- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Optimized Secretome Sample Preparation From High Volume Cell Culture Media for LC–MS/MS Proteomic Analysis
Published: Vol 15, Iss 24, Dec 20, 2025 DOI: 10.21769/BioProtoc.5542 Views: 1261
Reviewed by: Ralph Thomas BoettcherPrajita PandeyAnonymous reviewer(s)

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
The cellular secretome is a rich source of biomarkers and extracellular signaling molecules, but proteomic profiling remains challenging, especially when processing culture volumes greater than 5 mL. Low protein abundance, high serum contamination, and sample loss during preparation limit reproducibility and sensitivity in mass spectrometry–based workflows. Here, we present an optimized and scalable protocol that integrates (i) 50 kDa molecular weight cutoff ultrafiltration, (ii) spin column depletion of abundant serum proteins, and (iii) acetone/TCA precipitation for protein recovery. This workflow enables balanced recovery of both low- and high-molecular-weight proteins while reducing background from serum albumin, thereby improving sensitivity, reproducibility, and dynamic range for LC–MS/MS analysis. Validated in human mesenchymal stromal cell cultures, the protocol is broadly applicable across diverse cell types and experimental designs, making it well-suited for biomarker discovery and extracellular proteomics.
Key features
• Enables efficient concentration and cleanup of ≥5–500 mL of conditioned media, suitable for low-abundance secreted protein recovery.
• Combines 50 kDa ultrafiltration, optional HSA/IgG depletion, and acetone/TCA precipitation for robust removal of serum contaminants and improved signal-to-noise.
• Adaptable to various mammalian cell types and serum-free or serum-containing media; scalable for adherent and suspension cultures.
Keywords: SecretomeBackground
The secretome is a complete set of proteins secreted by cells into the extracellular space [1]. In a cell culture system, the secretome can be profiled from conditioned medium, which is a cell culture medium containing secreted proteins and signaling factors released by the cells during incubation under defined conditions [2]. Mass spectrometry–based proteomic analysis of the secretome is an established strategy for studying extracellular communication, discovering biomarkers, and identifying therapeutic targets [3,4]. In parallel, large-scale mapping of the human proteome highlights the importance of capturing both abundant housekeeping proteins and rare, tissue-specific factors in secretome studies [5]. However, secretome proteins are often present at very low concentrations and are masked by highly abundant serum proteins such as albumin and immunoglobulins [6], which complicates mass spectrometry analysis.
These challenges intensify when processing large volume samples (>5 mL), as often required to obtain sufficient secretome, particularly with primary cells or low-secreting cultures. Without rigorous sample processing, low-abundance proteins like cytokines and chemokines are frequently lost, while high-abundance proteins overshadow the results [6].
Early comparative work established in-gel digestion followed by LC–MS/MS as a robust strategy for protein identification, but this approach is labor-intensive, biased toward higher-abundance proteins, and suboptimal for low-molecular-weight cytokines [7]. More recently, systematic benchmarking studies demonstrated that acetone precipitation with in-solution digestion improved reproducibility and enhanced recovery of small-secreted proteins [8]. However, these protocols were primarily optimized for small input volumes (≤ 2 mL) and did not adequately address challenges of serum protein contamination or scalability.
To overcome these limitations, we developed an optimized protocol specifically designed for high-volume secretome samples (>5 mL) [9]. This workflow integrates three complementary steps: (i) 50 kDa ultrafiltration to generate both concentrate and filtrate fractions, (ii) spin column depletion [10] of abundant serum proteins, and (iii) acetone/TCA precipitation to maximize protein recovery. By processing both the filtrate and depleted concentrate in parallel, the protocol ensures balanced recovery of low-abundance cytokines and chemokines alongside high-molecular-weight extracellular matrix proteins, while minimizing background interference and serum protein contamination. The overall workflow is illustrated in Figure 1.
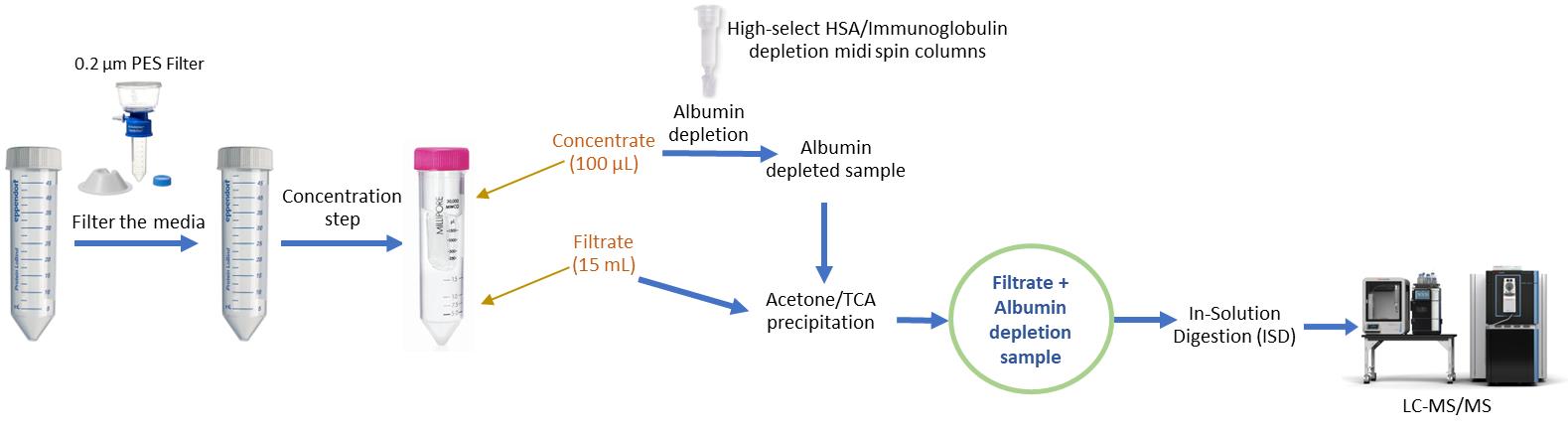
Figure 1. Workflow for optimized secretome preparation for LC–MS/MS analysis. Conditioned medium is first clarified by centrifugation (500× g, 5 min, 4 °C) and filtered through a 0.22 μm low-protein-binding filter to remove cells and debris. The clarified medium is then concentrated using Amicon Ultra-15 centrifugal filters (50 kDa MWCO), yielding approximately 100 μL of concentrate and 15 mL of filtrate, depending on the initial sample volume. The concentrate is subjected to HSA/IgG depletion to eliminate abundant serum proteins. The depleted concentrate and filtrate are then combined to generate a comprehensive secretome sample, which is subsequently precipitated, digested, and analyzed by LC–MS/MS. This workflow maximizes recovery of both low- and high-molecular-weight secreted proteins while minimizing background from serum components.
This method demonstrates improved sensitivity, reproducibility, and scalability compared to previous protocols. It maintains the identification capabilities of acetone precipitation workflows and extends their use to high-volume samples, which are often necessary for primary or low-secreting cultures. Additionally, it overcomes the reproducibility and coverage limitations associated with in-gel digestion-based approaches and outperforms ultrafiltration-only methods that do not efficiently recover low-molecular-weight proteins. Therefore, this optimized workflow offers a robust and adaptable solution for comprehensive secretome profiling, making it particularly advantageous for biomarker discovery pipelines that require broad dynamic range, scalability, and reproducibility.
Materials and reagents
Note: Catalog numbers and models are provided for reference; suitable alternatives may be used.
1. Amicon Ultra-15 centrifugal filter, 50 kDa MWCO (Merck Millipore, catalog number: UFC905008)
2. High-Select HSA/IgG depletion midi spin columns (Thermo Fisher Scientific, catalog number: A36367)
3. Chilled (-20 °C) acetone (Fisher Chemical, catalog number: A18-4)
4. Halt protease inhibitor cocktail (100×) (Thermo Scientific, catalog number: 1861278)
5. Trichloroacetic acid (TCA) (Fisher Chemical, catalog number: SA433-500)
6. Urea (Invitrogen, catalog number: 15505-035)
7. Ammonium bicarbonate (Fisher Chemical, catalog number: A643-500)
8. Dithiothreitol (DTT) (Fisher bioreagents, catalog number: BP172-5)
9. Iodoacetamide (IAA) (Sigma-Aldrich, catalog number: I1149 -5G)
10. MS-grade trypsin (Promega, catalog number: V5111)
11. Pierce C18 spin columns (Thermo Fisher Scientific, catalog number: 89870)
12. Formic acid (FA) (Thermo Scientific, catalog number: 28905)
13. Acetonitrile (ACN) (Fisher Chemical, catalog number: A955-4)
14. BCA Protein Assay kit (Thermo Fisher Scientific, catalog number: 23227)
15. Micro BCA Protein Assay kit (Thermo Fisher Scientific, catalog number: 23235)
16. Protein LoBind tube 2.0 mL (Eppendorf, catalog number: 022431102)
17. Centrifuge tube 50 mL flat cap (Thermo Fisher Scientific, catalog number: 05-539-13)
18. Nunc conical sterile polypropylene 15 mL centrifuge tubes (Thermo Scientific, catalog number: 339650)
19. Nalgene rapid-flow sterile disposable filter units with PES, CN, SFCA, or nylon membranes (Thermo Scientific, catalog number: 564-0020)
20. Mini-Protean TGX stain free gels (Bio-Rad, catalog number: 4568094)
21. Precision Plus protein dual color standards (Bio-Rad, catalog number: 1610374)
Equipment
1. Refrigerated benchtop centrifuge (Thermo Fisher Scientific, model: Sorvall X1R Pro-MD, Eppendorf, model: AG 5424)
2. SpeedVac concentrator (Thermo Scientific, model: Savant SPD111V, RVT5105)
3. NanoLC system (Thermo Scientific, model: Vanquish Neo)
4. Orbitrap mass spectrometer (Thermo Scientific, model: Orbitrap Exploris 240/480)
5. BCA reader (BioTek, model: Synergy/LX multi-mode reader)
6. Thermomixer (Eppendorf, model: Thermomixer C)
Procedure
Workflow overview
This protocol describes the processing of conditioned cell culture medium to isolate and identify secreted and extracellular proteins by LC–MS/MS. The workflow comprises three major stages:
1. Clarification and ultrafiltration: The conditioned medium is first clarified by centrifugation and 0.22 μm filtration to remove cell debris and dead cells. It is then concentrated and fractionated into >50 kDa (retentate/concentrate) and <50 kDa (filtrate) fractions using ultrafiltration.
2. Depletion: Removes highly abundant serum proteins—primarily albumin and immunoglobulins (IgG)—from the >50 kDa fraction using spin-column depletion.
3. Digestion: Precipitates, solubilizes, and digests the recovered proteins into peptides for LC–MS/MS analysis, enabling comprehensive profiling of the cellular secretome.
A key decision point in this workflow is whether to process the <50 kDa (filtrate) and >50 kDa (depleted concentrate) fractions separately or pooled. This decision depends on the composition of the starting culture medium; specifically, whether it is albumin-rich or serum-free.
• For albumin-rich media (e.g., containing FBS, HSA, or BSA supplements): Process the filtrate and depleted concentrate separately. The high abundance of serum albumin and immunoglobulins can suppress the detection of lower-abundance secreted peptides if the fractions are pooled.
• For serum-free media (also referred to as albumin-depleted, HSA-starved, or serum-starved media): The filtrate and depleted concentrate can be pooled after acetone/TCA precipitation to maximize recovery of both high- and low-molecular-weight secreted proteins. Pooling after precipitation ensures uniform cleanup and prevents differential precipitation losses.
A. Ultrafiltration of conditioned medium
This workflow was validated using human mesenchymal stromal cells (hMSCs) as in Wang et al. [9]. The method is applicable to other adherent or suspension mammalian cell types, but users should perform routine quality control (albumin band reduction at ~66 kDa by SDS-PAGE, low intracellular contamination in LC–MS/MS, consistent peptide yield) when first adopting a new model.
Notes:
1. Include a protease inhibitor cocktail (EDTA-free) in the conditioned medium prior to ultrafiltration to minimize protein degradation.
2. Maintain a constant centrifugation speed of 14,000× g for 20 min at 4 °C, regardless of the starting volume, to ensure consistent recovery and concentration of the conditioned medium.
3. When starting with serum-free medium, it is recommended to combine both the concentrate and filtrate fractions. This is because such a medium will have relatively low total protein content in addition to the absence of serum proteins, such as albumin and immunoglobulins. Combining both fractions ensures a more comprehensive recovery of secreted proteins, capturing both high- and low-molecular-weight components of the secretome.
4. Conditioned medium refers to the cell culture medium collected after incubation with cells, which contains proteins and other factors secreted into the extracellular environment.
5. Scaling above 15 mL: For total volumes >15 mL, use one Amicon Ultra-15 (50 kDa MWCO) per ≤15 mL aliquot (e.g., 30 mL → 2 filters; 45 mL → 3 filters). Spin each at 14,000× g for 20 min at 4 °C. Avoid serially reloading a single unit, as repeated loading can increase membrane fouling and introduce variability. After ultrafiltration, proceed with depletion/precipitation in parallel, then pool at the protein stage (combine acetone/TCA pellets or their unified resuspension) before reduction/alkylation/digestion.
1. After collecting the conditioned medium, centrifuge at 500× g for 5 min at 4 °C to pellet any detached cells.
2. Transfer the supernatant carefully into a new tube.
3. Pass the clarified medium through a 0.22 μm PVDF or PES filter to remove any remaining particulates.
4. Proceed immediately to ultrafiltration using a 50 kDa MWCO Amicon filter.
5. Centrifuge for 14,000× g for 20 min at 4 °C.
6. At this point, you have collected filtrate at the bottom, where the volume would generally be 10–15 mL, and concentrate on the top, which would generally be 100 μL.
a. Concentrate: proteins >50 kDa retained above the membrane (top chamber).
b. Filtrate: proteins <50 kDa collected in the bottom chamber.
7. The filtrate can be subjected to acetone/TCA in a 50 mL conical tube (see section C). The concentrate should be processed for depletion (see section B).
B. Depletion of abundant protein
Notes:
1. It is recommended to use only 10–20 μL of the concentrate when processing albumin-rich medium to prevent saturation of the depletion column. In contrast, for serum-free medium, the entire concentrate (approximately 100 μL) can be applied to the column without risk of overloading.
2. A simple quality control check can be performed to assess the efficiency of abundant protein depletion by running an SDS-PAGE gel and examining the region corresponding to 65–70 kDa.
1. Apply the concentrate (>50 kDa fraction) to a High-Select HSA/IgG depletion spin column and perform depletion according to the manufacturer’s instructions provided with the High-Select HSA/Immunoglobulin Depletion Resin kit.
2. The flowthrough (depleted sample) should be collected and subsequently processed for acetone/TCA precipitation prior to digestion.
C. Protein precipitation, solubilization, and digestion
Notes:
1. It is not essential to determine protein concentration if the downstream analysis is limited to protein identification by LC–MS/MS. However, for label-free quantification (LFQ) or comparative studies, it is advisable to measure the protein concentration (using BCA or Bradford assay) to ensure equal sample loading across conditions and improve quantitative accuracy.
2. Once you collect the filtrate (<50 kDa) and depleted concentrate (>50 kDa) after following the HSA/immunoglobulin depletion protocol, proceed as follows:
a. For albumin/HSA-rich media, it is recommended to process the concentrate and filtrate separately to prevent high-abundance serum proteins from masking low-abundance secreted peptides. If pooling is preferred for comprehensive analysis, a data-independent acquisition (DIA) workflow is advised to maintain quantitative balance across a wide dynamic range.
b. For HSA-depleted or serum-free (albumin-starved) media, the two fractions can be pooled after acetone/TCA precipitation to maximize recovery of both high- and low-molecular-weight secreted proteins. All buffers used in this protocol are MS-compatible and detergent-free to prevent ion suppression and improve peptide recovery during LC–MS/MS analysis.
This approach ensures optimal sensitivity and reproducibility across different culture conditions and media formulations.
3. Ensure that the final urea concentration is ≤2 M and the pH remains near 8.0, as higher urea or acidic conditions can inhibit trypsin activity and decrease peptide yield.
1. Prepare precipitation solution: Mix prechilled acetone (stored at -20 °C) and TCA in a 4:1 ratio (v/v). Acetone is added first, followed by TCA.
2. To each fraction, add 4 times the sample volume of the ice-cold acetone:TCA solution. Mix gently.
3. Incubate overnight at -20 °C.
4. Centrifuge at 14,000× g for 20 min at 4 °C.
5. Carefully discard the supernatant without disturbing the pellet.
6. Wash the pellet by adding 1 mL of ice-cold acetone. Vortex briefly to loosen the pellet.
7. Centrifuge at 14,000× g for 20 min at 4 °C. Repeat once.
8. Discard the supernatant carefully. If the pellet is not visible, leave around 10–20 μL behind to avoid accidental loss.
9. Air dry the pellet in the fume hood until residual acetone evaporates.
10. Prepare urea resuspension buffer (8 M urea in 400 mM ammonium bicarbonate, pH ~8).
11. Resuspend the dried pellet in 200 μL of 8 M urea and 400 mM ammonium bicarbonate. Make sure the pH is ~8. (Protein concentration can be determined with a Pierce BCA or Micro BCA Protein Assay kit.)
12. Add DTT to a final concentration of 10 mM. Incubate on a thermomixer at 55 °C for 45 min with gentle shaking.
13. Add IAA to a final concentration of 20 mM. Incubate at room temperature for 45 min in a dark environment.
14. Dilute the sample by adding an appropriate volume of 100 mM ammonium bicarbonate to reduce the urea concentration to 2 M (check that the pH is around 7.0–8.0).
15. Add MS-grade trypsin at a 1:50 enzyme-to-protein ratio. Incubate overnight at 37 °C.
16. Stop digestion by adding formic acid to a final concentration of 0.5%.
17. Desalt peptides using Pierce C18 spin columns as per the manufacturer’s protocol. Dry the elutes in a SpeedVac.
18. After desalting, dried peptides can be stored at -20 °C for short-term storage or at -80 °C for long-term storage.
19. Reconstitute peptides in 30 μL or 1 μg/μL of 5% acetonitrile and 0.1% formic acid for LC–MS/MS analysis.
Data analysis
Notes:
1. For label-free quantification, normalize intensities across both fractions (filtrate and depleted concentrate) to enable comparison.
2. If fractions are pooled, be aware that normalization may reduce or mask differences between the filtrate and depleted concentrate.
3. A minimum of three biological replicates is recommended for reliable statistical analysis.
4. DIA-NN is recommended for pooled samples.
Raw LC–MS/MS data can be processed using software such as MaxQuant, Proteome Discoverer, Fragpipe, or DIA-NN, depending on acquisition mode. For label-free quantification, ensure normalization across fractions and replicates. Statistical analysis can be performed using Perseus or R-based pipelines for differential protein expression.
Quality control and recovery assessment
Protein recovery and depletion efficiency may vary slightly depending on the sample composition, serum content, and spin-column performance. To monitor sample integrity and assess recovery, small aliquots can be collected from key workflow stages (post-filtration, post-depletion, and post-precipitation) and analyzed by SDS-PAGE to visualize total protein content and confirm the removal of high-abundance serum proteins (notably albumin at ~66 kDa).
Total peptide yield after digestion can be estimated using a BCA or micro-BCA assay, providing a practical indication of recovery consistency across biological replicates. This workflow typically yields 400–1,500 identified proteins per secretome dataset, depending on the cell type, serum condition, and experimental design. These recovery ranges are consistent with values reported in other published secretome studies.
A successful experiment is characterized by:
• Clear reduction or disappearance of serum-derived bands (e.g., albumin) in SDS-PAGE after depletion.
• Low representation of intracellular or nuclear proteins in LC–MS/MS results, indicating minimal cell lysis or contamination.
• Consistent total peptide yield and stable LC–MS signal intensity across technical and biological replicates.
Validation of protocol
This protocol was validated through multiple secretome experiments performed at the Northwestern Proteomics Core Facility under different biological and culture conditions. The workflow was originally applied to human mesenchymal stromal cells (hMSCs) in Wang et al. [9], where it demonstrated reproducible peptide recovery, efficient depletion of abundant serum proteins, and robust identification of secreted and extracellular matrix proteins.
Further validations have been conducted across different cell types and treatment conditions at the core to assess reproducibility and performance consistency; these studies have yielded comparable results, typically identifying ~400–1,500 proteins using data-dependent acquisition per secretome sample, depending on cell type, medium composition, and acquisition parameters. Collectively, these outcomes confirm that the protocol is robust, reproducible, and broadly applicable for secretome analysis across diverse mammalian cell systems.
General notes and troubleshooting
When adapting this workflow to new cell lines or primary cells, it is important to verify performance through key quality control parameters:
1. Efficient depletion of abundant serum proteins, confirmed by a reduced ~66 kDa albumin band in SDS-PAGE.
2. Low intracellular contamination, reflected by minimal detection of nuclear or cytosolic markers in LC–MS/MS data.
3. Consistent peptide yield across at least three biological replicates. Meeting these criteria indicates successful transfer of the protocol without requiring re-optimization.
This protocol separates the secretome into >50 kDa and <50 kDa fractions, each containing a mixture of secreted and serum-derived proteins. The major analytical challenge lies in minimizing serum protein interference while ensuring reproducible recovery of low-abundance proteins. Therefore, close attention should be paid to depletion efficiency, acetone/TCA precipitation conditions, and desalting consistency. Common issues and their solutions are summarized in Troubleshooting (Table 1).
Finally, this protocol is designed specifically for LC–MS/MS-based proteomic analysis, focusing on the standardized downstream processing of conditioned medium rather than on upstream cell culture optimization. The exact cell density, culture duration, and conditioning parameters are determined by the user’s experimental design, while this protocol ensures reproducible, high-quality sample preparation for mass spectrometry.
Troubleshooting
Table 1. Troubleshooting guide
| Problem | Possible cause | Solution |
| Low protein yield | Incomplete protein precipitation | Maintain a correct acetone:TCA ratio and incubate at -20 °C for enough time |
| High serum background | Inadequate depletion of abundant proteins | Confirm complete spin-column depletion; increase wash volume if necessary |
| Poor digestion efficiency | Excess urea concentration or insufficient time | Ensure urea <2 M before adding trypsin; extend digestion time if required |
| Peptide loss during desalting | Improper column equilibration or over-drying | Equilibrate desalting columns correctly and avoid over-drying peptides |
Acknowledgments
We gratefully acknowledge the support of our affiliated research core facilities. Proteomics analyses were performed at the Northwestern Proteomics Core Facility, which is supported by the NCI Cancer Center Support Grant P30 CA060553 awarded to the Robert H. Lurie Comprehensive Cancer Center, an instrumentation award (S10OD025194) from the NIH Office of the Director, and the National Resource for Translational and Developmental Proteomics funded by P41 GM108569. This protocol was first acknowledged in Wang et al. [9].
Authors’ contributions
Conceptualization, B.B.M. Investigation, B.B.M., R.G., and R.A.P. Instrumentation, R.A.P, Writing—Original Draft, R.G., J.K.C., and B.B.M. Writing—Review & Editing, B.B.M. P.A.F., R.A.P and J.K.C. Funding acquisition, B.B.M. and R.G. Supervision, B.B.M and P.A.F.
Funding: The authors did not receive any specific grant or financial support for the preparation of this manuscript. The Proteomics Core Facility used in this study is supported by the NCI Cancer Center Support Grant P30 CA060553, the NIH Office of the Director instrumentation grant S10OD025194, and the National Resource for Translational and Developmental Proteomics funded by P41 GM108569, which are acknowledged separately.
Competing interests
The authors declare no competing financial or non-financial interests related to this work. The protocol was developed and validated as part of standard service and method development activities within the Northwestern Proteomics Core Facility, with no commercial or proprietary bias.
References
- Knecht, S., Eberl, H. C., Kreisz, N., Ugwu, U. J., Starikova, T., Kuster, B. and Wilhelm, S. (2023). An Introduction to Analytical Challenges, Approaches, and Applications in Mass Spectrometry-Based Secretomics. Mol Cell Proteomics. 22(9): 100636. https://doi:10.1016/j.mcpro.2023.100636
- Ding, Z, Pan, X, Wang, X, Xie, H. and Ye, Q. (2021). Interactions between induced pluripotent stem cells and stem cell niche augment osteogenesis and bone regeneration. Smart Mater Med. 2: 196–208 https://doi.org/10.1016/j.smaim.2021.07.002
- Rajcevic, U., Niclou, S. P. and Jimenez, C. R. (2009). Proteomics strategies for target identification and biomarker discovery in cancer. Front Biosci. 14(9): 3292–3303. https://doi.org/10.2741/3452
- Paulitschke, V., Kunstfeld, R. and Gerner, C. (2010). Secretome Proteomics, a Novel Tool for Biomarkers Discovery and for Guiding Biomodulatory Therapy Approaches. In: Reichle, A. (Ed.). From Molecular to Modular Tumor Therapy. The Tumor Microenvironment, vol 3. Springer, Dordrecht. https://doi.org/10.1007/978-90-481-9531-2_21
- Kim, M.-S., Pinto, S. M., Getnet, D., Nirujogi, R. S., Manda, S. S., Chaerkady, R. and Pandey, A. (2014). A draft map of the human proteome. Nature. 509(7502): 575–581. https://doi.org/10.1038/nature13302
- Luque-Garcia, J. L. and Neubert, T. A. (2007). Sample preparation for serum/plasma profiling and biomarker identification by mass spectrometry. J Chromatogr A. 1153: 259–276. https://doi.org/10.1016/j.chroma.2006.11.054
- Piersma, S. R., Fiedler, U., Span, S., Lingnau, A., Pham, T. V., Hoffmann, S., Kubbutat, M. H. G. and Jiménez, C. R. (2010). Workflow Comparison for Label-Free, Quantitative Secretome Proteomics for Cancer Biomarker Discovery: Method Evaluation, Differential Analysis, and Verification in Serum. J Proteome Res. 9(4): 1913–1922. https://doi.org/10.1021/pr901072h
- Almeida-Marques, C., Rolfs, F., Piersma, S. R., Bijnsdorp, I. V., Pham, T. V., Knol, J. C. and Jimenez, C. R. (2024). Secretome processing for proteomics: A methods comparison. Proteomics. 24(7): 2300262. https://doi.org/10.1002/pmic.202300262
- Wang, X., Li, Y., Lin, Z., Pla, I., Gajjela, R., Mattamana, B. B., Joshi, M., Liu, Y., Wang, H., Zun, A. B., et al. (2025). Microtopography-induced changes in cell nucleus morphology enhance bone regeneration by modulating the cellular secretome. Nat Commun. 16(1): 6444. https://doi.org/10.1038/s41467-025-60760-y
- Cao, X., Sandberg, A., Araújo, J. E., Cvetkovski, F., Berglund, E., Eriksson, L. E. and Pernemalm, M. (2021). Evaluation of Spin Columns for Human Plasma Depletion to Facilitate MS-Based Proteomics Analysis of Plasma. J Proteome Res. 20(9): 4610–4620. https://doi.org/10.1021/acs.jproteome.1c00378
Article Information
Publication history
Received: Sep 16, 2025
Accepted: Nov 3, 2025
Available online: Nov 20, 2025
Published: Dec 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Baby Mattamana, B., Gajjela, R., K.C., J., Parish, R. A. and Faull, P. A. (2025). Optimized Secretome Sample Preparation From High Volume Cell Culture Media for LC–MS/MS Proteomic Analysis. Bio-protocol 15(24): e5542. DOI: 10.21769/BioProtoc.5542.
Category
Systems Biology > Proteomics > Secretome
Biochemistry > Protein > Isolation and purification
Biological Sciences > Biological techniques > Mass spectrometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.