- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient Fluorescent Labeling of Human Trophoblast Stem Cells via a CRISPR/Cas9-Mediated Knock-In Approach in a Safe Harbor Locus
Published: Vol 16, Iss 1, Jan 5, 2026 DOI: 10.21769/BioProtoc.5561 Views: 307
Reviewed by: Rajesh RanjanAnonymous reviewer(s)

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Labeling cells with reporter genes allows researchers to visually identify specific cells and observe how they interact with each other in dynamic biological systems. Even though various labeling methods are now available, a specific description of gene knock-in labeling methods for human trophoblast stem cells (hTSCs) has not been reported. Here, we present a streamlined protocol for labeling hTSCs with the green fluorescent protein (GFP) reporter gene via CRISPR/Cas9-mediated knock-in of the gene into the adeno-associated virus site 1 (AAVS1) safe harbor locus. A commonly used hTSC cell line, CT29, was transfected with a dual plasmid system encoding the Cas9 endonuclease and an AAVS1-targeted guide RNA in one plasmid and a donor plasmid encoding a puromycin resistance gene and GFP reporter gene flanked by AAVS1 homology arms. Puromycin-resistant clonal cells were isolated, and AAVS1 integration was confirmed via PCR and sequencing of the PCR products. The labeled cells are proliferative and can give rise to extravillous cytotrophoblast cells (EVT) and the syncytiotrophoblast (ST). To our knowledge, this is the first report using the CRISPR/Cas9 system for AAVS1 integration of a reporter gene in human trophoblast stem cells. It provides an efficient tool to facilitate the study of human trophoblast development and function in co-culture systems and will be highly useful in developing clinical gene therapy-related plasmid constructs.
Key features
• First report to constitutively express a fluorescent label in hTSCs by applying a CRISPR/Cas9 knock-in approach and an AAVS1 safe harbor locus.
• Provides an efficient tool to facilitate the study of human trophoblast development and function, particularly in heterologous co-culture systems.
• Offers an approach for developing clinical gene therapy–related plasmid constructs that allow insertion of therapeutic genes without associated disruption of essential genes.
• Widely applicable approach to label other human cell lines.
Keywords: hTSCsGraphical overview
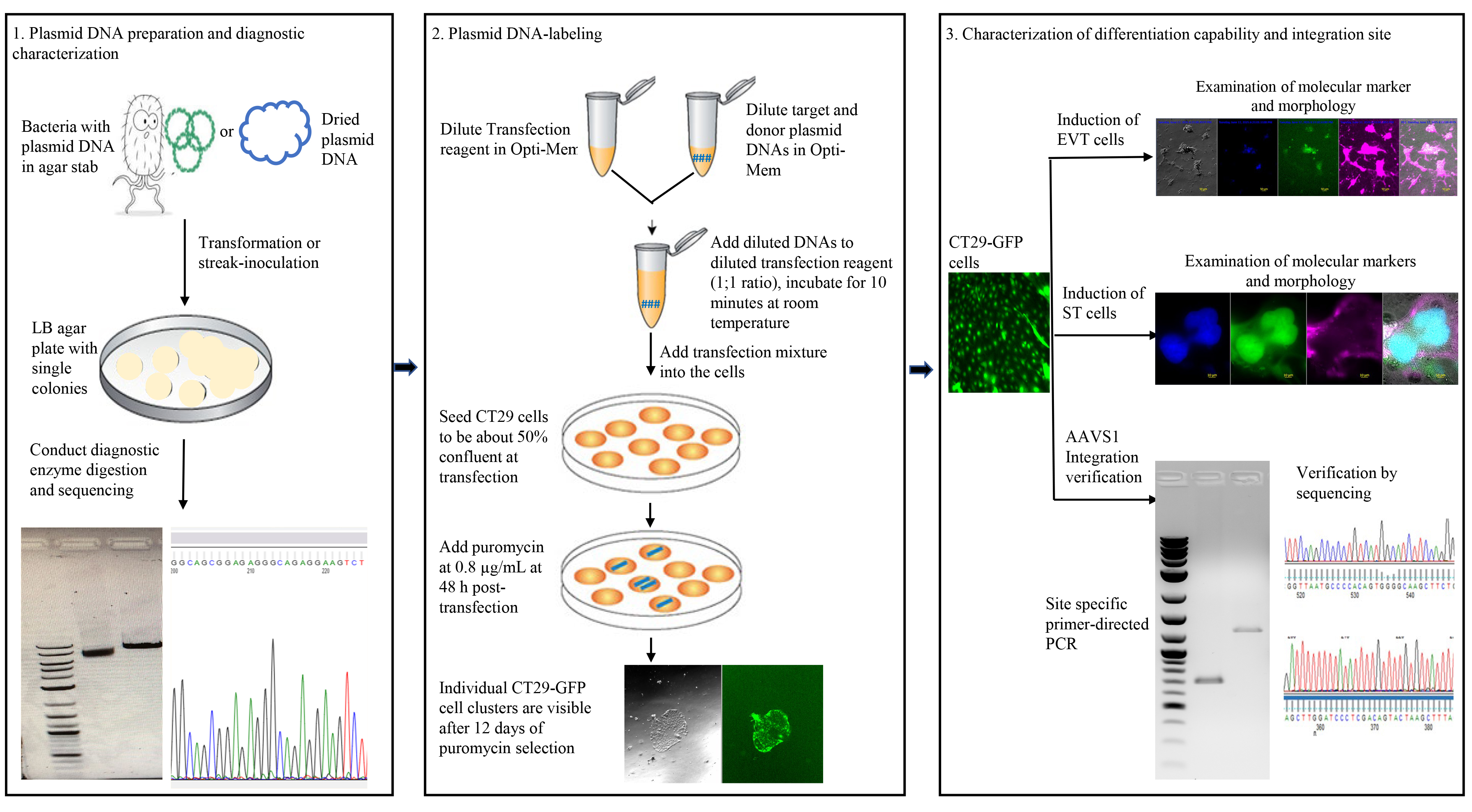
Background
Placental trophoblast cells are formed during the earliest stages of pregnancy and play a pivotal role in the complex interactions between the fetus and the mother. These early stages of human pregnancy are difficult to study due to inherent biological as well as legal and ethical factors; as such, in vitro modeling of human embryo implantation and placentation is necessary. In the human placenta, three major trophoblast subpopulations exist, which include cytotrophoblast cells (CT), extravillous cytotrophoblast cells (EVT), and the syncytiotrophoblast (ST) [1–3]. CT cells are capable of self-renewal and represent the undifferentiated progenitors of EVT and ST. Differentiated trophoblast types interact with many maternal and fetal cells types, including endometrial epithelial and stromal cells, maternal decidual macrophages, uterine natural killer cells, regulatory T cells, and fetal Hofbauer cells [4–6]. Coordinated proliferation and differentiation of trophoblast cells and appropriate interactions with these other cells and tissues are vital for a successful pregnancy. Impaired trophoblast development and function can lead to various pregnancy complications, including miscarriage, pre-eclampsia, and intrauterine growth restriction [7].
Labeling cells with reporter genes like the green fluorescent protein (GFP) gene allows researchers to easily identify cells under a microscope, making such labeling a crucial tool to study multicell interactions and to track the fate of specific cell populations during development or in response to stimuli. This is particularly useful when studying trophoblast development and placentation in vitro, and more specifically in heterologous cell co-culture models representing embryo implantation. Reporter gene cell labeling commonly involves integrating the reporter genes into the genomes of the cells to be studied in order to stably produce the reporter proteins throughout the lifetime of the cell and in any subsequent daughter cells. Over the years, many labeling methods have been developed, each with its own strengths, weaknesses, and specific applications. For instance, lentiviral vectors have often been used for transgene integration because they can carry large transgenes and integrate into both dividing and non-dividing cells, leading to stable and long-term expression. However, lentiviral vectors insert their genetic material at multiple random locations within the host genome, which has the potential to cause unwanted off-target effects such as insertional mutagenesis or oncogene activation [8]. Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are both well-established protein-based gene editing tools, but their design and construction are time-consuming, expensive, and challenging, as unique nuclease sequences must be generated for every genomic target. Alternatively, use of clustered regularly interspaced short palindromic repeats/Cas9 (CRISPR/Cas9), which was developed by several groups in 2013, has revolutionized gene editing by offering a method that is quicker, cheaper, and easier to design than previous approaches [9–11]. Importantly, CRISPR/Cas9 has also been shown to have higher efficiency and high specificity when compared to previously used gene editing tools, making it a promising approach for targeted integration of reporter genes [9–11].
Human trophoblast stem cells (hTSCs) are a specialized stem cell type that can be challenging to label using genetic tools because they are not robustly proliferative in culture and require a highly controlled microenvironment, including specific combinations of growth factors and signaling pathway modulators, to prevent differentiation in vitro. Further, their reluctance to grow under sparse culture densities makes certain genetic engineering methods that require cloning particularly difficult. While various cell-labeling techniques are available, and the CRISPR/Cas9-based approach has been used to perform gene knockout and transcriptional activation in hTSCs [12], there are few published studies specifically detailing the application of gene knock-in labeling methods in hTSCs. As a part of our broader and in-depth studies, we have developed a protocol to label hTSCs with the GFP reporter gene via CRISPR/Cas9-mediated knock-in into the adeno-associated virus site 1 (AAVS1). Here, AAVS1 lies within the first intron of the constitutively expressed PPP1R12C gene and acts as an important safe harbor where exogenous pieces of DNA can integrate and function in a predictable manner without causing adverse and unpredictable changes to the host genome or substantial risk to the host cell [13–15]. The resultant labeled hTSC line is an efficient tool to facilitate the study of human trophoblast development and function. Furthermore, this knock-in genome editing system should prove highly useful in a wide range of preclinical and potentially clinical gene therapy studies by offering a reliable method to develop genetically engineered plasmid constructs that can be introduced into human trophoblast progenitors.
Materials and reagents
Biological materials
1. CT29, a human trophoblast stem cell line derived from first-trimester placental cytotrophoblast cells (RIKEN BRC cell bank, catalog number: RCB4937), kindly provided by Dr. Okae [1] and Dr. Liping Feng at Duke University
Note: It is often recommended to use low-passage cells. The passage numbers we used for the CT29 cell line were between 19 and 25, and mycoplasma testing was performed periodically.
2. pCas-Guide-AAVS1, an all-in-one AAVS1 gRNA plasmid, delivered as a lyophilized 10 μg product (Origene, catalog number: GE100023)
3. pAAVS1-P-CAG-GFP, a donor plasmid for AAVS1 targeting and constitutive GFP expression, delivered as a bacterial agar slab (Addgene, catalog number: 80491)
Note: These are both high-copy plasmids with typical yields being 10–30 μg from a 1–5 mL bacterial culture volume when purified with a plasmid miniprep kit. We used DH5α competent cells to do the routine transformation.
4. 3′-junction directed primers: 5′-TCATTGCAATAGTGTGTTGG-3′ and 5′-AGCGGCCGCGAATTCGCCCTTAG-3′
5. 5′-junction directed primers:5′-CTGCACCACGTGATGTCCTCTG-3′ and 5′-GTGGGCTTGTACTCGGTCATC-3′
Note: These primers were designed by using the Oligonucleotide Properties Calculator (see Software) and were ordered from Integrated DNA Technologies. Standard desalting was performed by the vendor to purify them.
Reagents
1. DMEM/F12 (Thermo Fisher Scientific, catalog number: 11320033)
2. ITS-X (Thermo Fisher Scientific, catalog number: 51500056)
3. CHIR-99021 (Reprocell, catalog number: 04-0004); prepare at 3 mM in DMSO and store at -20 °C as 533 μL aliquots
4. Y-27632 (Reprocell, catalog number: 04-0012-02); prepare at 10 mM in MBG water and store at -20 °C as 50 μL aliquots
5. SB431542 (Reprocell, catalog number: 04-0010); make 10 mM SB431542 in DMSO and store as 80 μL aliquots at -20 °C
6. A83-01 (Reprocell, catalog number: 04-0014); make 5 mM A83-01 in DMSO and store as 80 μL aliquots at -20 °C
7. Valproic acid (VPA) (Sigma-Aldrich, catalog number: P4543); prepare 300 mM in sterile MBG water and store at 4 °C after aliquoted
8. KnockOut serum replacement (KOSR) (Thermo Fisher Scientific, catalog number: 10828010)
9. NRG1-beta 1, human (MCE, catalog number: HY-P7365); prepare at 200 μg/mL in 0.2% FBS in PBS and store in 25 μL aliquots at -20 °C
10. Collagen IV mouse (Fisher Scientific, catalog number: CB-40233)
Note: Collagen IV should be stored at -20 °C, and handling should always occur on ice. Collagen IV can be thawed by placing it on ice and slowly mixing it with an ice-cold buffer under sterile conditions.
11. BSA (Sigma-Aldrich, catalog number: A7906)
12. FBS (Sigma-Aldrich, catalog number: F2442)
13. EGF, human (hEGF) (Sigma-Aldrich, catalog number: E9644); dissolve in 0.2% FBS in PBS at 100 μg/mL and then aliquot at 250 μL and store at -20 °C
14. Foskolin (Sigma-Aldrich, catalog number: F6886); store powder at room temperature; store 5 mM DMSO solution at -20 °C as 500 μL aliquots
15. 2-Mercaptoethanol (2-ME) (Thermo Fisher Scientific, catalog number: 21985-023)
16. L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma-Aldrich, catalog number: A8960); prepare 30 mg/mL in MBG water and store at -20 °C after aliquoted
17. Penicillin-streptomycin (Pen/Strep, 10,000 U/mL) (Thermo Fisher Scientific, catalog number: 15140122)
18. Matrigel matrix (Fisher Scientific, catalog number: CB-40234C)
Note: Matrigel should be stored at -20 °C, and handling should always occur on ice. To thaw, place Matrigel overnight in a 2–8 °C ice bath or 4 °C refrigerator. Always use chilled labware and tips, keep Matrigel on ice, and aliquot after the first thaw to minimize freeze-thaw cycles. Matrigel is not growth factor-reduced (GFR) and contains several growth factors.
19. Certified molecular biology agarose (Bio-Rad, catalog number: 1613101)
20. Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: D8418)
21. 10× phosphate-buffered saline (PBS), without calcium and magnesium (Corning, catalog number: 46-013-CM)
22. 10× TAE buffer (Corning, catalog number: 46-010-CM)
23. Molecular biology grade (MBG) water (Corning, catalog number: 46-000-CM)
24. HyClone cell culture grade water (Cytiva, catalog number: SH30529.02)
25. HyClone Dulbecco's phosphate-buffered saline (DPBS) without calcium, magnesium (Cytiva, catalog number: SH30028-03)
26. Miller’s LB broth (Corning, catalog number: 46-050-CM)
27. TrypLE Express enzyme (Thermo Fisher Scientific, catalog number: 12604-021)
28. Lipofectamine Stem transfection reagent (Thermo Fisher Scientific, catalog number: STEM00008)
29. Opti-MEM 1 (Thermo Fisher Scientific, catalog number: 31985062)
30. CELLBANKER 1, ready-to-use serum-containing cell cryopreservation medium (Amsbio, catalog number: 11888)
Note: The volumes of the aliquots made for the reagents above are usually just enough to make the inhibitor cocktail or the complete medium for routine use (see Recipes). Frequent freeze-thaw cycles should be avoided when handling these reagents.
Solutions
1. Complete trophoblast stem cell (TS) growth media (see Recipes)
a. Basal media
b. Inhibitor cocktail
2. EVT differentiation medium (see Recipes)
a. Basal differentiation media
b. Initial differentiation media
c. Day 3 differentiation media
d. Day 6 differentiation media
3. ST (2D) differentiation medium (see Recipes)
a. Basal differentiation media
b. ST 2D complete differentiation media
Recipes
1. Complete trophoblast stem cell (TS) growth medium
Adapted from Okae et al. for routine use [1]. This medium is composed of basal medium, inhibitor cocktail, VPA, and Y-27632 stock solutions, and the recipes are shown below.
a. Basal media (500 mL)
| Reagent | Final concentration | Volume |
|---|---|---|
| DMEM/F12 | n/a | 486 mL |
| BSA, 30% prepared in sterile MBG water | 0.3% | 5 mL |
| ITS-X, 100× | 1% | 5 mL |
| FBS | 0.2% | 1 mL |
| Pen/strep (optional) (10,000 U/mL) | 0.5% | 2.5 mL |
| 2-ME, 55 mM | 0.1 mM | 900 μL |
| hEGF, 100 μg/mL | 50 ng/mL | 250 μL |
| L-ascorbic acid, 30 mg/mL | 1.5 μg/mL | 25 μL |
Store at 4 °C and use within 6 months.
b. Inhibitor cocktail (800 μL)
| Reagent | Final concentration | Volume |
|---|---|---|
| 3 mM CHIR-99021 | 2 mM | 533 μL |
| 5 mM A83-01 | 0.5 mM | 80 μL |
| 10 mM SB431542 | 1 mM | 80 μL |
| DMSO | n/a | 107 μL |
Store at -20 °C and use within 6 months.
c. Complete TS growth media (10 mL)
| Reagent | Final concentration | Volume |
|---|---|---|
| Basal media | n/a | 10 mL |
| Inhibitor cocktail | n/a | 10 μL |
| 300 mM VPA | 0.8 mM | 26.6 μL |
| 10 mM Y-27632 | 5 μM | 5 μL |
Store at 4 °C and use within 2 weeks.
2. EVT differentiation medium
Adapted from Okae et al. [1]. Medium compositions for performing EVT differentiation at different time points are slightly different. The recipes are shown below.
a. Basal differentiation media (50 mL)
| Reagent | Final concentration | Volume |
|---|---|---|
| DMEM/F12 | n/a | 48.2 mL |
| BSA, 30% | 0.3% | 0.5 mL |
| ITS-X, 100× | 1% | 0.5 mL |
| Pen/strep (optional) (10,000 U/mL) | 0.5% | 0.25 mL |
| 2-ME, 55 mM | 0.1 mM | 91 μL |
| A83-01, 5 mM | 7.5 μM | 75 μL |
| Y-27632, 10 mM | 2.5 μM | 12.5 μL |
Store at 4 °C and use within 6 months.
b. Initial differentiation media (12 mL)
| Reagent | Final concentration | Volume |
|---|---|---|
| Basal differentiation media | n/a | 12 mL |
| KnockOut serum replacement | 4% | 480 μL |
| NRG1-beta 1 | 100 ng/mL | 6 μL |
| Matrigel matrix (see the note below) | 2% | 240 μL |
Make fresh and prepare on demand.
c. Day 3 differentiation media
| Reagent | Final concentration | Volume |
|---|---|---|
| Basal differentiation media | n/a | 12 mL |
| KnockOut serum replacement | 4% | 480 μL |
| Matrigel matrix (see the note below) | 0.5% | 60 μL |
Make fresh and prepare on demand.
d. Day 6 differentiation media
| Reagent | Final concentration | Volume |
|---|---|---|
| Basal differentiation media | n/a | 12 mL |
| Matrigel matrix (see the note below) | 0.5% | 60 μL |
Make fresh and prepare on demand.
Note: Other required components should be added first in a tube and keep the tube on ice for a few minutes, add Matrigel last to the mixture, and then mix gently and quickly before plating.
3. ST (2D) differentiation medium
Adapted from Okae et al. [1]. This medium is composed of basal differentiation media, KOSR, Y-27632, and forskolin stock solutions, and the recipes are shown below.
a. Basal differentiation media (500 mL)
| Reagent | Final concentration | Volume |
|---|---|---|
| DMEM/F12 | n/a | 486 mL |
| BSA, 30% | 0.3% | 5 mL |
| ITS-X, 100× | 1% | 5 mL |
| Pen/strep (optional) (10,000 U/mL) | 0.5% | 2.5 mL |
| 2-ME, 55 mM | 0.1 mM | 900 μL |
Store at 4 °C and use within 6 months.
b. ST 2D complete differentiation media (12 mL)
| Reagent | Final concentration | Volume |
|---|---|---|
| Basal differentiation medium | n/a | 12 mL |
| Y-27632, 10 mM | 2.5 μM | 3 μL |
| KnockOut serum replacement | 4% | 480 μL |
| Forskolin, 5 mM | 2 μM | 4.8 μL |
Make fresh and prepare on demand.
Laboratory supplies
1. 8-strip PCR tubes and caps (Genesee Scientific, catalog number: 24-705)
2. 1.5 mL tubes (Sigma-Aldrich, catalog number: HS4323)
3. 15 mL and 50 mL disposable centrifuge tubes (VWR, catalog numbers: 89039-664 and 89039-660)
4. 16 G BD precision Glide needle (BD, catalog number: 305198)
5. 10 mL syringe (BD, catalog number: 309604)
6. 50 mL syringe (BD, catalog number: 309653)
7. 0.22 μm, 30 mm, PVDF syringe filter (Vista Lab Technologies, catalog number: 229743)
8. Corning® cell strainer, pore size 40 μm (Corning, Millipore Sigma, catalog number: CLS431750-50EA)
9. 250 mL and 500 mL filter systems (Corning, catalog numbers: 431096 and 430769)
10. VWR® disposable Petri dishes, 60 × 15 mm, mono Petri dishes (VWR International, catalog number: 25384-168)
11. Snap Cap low retention microcentrifuge tubes (Thermo Fisher Scientific, catalog number: 3448PK)
12. Adjustable volume pipettes: P100–1000 μL, P10–100 μL, P2–20 μL, and P0.5–10 μL
Equipment
1. Nanodrop 2000/2000C spectrophotometer (Thermo Fisher Scientific, catalog number: ND2000CLABTOP)
2. Gel electrophoresis assembly (Bio-Rad, model: 200/2.0)
3. Corning® Axygen® refrigerated microcentrifuge (Corning, Millipore Sigma, catalog number: AXY60105021)
4. Beckman Coulter Microfuge 18 centrifuge (Fisher Scientific, catalog number: NC2380892)
5. CO2 incubator for cell culture (Thermo Fisher Scientific, model: 3310)
6. Eppendorf 5331 MasterCycler gradient thermal cycler
7. Zeiss Axio Observer Z1 inverted fluorescence motorized microscope (Zeiss, catalog number: SM100331-MG1MOTC); objective type: EC Plan-Ncofluar 10×/0.30 Ph 1; filters: 450–490, 500–550
8. Sanger DNA sequencing facility (GENEWIZ, https://www.genewiz.com)
9. Automated cell counter (Fisher Scientific, catalog number: AMQAX2000)
Software and datasets
1. ZEN software for Carl Zeiss fluorescence microscope (Zen 2.3, blue edition, v2.3.69.1000): used to analyze all the fluorescent imaging pictures
2. Oligonucleotide Properties Calculator: used to design the primers (https://www.biosyn.com/gizmo/tools/oligo/oligonucleotide%20properties%20calculator.htm)
3. FinchTV (https://finchtv.software.informer.com): used to view the sequencing data
Procedure
Note: All steps are carried out at room temperature (RT) unless otherwise stated.
A. Plasmid DNA preparation and diagnostic characterization
1. Prepare plasmid DNAs
a. Reconstitute 10 μg of the lyophilized pCas-Guide-AAVS1 target plasmid DNA with 50 μL of MBG water, then use a standard protocol to perform E. coli transformation, single colony isolation, clonal bacterial culture, and plasmid DNA purification to expand the quantity of plasmid DNA. Remember to freeze a cryovial of cells to recover when needed for future use.
b. Streak bacteria containing the pAAVS1-P-CAG-GFP donor plasmid to isolate single colonies on an LB agar antibiotic plate. Then, get more plasmid DNA and freeze a cryovial of isolated bacterial cells as above.
2. Diagnostic characterization: It is strongly recommended that the prepared plasmid DNAs be verified by performing either diagnostic enzyme digestion or sequencing of important regions of the plasmids.
a. In our work, we performed enzyme digestions by choosing Xho I (New England Biolabs, catalog number: R0146S) or Not I (New England Biolabs, catalog number: R0189S) restriction enzymes, which have unique cutting sites for the target plasmid and donor plasmid, respectively.
b. We also performed a Sanger sequencing for both the target and donor plasmid by using a specific primer against the locus spanning the AAVS1 target sequence in the target plasmid and a specific primer recognizing the left AAVS1 homology arm of the donor plasmid.
Critical: It is recommended that you proceed only after the sequencing or enzyme digestion data clearly give confirmatory results.
B. Transfection of target and donor plasmid DNA-labeling of reporter gene
Note: All steps are performed in a laminar flow hood unless otherwise noted.
1. Culture cells: Retrieve the cells to be transfected or labeled from a cryo-vial kept in a liquid nitrogen tank and thaw at 37 °C in a water bath at least a week prior to being transfected. Seed thawed cells at a density of 2–5 × 105 cells per well in a 6-well plate precoated with 2–5 μg/mL collagen IV for 1 h at 37 °C and culture them in 2 mL of prewarmed complete TS growth medium. Replace the medium every two or three days; cells are ready to be passaged when they reach about 85% confluency. Dissociate with TrypLE for 8–15 min at 37 °C for passaging.
Critical: It is necessary to avoid seeding the cells at a very low density as hTSCs proliferate poorly at low density. We recommend seeding 2–5 × 105 hTSCs per well in a 6-well plate routinely.
Note: We suggest passaging the cells one day prior (day 0) to transfection to allow them to re-adhere to the culture wells and become actively dividing for optimal DNA uptake when transfection is performed on the following day. Passaging the day before transfection also allows the cells to recover from stress and acclimate. Transfection works best when cells are healthy, actively dividing, and at approximately 50% confluency level.
2. Transfecting CT29 hTSCs with plasmid DNAs (day 1).
a. Perform transfection (Table 1). By referring to the protocol provided by the transfection reagent vendor, a suggested procedure is shown below.
Table 1. Optimization of transfection by varying the proportional amount of transfection reagent to plasmid DNA
| Step | Component | Well number | |
| 1 | 2 | ||
| Dilute 2 amounts of Lipofectamine Stem reagent in Opti-MEM 1 medium | Opti-MEM 1 medium | 140 μL | 140 μL |
| Lipofectamine Stem reagent | 10 μL | 12 μL | |
| Dilute 2 amounts of DNA in Opti-MEM 1 medium | Opti-MEM 1 medium | 140 μL | 140 μL |
| Target plasmid DNA | 1,000 ng | 1,250 ng | |
| Donor plasmid DNA | 1,000 ng | 1,250 ng | |
| Add diluted DNA to diluted Lipofectamine Stem reagent (near 1:1 ratio) | Diluted DNA volume | 140–150 μL | 140–160 μL |
| Diluted Lipofectamine Stem reagent volume | 150 μL | 152 μL | |
| Incubate | Incubate for 10 min at room temperature | ||
| Add DNA-lipid complex to cells, and incubate and monitor transfected stem cells at 37 °C for 2 days | |||
b. Puromycin-based selection (day 3).
Note: Based on the findings in our pilot dose-response study and a reference publication [16], we initiated puromycin selection at 0.8 μg/mL at 48 h post-transfection and allowed it to continue for 12 days. The cells were then cultured for another 10 days in TS growth medium without puromycin. Pictures were taken at several time points and are shown below (Figure 1).

Figure 1. Representative images of different stages of CT29 cell labeling with GFP using CRISPR/Cas9 system. (A) Fluorescent image with GFP filter shows CT29 cells at 16 h post-transfection. (B) Fluorescent image with GFP filter shows CT29 cells at 48 h post-transfection. (C and C’) Images of the transfected CT29 cells following puromycin selection at 0.8 µg/mL for 12 days. The left and right panels show phase-contrast and GFP-labeled images, respectively. A few GFP-labeled cell clusters can sometimes be observed when a low magnification objective is used (D) phase-contrast; and (D’) GFP-labeled). (E and E’) Images of the phase-contrast (left panel), GFP-labeled (right panel) were taken when the puromycin resistant clonal cells in the original transfection plate are further cultured for 10 days in the same type of growth medium omitting the selection reagent following the 12-day selection period.
c. Maintain the labeled cells under routine culture conditions.
In our studies, we have found that the GFP-labeled CT29 (hereafter referred to as CT29-GFP) cells can be cultured in the same manner as unlabeled cells (Figure 2), suggesting that labeling hTSCs at the AAVS1 site with the CRISPR/Cas9 system does not negatively affect their culture requirements when they are plated at a sufficient cell density.
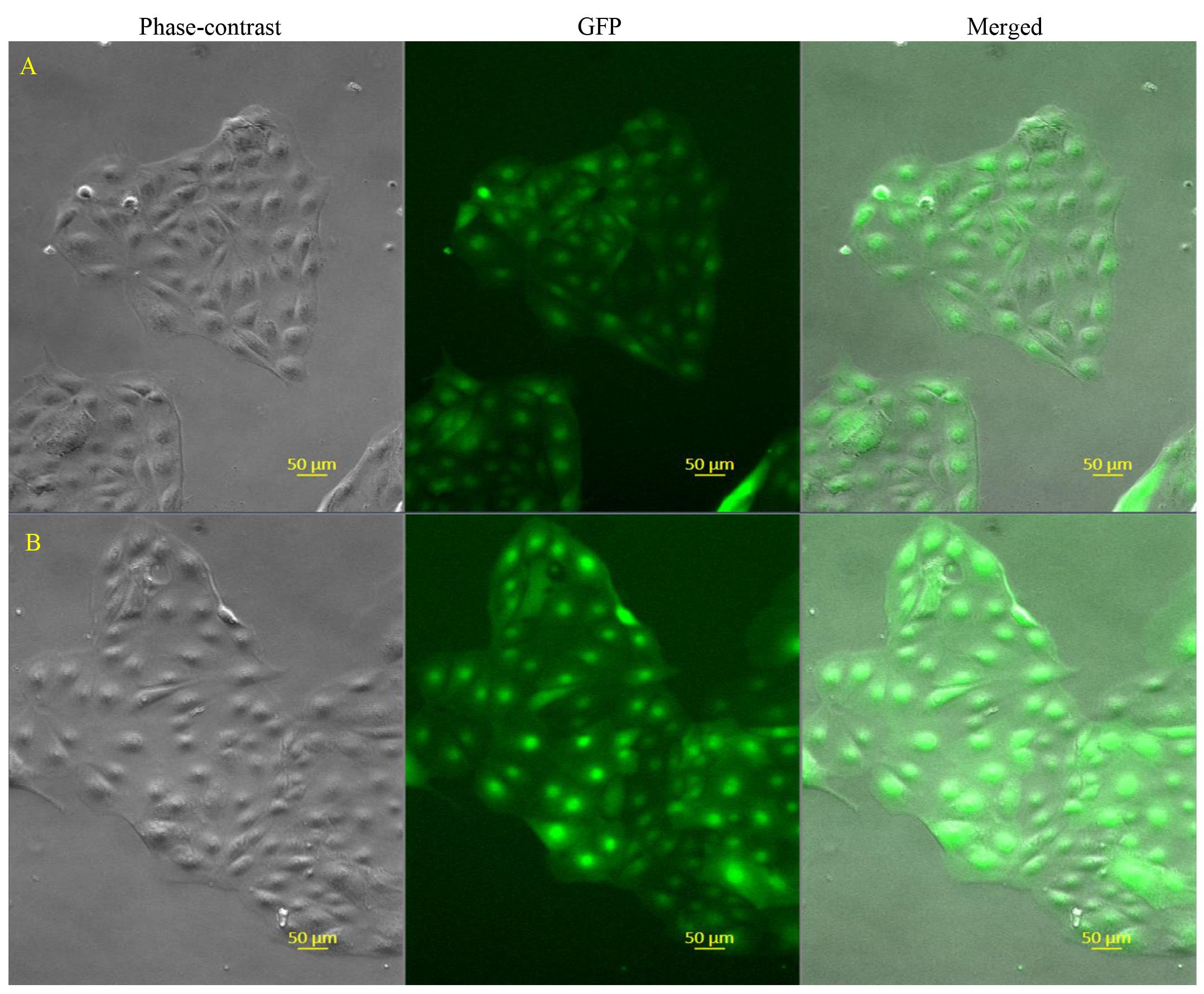
Figure 2. Representative images showing CT29-GFP cells maintained in TS growth medium. Frozen CT29-GFP cells were thawed and cultured in TS growth medium for 3 days and then passaged. Images from two distinct groups of cells (Cluster A and cluster B) were taken under a fluorescent microscope after passage and 3 days of culture to show more confirmative information. Phase-contrast imaging shows the morphology of live labeled CT29-GFP cells in culture. The green signal in the middle panel indicates the constitutive expressing of GFP by the labeled cells themselves. The third panel shows the merged signal.
Validation of protocol
Parts of this protocol have been used and validated in a recent publication [17].
In our experiments, we have validated the protocol by examining the labeled cells’ molecular markers and their functional capacity to differentiate into EVT and ST cells. Additionally, we confirmed the AAVS1 integration of the GFP reporter gene via PCR and the sequencing of the PCR products.
1. Perform routine immunofluorescence microscopy to check the expression profile of CK7 in the labeled CT29-GFP cells. CK7 is widely accepted as a reliable marker for identifying cells of the trophoblast lineage, and strong positive CK7 staining in our cells supported the trophoblast nature of the CT29-GFP cells after labeling (Figure 3).
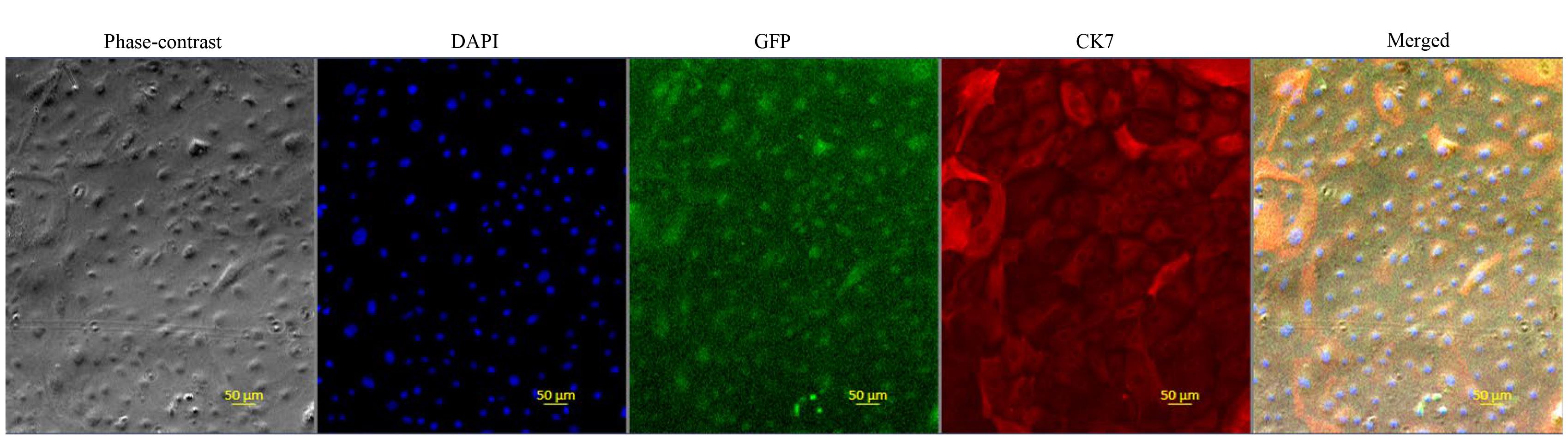
Figure 3. Morphological and molecular profiling of cultured CT29-GFP cells. Phase-contrast imaging shows the morphology of live labeled CT29-GFP cells in culture. Nuclei were stained with DAPI (blue). The green signal indicates the constitutive expression of GFP by the labeled cells themselves. Immunostaining for the CK7 trophoblast marker (red). The fifth panel shows merged DAPI and CK7 antibody staining.
2. Labeled CT29-GFP cells retain the functional capacity to differentiate into EVT cells (Figure 4) and ST (Figure 5) when cultured in the EVT and ST differentiation media (see Recipes) following standard protocols, as described in [1]. In Figure 4, we show that the labeled cells strongly express HLA-G, a well-established specific marker of invasive EVT cells. Moreover, the labeled CT29-GFP cells also have tumor-like outgrowth features, a depth-limited but crucial part of their differentiation that allows invasion into the maternal tissues during pregnancy.
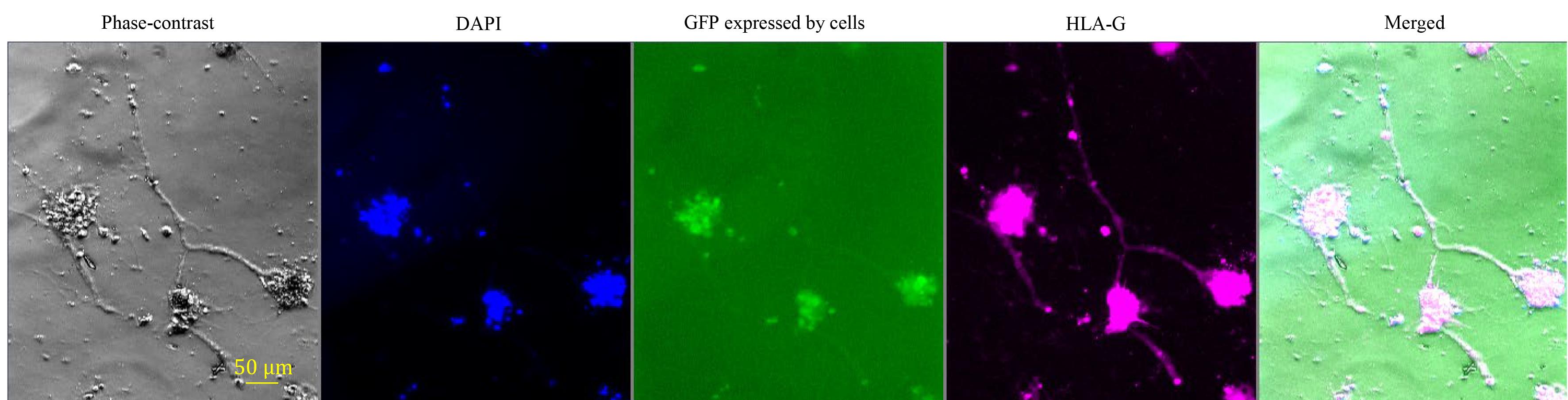
Figure 4. Immunofluorescence examination of the functional capacity of CT29-GFP cells to differentiate into EVT cells. Phase-contrast imaging visualizes spike-like morphological changes associated with the outgrowth of the live CT29-GFP cells differentiated toward the EVT lineage in culture. Nuclei were visualized with DAPI (blue). Green color demonstrates the constitutive expression of GFP by the labeled cells. HLA-G (an EVT marker) expression in the labeled cells was visualized in pink with an anti-HLA-G primary antibody. The fifth panel depicts merged DAPI and HLA-G antibody staining.
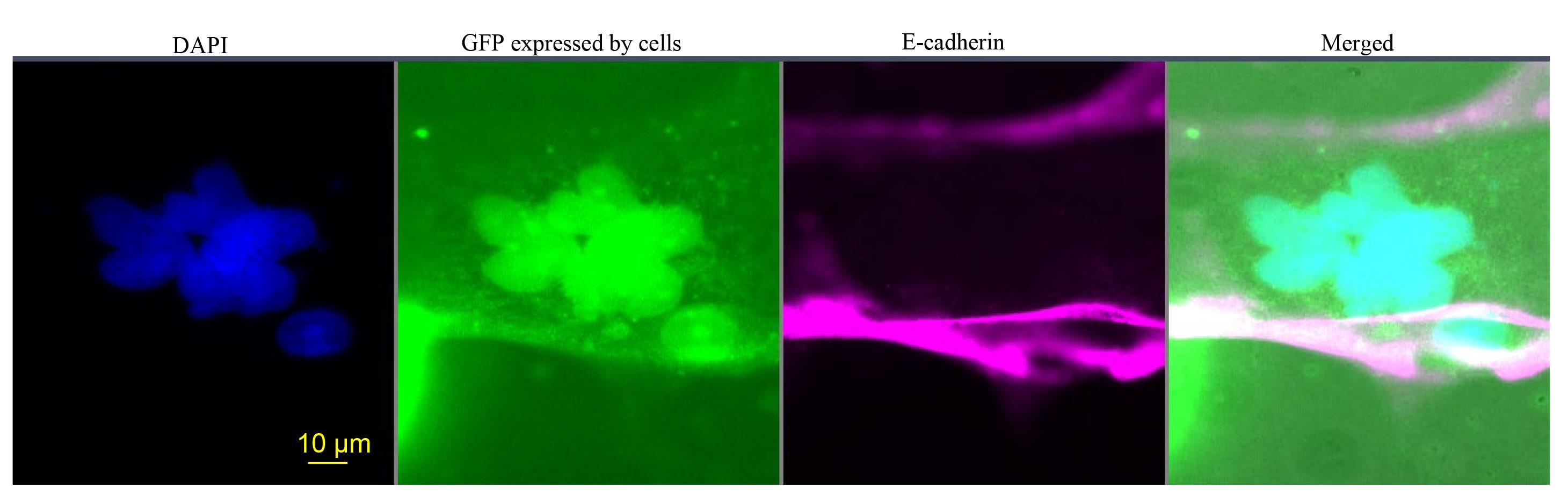
Figure 5. Immunofluorescence examination of the functional capacity of CT29-GFP cells to differentiate into fused ST cells. Nuclei were visualized with DAPI (blue). Green color demonstrates the constitutive expression of GFP by labeled cells. E-cadherin antibody labeling of the surface of multinucleated ST confirms the live trophoblast cell fusion (pink). The fourth panel merges nuclear DAPI and surface E-cadherin antibody staining.
Mononuclear trophoblast cells in culture express E-cadherin along their cell borders, with each trophoblast cell containing a single nucleus completely surrounded by an E-cadherin-expressing plasma membrane. The inclusion of more than one nucleus within this continuous layer of E-cadherin labeling is a hallmark of trophoblast cell fusion [17]. In Figure 5, the E-cadherin/DAPI/GFP image demonstrates the classical cell-fusion appearance of the syncytiotrophoblast, which is formed by a crucial and tightly regulated process of cellular differentiation and fusion. Figure 6 demonstrates the validation of correct GFP reporter gene integration within the AAVS1 locus.
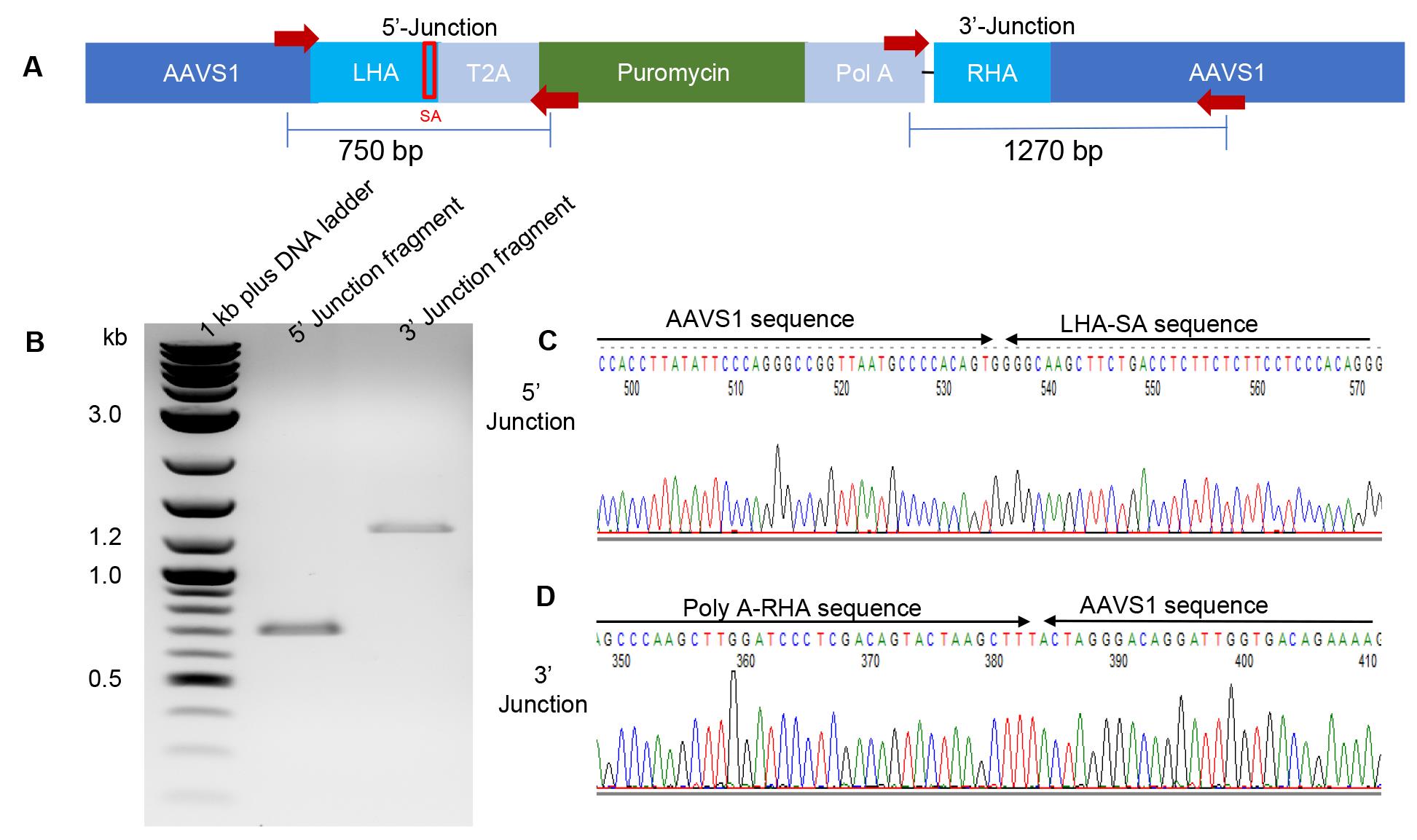
Figure 6. Reporter gene integration junction PCR analysis of CT29-GFP cells at the AAVS1 site. (A) The AAVS1 site after homology-directed repair-mediated genomic insertion of the donor cassette. The red arrows represent PCR primers and their respective product sizes. (B) PCR assessment of the mixed cell population with the specific primer pairs detected as single bands with the expected sizes deduced from the inserted sequence of the donor plasmid at the gRNA-targeted site. (C, D) Sequencing of the PCR products shows the 5′ junction between the AAVS1 site and the left homology arm (LHA) and the 3′ junction between the right homology arm (RHA) and the AAVS1 site, respectively.
General notes and troubleshooting
1. The described protocol generates genetically labeled cell lines without the requirement for manually isolating many individual colonies using the traditional cloning ring or cylinder. Sequencing of the PCR products from multiple cell populations demonstrated successful and accurate integration of the donor cassette into the AAVS1 safe harbor site using the CRISPR/Cas9 system. The overall labeling process can be very effective.
2. Our studies strongly suggest that knock-in of a fluorescent label (GFP) guided by the AAVS1-targeted CRISPR/Cas9 system does not disturb important functional downstream activities of the CT29-GFP cells, as evidenced by retained EVT and ST sublineage differentiation, making the labeled hTSCs a valuable tool for studying the development and function of the placenta, particularly in heterologous co-culture-related studies.
3. Matrigel polymerizes at 37 °C and stays in a liquid state at 4 °C. To prevent premature polymerization when preparing cell cultures, it is critical to handle it under icy conditions.
Acknowledgments
This research was supported by the Department of Obstetrics and Gynecology at Duke University (to D.J.S.) and a generous gift from the Szulick Family Foundation (to D.J.S.). We thank Dr. Yasheng Gao in the Duke Light Microscopy Core Facility for helping with the fluorescent microscopy.
Competing interests
The authors declare no competing interests.
Ethical considerations
All procedures were conducted in accordance with National Institutes of Health guidelines and with approval from the Institutional Biosafety Committee at Duke University.
References
- Okae, H., Toh, H., Sato, T., Hiura, H., Takahashi, S., Shirane, K., Kabayama, Y., Suyama, M., Sasaki, H., Arima, T., et al. (2018). Derivation of Human Trophoblast Stem Cells. Cell Stem Cell. 22(1): 50–63.e6. https://doi.org/10.1016/j.stem.2017.11.004
- Bischof, P. and Irminger-Finger, I. (2005). The human cytotrophoblastic cell, a mononuclear chameleon. Int J Biochem Cell Biol. 37(1): 1–16. https://doi.org/10.1016/j.biocel.2004.05.014
- James, J. L., Carter, A. M. and Chamley, L.W. (2012). Human placentation from nidation to 5 weeks of gestation. Part I: What do we know about formative placental development following implantation? Placenta. 33: 327–334. https://doi.org/10.1016/j.placenta.2012.01.020
- Ding, J., Zhang, Y., Cai, X., Diao, L., Yang, C. and Yang, J. (2021). Crosstalk Between Trophoblast and Macrophage at the Maternal-Fetal Interface: Current Status and Future Perspectives. Front Immunol. 12: e758281. https://doi.org/10.3389/fimmu.2021.758281
- Li, Q., Sharkey, A., Sheridan, M., Magistrati, E., Arutyunyan, A., Huhn, O., Sancho-Serra, C., Anderson, H., McGovern, N., Esposito, L., et al. (2024). Human uterine natural killer cells regulate differentiation of extravillous trophoblast early in pregnancy. Cell Stem Cell. 31(2): 181–195.e9. https://doi.org/10.1016/j.stem.2023.12.013
- Zambuto, S. G., Scott, A. K. and Oyen, M. L. (2024). Beyond 2D: Novel biomaterial approaches for modeling the placenta. Placenta. 157: 55–66. https://doi.org/10.1016/j.placenta.2024.03.006
- Norwitz, E. R. (2006). Defective implantation and placentation: laying the blueprint for pregnancy complications. Reprod Biomed Online. 13(4): 591–599. https://doi.org/10.1016/s1472-6483(10)60649-9
- Milone, M. C. and O’Doherty, U. (2018). Clinical use of lentiviral vectors. Leukemia. 32(7): 1529–1541. https://doi.org/10.1038/s41375-018-0106-0
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-Guided Human Genome Engineering via Cas9. Science. 339(6121): 823–826. https://doi.org/10.1126/science.1232033
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339(6121): 819–823. https://doi.org/10.1126/science.1231143
- Ding, Q., Regan, S. N., Xia, Y., Oostrom, L. A., Cowan, C. A. and Musunuru, K. (2013). Enhanced Efficiency of Human Pluripotent Stem Cell Genome Editing through Replacing TALENs with CRISPRs. Cell Stem Cell. 12(4): 393–394. https://doi.org/10.1016/j.stem.2013.03.006
- Wu, H., Wang, Y. and Wang, H. (2022). Workflow for Performing Genetic Manipulation in Human Trophoblast Stem Cells Using CRISPR/Cas9 Technology. Methods Mol Biol. 2767: 53–62. https://doi.org/10.1007/7651_2022_464
- Li, S. J., Luo, Y., Zhang, L. M., Yang, W. and Zhang, G. G. (2017). Targeted introduction and effective expression of hFIX at the AAVS1 locus in mesenchymal stem cells. Mol Med Rep. 15(3): 1313–1318. https://doi.org/10.3892/mmr.2017.6131
- Luo, Y., Liu, C., Cerbini, T., San, H., Lin, Y., Chen, G., Rao, M. S. and Zou, J. (2014). Stable Enhanced Green Fluorescent Protein Expression After Differentiation and Transplantation of Reporter Human Induced Pluripotent Stem Cells Generated by AAVS1 Transcription Activator-Like Effector Nucleases. Stem Cells Transl Med. 3(7): 821–835. https://doi.org/10.5966/sctm.2013-0212
- Papapetrou, E. P. and Schambach, A. (2016). Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol Ther. 24(4): 678–684. https://doi.org/10.1038/mt.2016.38
- Martens, Y. A., Xu, S., Tait, R., Li, G., Zhao, X. C., Lu, W., Liu, C. C., Kanekiyo, T., Bu, G., Zhao, J., et al. (2021). Generation and validation of APOE knockout human iPSC-derived cerebral organoids. STAR Protoc. 2(2): 100571. https://doi.org/10.1016/j.xpro.2021.100571
- Dubois, V. P., Zotova, D., Parkins, K. M., Swick, C., Hamilton, A. M., Kelly, J. J. and Ronald, J. A. (2018). Safe Harbor Targeted CRISPR-Cas9 Tools for Molecular-Genetic Imaging of Cells in Living Subjects. CRISPR J. 1(6): 440–449. https://doi.org/10.1089/crispr.2018.0030
Article Information
Publication history
Received: Oct 10, 2025
Accepted: Dec 2, 2025
Available online: Dec 25, 2025
Published: Jan 5, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Zhang, H., Zhou, J., Orsolini, M., Zhao, A., Takhirov, A. and Schust, D. J. (2026). Efficient Fluorescent Labeling of Human Trophoblast Stem Cells via a CRISPR/Cas9-Mediated Knock-In Approach in a Safe Harbor Locus. Bio-protocol 16(1): e5561. DOI: 10.21769/BioProtoc.5561.
Category
Stem Cell > Embryonic stem cell > Cell-based analysis
Molecular Biology > DNA > Chromosome engineering
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.