | 10 mL |
| Sucrose | 20 g/L | 10 g |
| Agar-agar | 2% (w/v) | 10 g |
| ddH2O | n/a | Add up to 500 mL |
| Total | n/a | 500 mL |
The pH of the minimal medium is about 5.8. No adjustment is necessary.
Autoclave at 112 °C. Pour into flasks or slant agar tubes on a sterile workbench. Allow to cool and set at RT. Store at 4 °C until needed.
4. Vogel’s minimal medium + sorbose-fructose-glucose + cyclosporin A (VMM+SGF+CsA)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 50× Vogel’s solution | 1× | 10 mL |
| Agar-agar | 2% (w/v) | 10 g |
| ddH2O | n/a | Add up to 475 mL |
| SGF* | 5% (v/v) | 25 mL |
| Cyclosporin A (5 mg/mL)* | 5 μg/mL | 500 μL |
| Total | n/a | 500 mL |
*Add after autoclaving.
Autoclave at 121 °C, mix, and pour into 92 mm × 16 mm Petri dishes in a sterile workbench. Allow to cool and set at RT. Store at 4 °C until needed.
5. Sorbose-fructose-glucose solution (SGF)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Sorbose | 20% (w/v) | 20 g |
| Fructose | 0.5% (w/v) | 0.5 g |
| Glucose | 0.5% (w/v) | 0.5 g |
| ddH2O | n/a | Add up to 100 mL |
| Total | n/a | 100 mL |
Autoclave at 112 °C. Let it cool down at RT. Store at 4 °C until needed.
6. Top agar
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 50× Vogel’s solution | 1× | 2 mL |
| Agarose | 1% (w/v) | 1 g |
| ddH2O | n/a | Add up to 95 mL |
| SGF* | 5% (v/v) | 5 mL |
| Cyclosporin A (5 mg/mL)* | 5 μg/mL | 100 μL |
| Total | n/a | 100 mL |
*Add after autoclaving.
Autoclave at 121 °C, add SGF, and keep at 45 °C to avoid solidification.
7. TPS buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 1 M Tris-HCl, pH 8.0 | 100 mM | 10 mL |
| 0.5 M EDTA, pH 8.0 | 10 mM | 2 mL |
| KCl | 1 M | 7.55 g |
| ddH2O | n/a | Add up to 100 mL |
| Total | n/a | 100 mL |
Autoclave at 121 °C or filter sterilize. Store at RT.
8. MKM medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Sucrose | 0.5% (w/v) | 2 g |
| 2× SC solution (Recipe 9) | 0.1× | 10 mL |
| 100 mM Na-Iodoacetate solution* | 1 mM | 2 mL |
| Agar-agar | 2% (w/v) | 4 g |
| ddH2O | n/a | Add up to 198 mL |
| Total | n/a | 200 mL |
*Add after autoclaving.
Autoclave at 112 °C. After cooling to 45 °C, add the Na-Iodoacetate and pour into 92 mm × 16 mm Petri dishes in a sterile workbench. Allow to cool and set at RT. Store at 4 °C until needed.
9. 2× Synthetic crossing (SC) solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| KNO3 | 19.8 mM | 400 mg |
| K2HPO4 | 8 mM | 280 mg |
| KH2PO4 | 7.34 mM | 200 mg |
| MgSO4·7H2O | 8 mM | 400 mg |
| NaCl | 3.4 mM | 40 mg |
| CaCl2·2H2O | 1.4 mM | 40 mg |
| Biotin | 0.04 mM | 2 mg |
| TES | 0.02% (v/v) | 40 μL |
| Chloroform* | 0.5% (v/v) | 1 mL |
| Autoclaved ddH2O | n/a | Add up to 200 mL |
| Total | n/a | 200 mL |
*Chloroform is added as a preservative and is optional.
Store at 4 °C in the dark.
10. 1 M Sorbitol + 0.25 % Tween 80
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Sorbitol | 1 M | 18,2 g |
| Tween 80 | 0.25 % (v/v) | 0.25 mL |
| ddH2O | n/a | Add up to 100 mL |
| Total | n/a | 100 mL |
Autoclave at 112 °C. Store at RT.
Laboratory supplies
1. Erlenmeyer Flask, wide neck, 200 mL (Roth, catalog number: X737.1)
2. 1.5 mL Eppendorf tube (Sarstedt, catalog number: 72.690.001)
3. 2 mL Eppendorf tube (Sarstedt, catalog number: 72.695.500)
4. 50 mL Sarstedt tube (Sarstedt, catalog number: 62.547.254)
5. 1,000 μL pipette tips (Sarstedt, catalog number: 70.3050.020)
6. 200 μL pipette tips (Sarstedt, catalog number: 70.3030.020)
7. 10 μL pipette tips (Sarstedt, catalog number: 70.3010)
8. Petri dish, 92 mm × 16 mm (Sarstedt, catalog number: 82.1473.001)
9. Electroporation cuvette, 0.2 cm (Roth, catalog number: PP39.1)
10. Gauze (from medical supplier)
11. Funnel 65 mm (Roth, catalog number: HY46.1)
12. Parafilm M (Roth, catalog number: CNP8.1)
13. Glass test tube with beaded rim, 180 mm × 18 mm (Roth, catalog number: C209.1)
14. Glass test tube with no rim, 130 mm × 16 mm (Roth, catalog number: C188.1)
15. Cellophane made from viscose (any online supplier)
16. Whatman paper 0.35 mm thick (Roth, catalog number: CL66.1)
Equipment
1. PCR machine (e.g., SensoQuest Labcycler, biolab, catalog number: 11-011-103-XXX)
2. Thoma counting chamber (Roth, catalog number: T732.1)
3. Centrifuge (e.g., Beckmann Coulter, model: Allegra X-30R)
4. Electroporation device (e.g., Bio-Rad, model: Genepulser II)
5. Pipettes (Eppendorf)
6. Lancet needle (Roth, catalog number: KY00.1)
7. Inoculation loop (Roth, catalog number: KL99.1)
8. Heating block (e.g., Eppendorf, model: Thermomixer comfort)
9. NanoDrop
Software and datasets
1. CHOP-CHOP tool (https://chopchop.cbu.uib.no/) [15]; online tool used for gRNA design
Procedure
A. Designing gRNA and duplex formation
1. Go to the website of the CHOP-CHOP tool (https://chopchop.cbu.uib.no/) for the design of the crRNA (see Figure 1A).

Figure 1. Example for designing a crRNA using the online tool CHOP-CHOP for the csr-1 gene of Neurospora crassa. (A) Define the parameters for the design of the crRNA. Define the target (csr-1), the organism (N. crassa), the endonuclease (Cas9), and the purpose of the editing (knock-out). (B) The result page, showing possible target sequences with the PAM in red, the genomic location as a number and on the gene (green: good; yellow: moderate; red: not good), the targeted strand (+/-), the GC content, self-complementarity, number of off-targets (MM0-MM3; 0 = no mismatches, 1 = one mismatch, and so on), and efficiency. (C) Detailed results for a chosen target sequence with information on the predicted editing site (light blue rectangle), oligonucleotides (purple) that can be used for the PCR analysis of the target site after editing, information on off-target sites, and prediction of possible outcomes according to Shen et al. [16].
2. Enter the RefSeq/ENSEMBL/gene ID/genomic coordinates or FASTA sequence of the gene to be edited in the Target field.
3. Choose the Neurospora crassa genome in the In field.
4. Choose CRISPR/Cas9 in the Using field.
5. In the For field, choose knock-out.
6. Then, click on Find Target Sites!
7. Various target sequences will be visualized with their location in the gene sequence as green (good), yellow (moderate), and red (bad) bars (see Figure 1B).
8. For a loss-of-function mutation, make sure that the gRNA binding site is:
a. Not too close to the start codon (at least 50 bp downstream) to avoid usage of alternative start codons.
b. Not too close to the stop codon (at least 300 bp upstream) to avoid generating a truncated but still functional protein.
c. Inside an exon.
Note: For detailed instructions on gRNA design with CHOP-CHOP, see Labun et al. [17].
9. In addition to the visualization, there is a table summarizing the target sequence features (see Figure 1B). Make sure the following parameters are given for your chosen sequence:
a. GC content of 40%–70%.
b. No self-complementarity.
c. No off-target effects (the MM0-MM4 values show the number of off-targets).
10. Select your desired gRNA sequence. This will lead to the individual results page with more information on the selected target sequence (see Figure 1C).
Critical: The sequence result also contains the PAM sequence. Keep in mind that the gRNA should not include the PAM.
Note: This protocol describes the use of crRNA and tracrRNA ordered from IDT Integrated DNA Technologies (https://eu.idtdna.com/pages/products/crispr-genome-editing/alt-r-crispr-cas9-system) to synthesize the gRNA. If you decide to use a different method for the synthesis of your gRNA, follow the respective instructions.
11. Order lyophilized Alt-RTM CRISPR-Cas9 crRNA (10 nmol) and Alt-RTM CRISPR-Cas9 tracrRNA (20 nmol) (IDT, catalog number: 1072533) at IDT.
Note: Ordering crRNA and tracrRNA separately is economically preferred over ordering a gRNA for each gene of interest. The tracrRNA can be used for several crRNAs, making it cheaper when editing more than one gene.
12. Reconstitute the crRNA and tracrRNA to 200 μM with IDT duplex buffer. In case of 10 nmol crRNA, add 50 μL of IDT duplex buffer. For 20 nmol tracrRNA, add 100 μL of IDT duplex buffer.
13. Mix the two RNAs in an equimolar ratio, e.g., 10 μL of each RNA, and incubate for 5 min at 95 °C in a heating block.
14. Then, let it cool down to room temperature to form the crRNA:tracrRNA duplex.
Pause point: Either use the gRNA directly for transformation or store it at -20 °C, where it remains stable for up to two years.
B. Isolation of macroconidia
1. Prepare a 200 mL flask with 50 mL of VMM+S agar and close it with a sterile cotton plug.
2. Inoculate the flask with some macroconidia of the N. crassa strain NcCas9SG and incubate for 14–20 days at 25 °C in a light/dark cycle (16 h light, 8 h dark) in a climate chamber.
3. After incubation, carefully pour 40 mL of 1 M sorbitol + 0.25% (v/v) Tween 80 into the flask. Let the solution run down the side of the flask to avoid stirring up the conidia.
4. Tightly seal the flask with parafilm and shake vigorously until all conidia are suspended.
5. Prepare a 50 mL Sarstedt tube with a sterile funnel lined with four layers of sterile gauze on top.
6. Pour the conidia suspension via the funnel and through the gauze into the Sarstedt tube to remove hyphae and agar from the suspension.
7. Centrifuge the suspension at 4,255× g for 5 min, either in a swing-out or fixed-angle rotor.
8. Remove the supernatant.
9. To wash the Tween out, fully resuspend the pellet in 50 mL of 1 M sorbitol by gently shaking the tube.
10. Centrifuge the suspension at 4,255× g for 5 min, either in a swing-out or fixed-angle rotor, and remove the supernatant.
11. Repeat steps B8–10 twice (three washing steps in total).
12. After the third washing step, resuspend the conidia in 500 μL of 1 M sorbitol and transfer them into a 2 mL Eppendorf tube.
13. Dilute the conidia 1:100 and determine their concentration by using a Thoma counting chamber. Count the conidia of four diagonally arranged big squares. To calculate the amount of conidia for 1 mL, use this formula: n/16 × 106 spores (see Figure 2).
14. Adjust the concentration to 2.5 × 109 spores/mL.
Pause point: The spores stay viable for up to three days when kept at 4 °C.
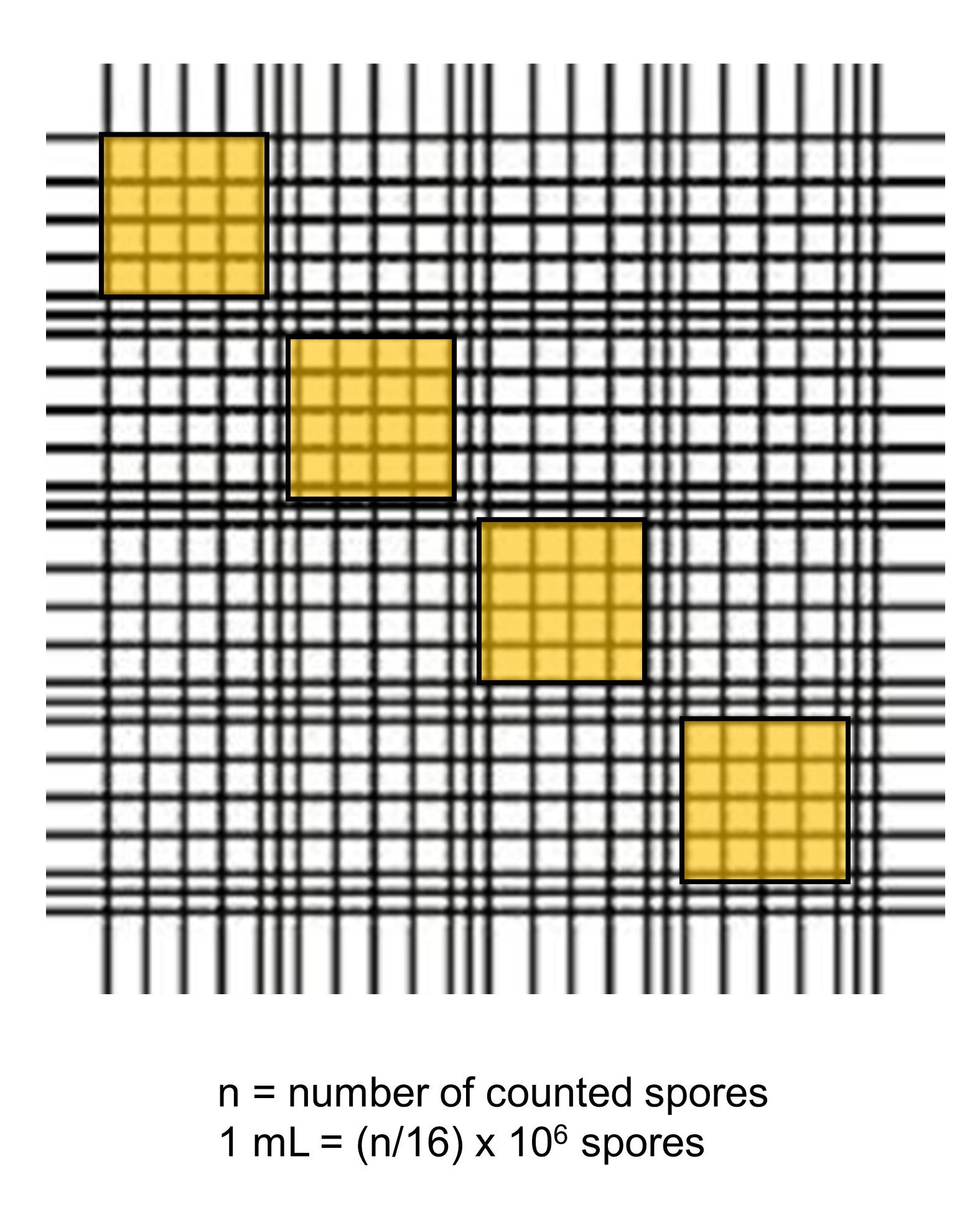
Figure 2. Scheme of the Thoma counting chamber. In yellow are the squares that should be counted. Then, use the given formula to determine the amount of conidia per milliliter.
C. Transformation of macroconidia with gRNA
1. Prechill the 0.2 cm electroporation cuvettes on ice.
2. Transfer 40 μL of the 2.5 × 109 spores/mL into a 1.5 mL Eppendorf cup for each transformation assay.
3. Mix the 40 μL suspension with 2 μL of 100 μM gRNA targeting the csr-1 gene (as a selection marker) and 2 μL of 100 μM target gRNA.
4. As a control, mix 40 μL of suspension with 2 μL of sterile H2O.
5. All samples need to be incubated on ice for 5 min. Then, transfer the samples into the chilled electroporation cuvette.
6. Use the following conditions for electroporation with the Bio-Rad Gene Pulser: 1.5 kV, 25 μF, and 600 Ω.
7. After the pulse, add 1 mL of 1 M sorbitol (prechilled) into the cuvette and mix carefully with a pipette. Leave the cuvettes at RT for at least 10 min before plating on VMM+SGF+CsA.
8. For plating, prepare top agar in glass test tubes (8 mL/tube) and keep it at 45 °C in a water bath.
Critical: The conidia are not viable at temperatures >45 °C.
9. Dilute the electroporated suspension 1:100 with 1 M sorbitol before plating.
10. Mix 200 μL of the dilution with 8 mL of top agar in the test tube by vortexing, then pour the top agar containing the macroconidia on the VMM+SGF+CsA plate.
11. Incubate the plate for 3–5 days at 25 °C under a light/dark cycle in the climate chamber until colonies form (see Figure 3). The average editing efficiency of this protocol lies between 7.35% and 11.89% (see [10]).
12. Transfer the colonies to slant agar tubes containing VMM+S medium and incubate in the climate chamber (25 °C, light/dark cycle of 8/16 h) for 3–5 days until macroconidia form.
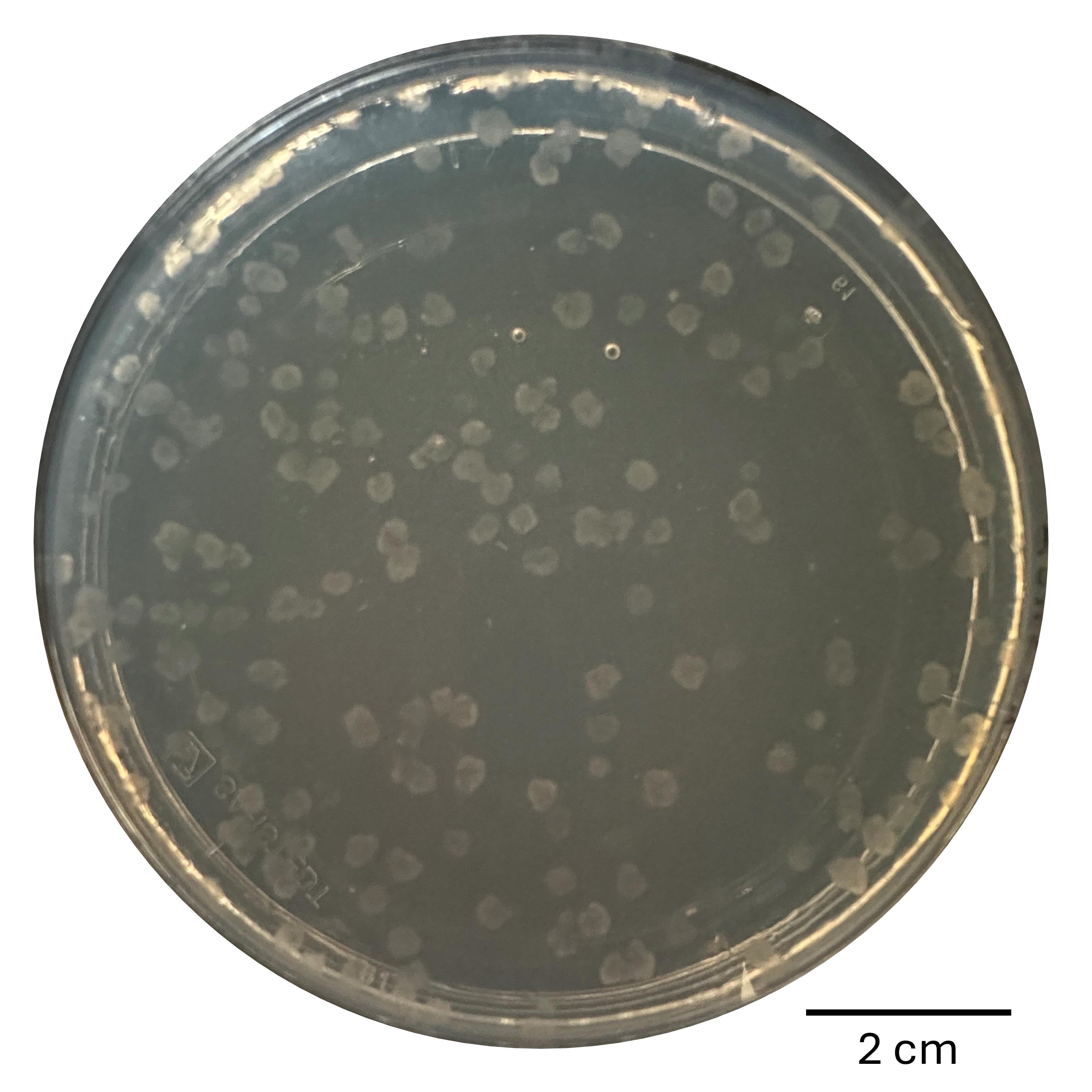
Figure 3. Example of colonies of N. crassa after transformation, plated on VMM+SGF+CsA. Cut out single colonies and transfer them onto slant agar tubes for further analysis.
D. Screening of editing event
1. Isolate DNA from the putative transformants using the quick method described here.
2. Collect conidia from the slant agar tubes with a sterile inoculation loop and transfer the conidia to a 1.5 mL Eppendorf cup filled with 160 μL of TPS buffer.
3. Incubate for 10 min in a heating block set to 99 °C. Alternatively, incubation for 10 min in a boiling water bath is also possible.
4. After incubation, transfer the samples immediately to ice.
5. Keep them on ice for at least 10 min.
6. Centrifuge the samples at 15,000× g for 5 min at RT.
7. Transfer 140 μL of the supernatant into a new 1.5 mL Eppendorf cup filled with 100 μL of sterile ddH2O, 6 μL of 4 M NaCl, and 480 μL of EtOH p.a.
8. Incubate at -20 °C for at least 20 min to precipitate the DNA.
Note: The precipitation of the DNA can be prolonged overnight.
9. Centrifuge the samples at 15,000× g for 15 min at 4 °C and discard the supernatant.
10. Wash the pellet with 800 μL of 70% EtOH by centrifuging at 15,000× g for 20 min at 4 °C.
11. Discard the supernatant and resuspend the pellet in 35 μL of 5 mM Tris-HCl, pH 8.0.
12. Measure the DNA concentration with a NanoDrop and bring the concentration to 15 ng/μL.
13. Use 15 ng of DNA for a PCR with your favored polymerase using oligonucleotides flanking the mutation site.
Note: Either use oligonucleotides designed by you or use one of the pairs recommended by CHOP-CHOP when designing the gRNA (see Figure 1C).
14. Sequence the obtained PCR product and check for mutations at the desired mutation site.
Note: In case of heterokaryons, proceed with microconidia passage (see section E) to obtain homokaryons.
E. Microconidia passage
1. Prepare MKM plates and cut the cellophane into circles that fit exactly inside the Petri dish (92 mm in diameter). Boil the cellophane in 800 mL of 1% KOH for 5 min to remove soluble impurities.
2. Cut Whatman paper (30 × 30 cm) and soak it in ddH2O. Lay the boiled cellophane circles on the lower part of the Whatman paper next to each other and fold the paper over the cellophane so that it is wrapped in the paper. Put the package in a 1 L beaker with 50 mL of ddH2O and autoclave at 121 °C for 20 min.
3. Layer the autoclaved cellophane circle on top of the MKM medium using sterile forceps. Work in a sterile workbench. Make sure to evenly distribute the cellophane. Make sure that no air is trapped between the cellophane and the agar.
Critical: Make sure that the cellophane stays humid throughout the procedure. Otherwise, the handling is more difficult.
4. Now, take macroconidia from a slant agar culture with an inoculation loop and transfer them into a 1.5 mL Eppendorf cup filled with 50 μL of sterile ddH2O.
5. Take 1.5 μL of the conidia suspension. Using the pipette tip, pierce a hole in the middle of the cellophane circle layered on top of the MKM medium and transfer the suspension between the agar and the cellophane.
6. Wrap parafilm around the Petri dish to keep the inside humid and incubate the plates for 7–12 days inside a climate chamber at 25 °C in a light/dark cycle (16 h light, 8 h dark).
7. After this time, remove the cellophane carefully from the plates and keep the plates one more day in the climate chamber at 25 °C in a light/dark cycle (16 h light, 8 h dark).
8. Now, use an inoculation loop and dip it in sterile ddH2O so that the inner circle of the loop is filled with water. Swipe across the grown mycelia on the plate. The microconidia will be collected inside the water.
9. Transfer the microconidia on VMM+SGF plates to obtain single homokaryotic colonies.
Data analysis
The typical mutation frequency depends on the gRNA used for editing and the accessibility of the target sequence within the genomic DNA. When designing the gRNA with the CHOP-CHOP tool, mutation frequencies are predicted from Shen et al. [16]. Table 1 shows experimental mutation frequencies for two different gRNAs targeting the csr-1 gene using the experimental data from [10]. Target site 1 refers to a gRNA targeting the minus strand in the third exon of csr-1, and target site 2 refers to a gRNA targeting the plus strand of the fourth exon of csr-1.
Table 1. Example of different mutation frequencies for two gRNAs targeting the csr-1 gene from [10].
| Type of mutation | Frequency for target site 1 | Frequency for target site 2 |
|---|---|---|
| 1 bp insertion | 50% | 30.18% |
| 2 bp insertion | 0% | 7.5% |
| >2 bp insertion | 0% | 1.9% |
| 1 bp deletion | 12.5% | 22.6% |
| 2 bp deletion | 18.75% | 20.8% |
| >2 bp deletion | 18.75% | 17% |
Usually, 20 colonies were screened to find a successful edit of the gene of interest.
Validation of protocol
This protocol has been used and validated in the following research article:
Grüttner and Kempken. [10] A user-friendly CRISPR/Cas9 system for mutagenesis of Neurospora crassa. Scientific Reports. 14(1): 20469. https://doi.org/10.1038/s41598-024-71540-x
General notes and troubleshooting
Troubleshooting
Problem 1: Low transformation efficiency.
Possible causes: Condition of macroconidia (might be too old), or the top agar was too hot.
Solution 1: Make sure the macroconidia are not too old. After isolation of the macroconidia, the transformation efficiency is reduced drastically if stored for longer than three days. Best use them right after isolation.
Solution 2: Isolate macroconidia no later than 20 days after inoculation of the Erlenmeyer flask.
Solution 3: Make sure the temperature of the top agar for plating after the electroporation is not above 45 °C.
Problem 2: No editing event can be detected in the sequencing results after PCR of the putatively edited region.
Possible cause: The used target sequence for the gRNA was not optimal.
Solution 1: Follow the detailed instructions for gRNA design in Labun et al. [17] and/or the CHOP-CHOP website.
Solution 2: Make sure that the PAM sequence is not included in the sequence of the crRNA.
Solution 3: Use more gRNA for the electroporation.
Acknowledgments
Authors’ contribution: Conceptualization, S.G.; Investigation, S.G.; Writing—Original Draft, S.G.; Writing—Review & Editing, S.G.; Supervision, S.G.
There are no funding sources that supported the work. This protocol is based on a previously published article by Grüttner and Kempken [10].
Competing interests
The author declares no conflicts of interest.
References
- Roche, C. M., Loros, J. J., McCluskey, K. and Glass, N. L. (2014). Neurospora crassa: looking back and looking forward at a model microbe. Am J Bot. 101(12): 2022–2035. https://doi.org/10.3732/ajb.1400377
- Collopy, P. D., Colot, H. V., Park, G., Ringelberg, C., Crew, C. M., Borkovich, K. A. and Dunlap, J. C. (2010). High-throughput construction of gene deletion cassettes for generation of Neurospora crassa knockout strains. Methods Mol Biol. 638: 33–40. https://doi.org/10.1007/978-1-60761-611-5_3
- Colot, H. V., Park, G., Turner, G. E., Ringelberg, C., Crew, C. M., Litvinkova, L., Weiss, R. L., Borkovich, K. A. and Dunlap, J. C. (2006). A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci USA. 103(27): 10352–10357. https://doi.org/10.1073/pnas.0601456103
- Ninomiya, Y., Suzuki, K., Ishii, C. and Inoue, H. (2004). Highly efficient gene replacements in Neurospora strains deficient for nonhomologous end-joining. Proc Natl Acad Sci USA. 101(33): 12248–12253. https://doi.org/10.1073/pnas.0402780101
- Park, G., Servin, J. A., Turner, G. E., Altamirano, L., Colot, H. V., Collopy, P., Litvinkova, L., Li, L., Jones, C. A., Diala, F.-G., et al. (2011). Global analysis of serine-threonine protein kinase genes in Neurospora crassa. Eukaryot Cell. 10(11): 1553–1564. https://doi.org/10.1128/EC.05140-11
- Park, G., Colot, H. V., Collopy, P. D., Krystofova, S., Crew, C., Ringelberg, C., Litvinkova, L., Altamirano, L., Li, L., Curilla, S., et al. (2011). High-throughput production of gene replacement mutants in Neurospora crassa. Methods Mol Biol. 722: 179–189. https://doi.org/10.1007/978-1-61779-040-9_13
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337(6096): 816–821. https://doi.org/10.1126/science.1225829
- Schuster, M. and Kahmann, R. (2019). CRISPR-Cas9 genome editing approaches in filamentous fungi and oomycetes. Fungal Genet Biol. 130: 43–53. https://doi.org/10.1016/j.fgb.2019.04.016
- Matsu-Ura, T., Baek, M., Kwon, J. and Hong, C. (2015). Efficient gene editing in Neurospora crassa with CRISPR technology. Fungal Biol Biotechnol. 2: 4. https://doi.org/10.1186/s40694-015-0015-1
- Grüttner, S. and Kempken, F. (2024). A user-friendly CRISPR/Cas9 system for mutagenesis of Neurospora crassa. Sci Rep. 14(1): 20469. https://doi.org/10.1038/s41598-024-71540-x
- Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K. and Sander, J. D. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 31(9): 822–826. https://doi.org/10.1038/nbt.2623
- Al Abdallah, Q., Souza, A. C. O., Martin-Vicente, A., Ge, W. and Fortwendel, J. R. (2018). Whole-genome sequencing reveals highly specific gene targeting by in vitro assembled Cas9-ribonucleoprotein complexes in Aspergillus fumigatus. Fungal Biol Biotechnol. 5: 11. https://doi.org/10.1186/s40694-018-0057-2
- Schuster, M., Schweizer, G., Reissmann, S. and Kahmann, R. (2016). Genome editing in Ustilago maydis using the CRISPR-Cas system. Fungal Genet Biol. 89: 3–9. https://doi.org/10.1016/j.fgb.2015.09.001
- Bardiya, N. and Shiu, P. K. T. (2007). Cyclosporin A-resistance based gene placement system for Neurospora crassa. Fungal Genet Biol. 44(5): 307–314. https://doi.org/10.1016/j.fgb.2006.12.011
- Labun, K., Montague, T. G., Krause, M., Torres Cleuren, Y. N., Tjeldnes, H. and Valen, E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47(W1): W171–W174. https://doi.org/10.1093/nar/gkz365
- Shen, M. W., Arbab, M., Hsu, J. Y., Worstell, D., Culbertson, S. J., Krabbe, O., Cassa, C. A., Liu, D. R., Gifford, D. K. and Sherwood, R. I. (2018). Predictable and precise template-free CRISPR editing of pathogenic variants. Nature. 563(7733): 646–651. https://doi.org/10.1038/s41586-018-0686-x
- Labun, K., Krause, M., Torres Cleuren, Y. and Valen, E. (2021). CRISPR Genome Editing Made Easy Through the CHOPCHOP Website. Curr Protoc. 1(4): e46. https://doi.org/10.1002/cpz1.46
Article Information
Publication history
Received: Oct 15, 2025
Accepted: Dec 7, 2025
Available online: Dec 15, 2025
Published: Jan 5, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Grüttner, S. (2026). Creating Loss-of-Function Mutants of Neurospora crassa Using a Novel CRISPR/Cas9 System. Bio-protocol 16(1): e5562. DOI: 10.21769/BioProtoc.5562.
Category
Microbiology > Microbial genetics > CRISPR-Cas9
Molecular Biology > DNA > Mutagenesis
Microbiology > Microbial genetics > Genome editing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.