- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Revisiting Primary Microglia Isolation Protocol: An Improved Method for Microglia Extraction
Published: Vol 15, Iss 23, Dec 5, 2025 DOI: 10.21769/BioProtoc.5530 Views: 2133
Reviewed by: Elena A. OstrakhovitchEmmanuelle BerretAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2025
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Microglia, the resident immune cells of the central nervous system, play a crucial role in maintaining neural homeostasis and in regulating neurodevelopment, neuroinflammation, tissue repair, and neurotoxicity. They are also key contributors to the pathogenesis of various neurodegenerative disorders, underscoring the need for in vitro models that accurately recapitulate disease-relevant conditions. Among the available isolation methods, the classical mixed glial culture shaking technique remains the most commonly employed, while alternatives such as magnetic bead separation and fluorescence-activated cell sorting (FACS) offer higher purity but are often constrained by technical complexity and cost. In this study, we refined the traditional shaking method by supplementing specific cytokines during culture to enhance microglial viability and proliferation. Our optimized protocol produced primary microglia with higher purity, greater yield, and improved viability compared with the conventional approach, thereby increasing experimental efficiency while substantially reducing time, animal usage, and overall cost.
Key features
• The microglial cells obtained using this protocol achieve a purity of approximately 90%.
• This protocol maximizes the viability of primary microglial cells.
• The entire procedure requires a minimum of 9 days to complete.
• Antibiotic or antifungal solutions are not used in this protocol.
Keywords: Cell cultureGraphical overview
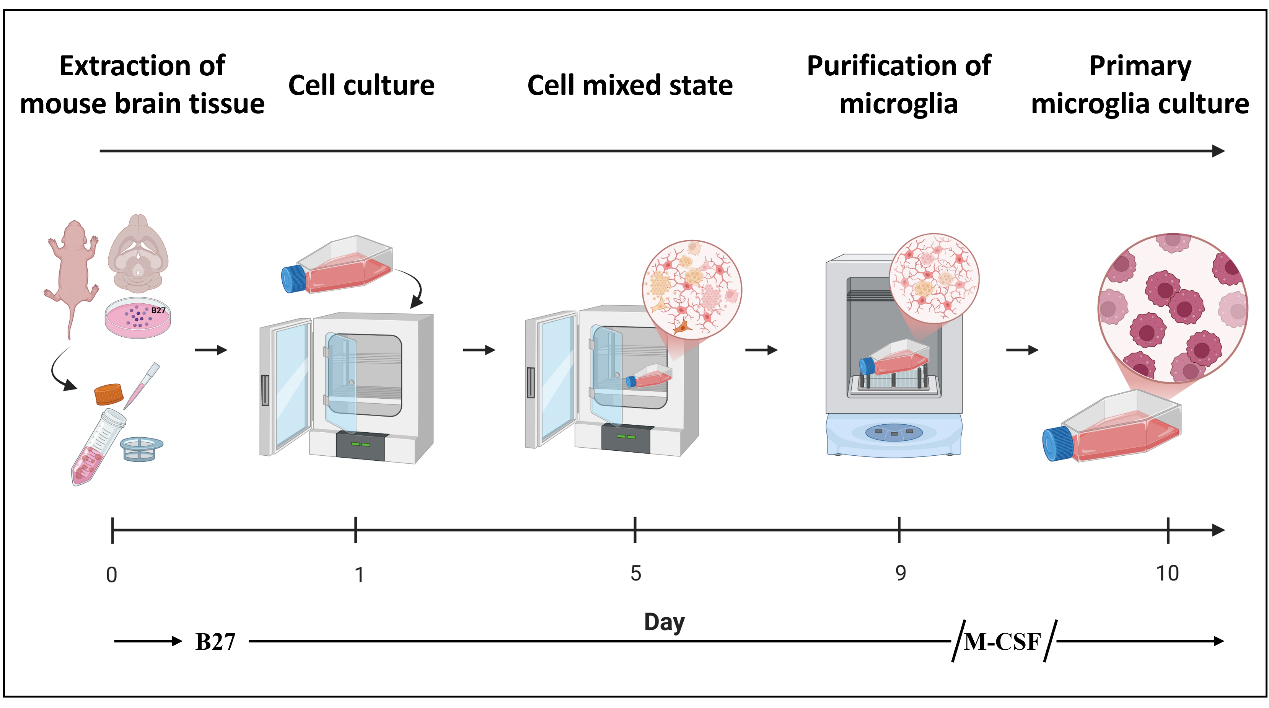
Flowchart of the isolation procedure for primary microglial cells from mice
Background
Microglia are the resident immune effector cells of the central nervous system (CNS), endowed with essential functions such as phagocytosis of pathogens and clearance of apoptotic cells. They are primarily responsible for maintaining neural homeostasis, mediating inflammatory responses, and contributing to tissue repair. Increasing evidence has highlighted their pivotal roles in pathological conditions, including neurodegenerative diseases, brain injury, and psychiatric disorders, which have become major focuses of contemporary neuroscience research [1–4]. Although several established in vitro models, such as the murine BV2 microglial cell line, are widely used, they exhibit intrinsic limitations as experimental systems [5]. In detail, the immortalization of BV2 cells may lead to aberrant gene expression, dysregulated signaling pathways, and altered metabolic states. Compared with primary microglia, BV2 cells lack key homeostatic features and immune functionalities, making them less suitable for accurately modeling the physiological responses of microglia in Parkinson’s disease (PD) [6]. Consequently, obtaining primary microglia with both high yield and high purity is of great importance for mechanistic studies in molecular neuroscience. Furthermore, in vitro experiments employing purified primary microglia provide a controllable and physiologically relevant model for dissecting activation pathways and intercellular interactions, thereby facilitating a deeper understanding of their functions under diverse pathological states.
Several approaches have been developed for the isolation of primary microglia from mice, including differential adherence, magnetic-activated cell sorting (MACS), fluorescence-activated cell sorting (FACS), and the mixed glial culture shaking method [7–9]. Among them, MACS and FACS offer high specificity and yield microglia of greater purity; however, these methods are limited by their high cost and relatively low overall recovery. In contrast, differential adherence and shaking methods are technically straightforward and cost-effective for routine use, yet they typically produce cells of lower purity. Therefore, developing an isolation protocol that allows rapid, efficient, and cost-effective acquisition of highly purified primary microglia would be highly valuable for experimental model establishment and subsequent mechanistic studies.
In this protocol, we optimized the traditional mixed glial culture shaking method for microglial isolation. During the initial brain tissue dissociation and early mixed glial culture stage, the neuronal supplement B27 was added to enhance cell survival and preserve microglial function [10,11]. In the subsequent culture phase, macrophage colony-stimulating factor (M-CSF) was supplemented to promote microglial proliferation and expansion [12,13]. This modified protocol not only produced microglia with higher purity but also markedly shortened the isolation timeline, enabling the acquisition of usable primary microglia within as few as 10 days. Moreover, no antibiotic or antifungal agents were used in this protocol. Importantly, the improved method yielded sufficient numbers of more viable cells to support common biochemical analyses—including western blot, polymerase chain reaction (PCR), and proteomic studies—without requiring large numbers of neonatal mice. In addition, the functional competence of the isolated primary microglia was validated through co-culture with lipopolysaccharide (LPS) stimulation (1 μg/mL) [14].
Materials and reagents
Biological materials
1. C57BL/6-Sncatm2.1(SNCA*A53T)Mdk/J (The Jackson Laboratory, strain: #039167)
2. C57BL/6J (The Jackson Laboratory, strain: #000664)
Reagents
1. Phosphate-buffered saline (PBS) 1×, pH 7.2–7.4 (Solarbio, catalog number: P1020)
2. Dulbecco’s modified Eagle medium/nutrient mixture F-12 (DMEM/F-12), 1:1, 1×, supplemented with 2 mM L-glutamine (Gibco, catalog number: 11320033)
3. Trypsin-EDTA solution, 0.25%, 1× (NCM Biotech, catalog number: C100C1)
4. 4% paraformaldehyde fixative solution (Solarbio, catalog number: P1110)
5. B-27TM supplement (50×), serum-free (Thermo Fisher, catalog number: 17504044)
6. Macrophage colony-stimulating factor (M-CSF) (Sigma-Aldrich, catalog number: SRP3221)
7. Fetal bovine serum (FBS), qualified, heat-inactivated (Thermo Fisher, catalog number: 10091148)
Solutions
1. Wash buffer (see Recipes)
2. DMEM/F12 + 10% FBS (see Recipes)
3. DMEM/F12 + 20% FBS (see Recipes)
Recipes
1. Wash buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DMED/F12 | n/a | 49 mL |
| B27 (50×) | 1× | 1 mL |
| Total | n/a | 50 mL |
2. DMEM/F12 + 10% FBS
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DMED/F12 | n/a | 45 mL |
| FBS | 2.5 mL/L | 5 mL |
| Total | n/a | 50 mL |
3. DMEM/F12 + 20% FBS
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DMED/F12 | n/a | 40 mL |
| FBS | 5 mL/L | 10 mL |
| Total | n/a | 50 mL |
Laboratory supplies
1. 6-cm cell culture dish (JETBIOFIL, catalog number: TCD010060)
2. 12-well cell culture plate (JETBIOFIL, catalog number: TCP001012)
3. 15-mL centrifuge tube (JETBIOFIL, catalog number: CFT411150)
4. T25 cell culture flask (JETBIOFIL, catalog number: TCF002025)
5. 70-μm cell strainer (JETBIOFIL, catalog number: CSS013070)
6. Glass coverslips (Biosharp, catalog number: BS-18-RC)
Equipment
1. BB15 CO2 incubator (Thermo Scientific, catalog number: 51023126)
2. Multifuge X1 Pro centrifuge (Thermo Scientific, catalog number: 75009750)
3. CS-200 orbital shaker (Yooning, catalog number: CS-200)
4. Dissecting scissors (Fine Science Tools, catalog number: 14160-10)
5. Spring scissors (Fine Science Tools, catalog number: 15018-10)
6. Ophthalmic forceps (Fine Science Tools, catalog number: 11053-10)
7. Dissecting forceps (Fine Science Tools, catalog number: 11000-12)
8. Tissue forceps (Fine Science Tools, catalog number: 11021-12)
9. Scalpel handle and blades (Integra Miltex, catalog number: 4-7)
Procedure
A. Isolation of primary mouse glial cells
1. Mixed glial cell preparation: One neonatal (postnatal day 1–2) SNCA A53T transgenic mouse was euthanized by cervical dislocation and surface-sterilized in 75% ethanol for 5 min.
2. Place the sterilized mouse on a sterile surgical platform and perform all procedures on ice under aseptic conditions. Make a midline incision along the scalp and skull to expose the brain tissue. Using sterile forceps, gently separate and carefully remove the whole brain intact. Immediately transfer the excised brain into pre-chilled PBS and incubate for 30 s to remove residual blood and debris. Throughout the procedure, strict aseptic technique should be maintained.
3. Transfer the brain tissue into a 6-cm culture dish containing 5 mL of DMEM/F12 medium supplemented with 2.5 mM L-glutamine and 1× B-27 supplement.
Note: The B-27 supplement is widely used in culture systems for the differentiation of induced pluripotent stem cells (iPSCs) into neurons and their supporting cells, including microglia. In three-dimensional or multicellular co-culture models, B-27 primarily serves as a culture additive that supports neuronal survival and maturation, while providing a stable, serum-free environment conducive to neuronal maintenance [15]. Because the formulation of B-27 contains multiple antioxidants and hormone-like components (such as vitamin E and corticosterone), it can reduce oxidative stress and inflammatory signaling within the culture system. Consequently, B-27 helps microglia maintain a resting, ramified morphology and prevents excessive activation [7,16]. Therefore, in culture systems containing B-27, microglia typically exhibit low-inflammatory, homeostatic characteristics, which contribute to the establishment of a neuroprotective microenvironment.
4. Under a dissection microscope, carefully remove the meninges and surface blood vessels using sterile dissecting scissors and ophthalmic forceps. Place the brain with the dorsal side facing upward and divide it along the midline into two hemispheres. Under the microscope, identify the cerebral cortex (a thin, grayish-white layer on the surface) and the underlying hippocampus (located on the medial side of each hemisphere, with a curved “C” shape). Gently separate along the interface between the cortex and the white matter to remove the cortex and expose the hippocampus. Carefully dissect out the hippocampus along its outer boundary. Transfer the isolated cortical and hippocampal tissues into pre-labeled 1.5 mL microcentrifuge tubes containing PBS and keep them on ice. The entire dissection process should be completed within 10 min to ensure tissue viability.
5. Transfer the 1.5 mL microcentrifuge tubes from the sterile surgical bench to a biosafety cabinet. After washing the tissues with PBS 1–2 times, digest them with 1 mL of 0.25% trypsin at 37 °C for 3 min with gentle pipetting, followed by neutralization with 1 mL of DMEM/F12 medium containing 10% FBS.
6. In a biosafety cabinet, pass the homogenate through a 70-μm cell strainer, collect the filtrate into a clean 15 mL tube, and centrifuge at 300× g for 5 min. Discard the supernatant and resuspend the pellet in 3 mL of DMEM/F12 supplemented with 20% FBS. Then, seed into a T25 culture flask and maintain at 37 °C in a humidified 5% CO2 incubator for 24 h (Figure 1A).
7. After 24 h, numerous floating cell aggregates and debris are observed. Remove the supernatant and wash the culture three times with PBS (Figure 1B), followed by the addition of 5 mL of DMEM/F12 + 20% FBS and further incubation for 5 days.
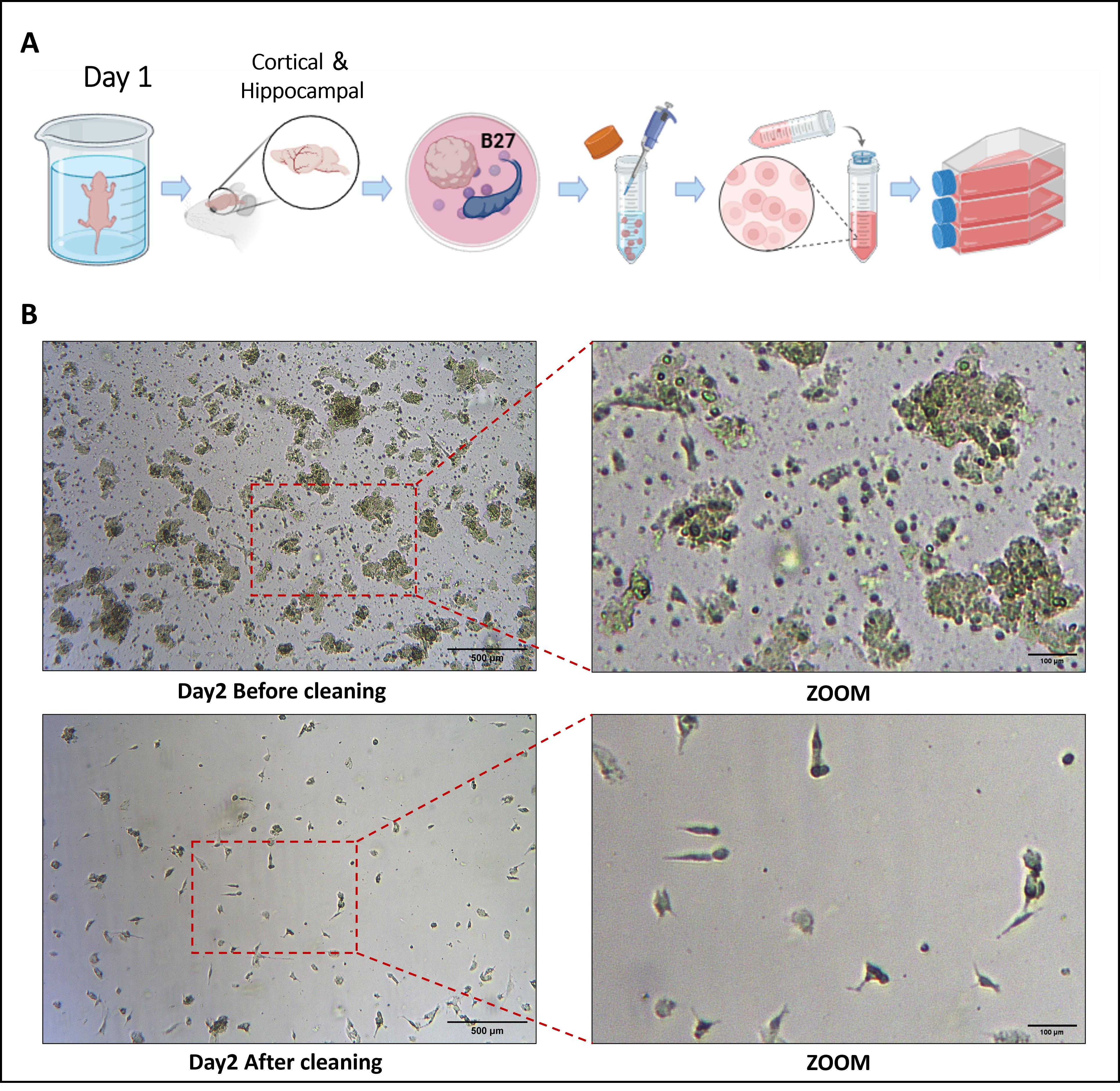
Figure 1. Morphological characteristics of brain tissue–derived cells from nude mice observed under a microscope before and after medium change on day 2 of culture. (A) Day 1: Schematic workflow for isolating primary cells from mice; (B) Day 2: Cell morphology before and after medium change under light microscopy.
8. By day 5, cells exhibit stratified growth: astrocytes and fibroblasts predominate in the adherent layer, while microglia appear as round or oval cells in the upper layer. Following medium replacement, add M-CSF (20 ng/mL) to promote microglial proliferation and maintain the cultures until day 8.
Notes:
1. Since antibiotics and antifungal agents are not used during the culture process, strict aseptic techniques must be maintained throughout dissection. All surgical instruments should be thoroughly sterilized before use.
2. The entire dissection should be performed at low temperature to preserve cell viability.
3. The duration of trypsin digestion should not be prolonged to avoid cell damage.
B. Isolation and purification of primary microglia
1. By day 8 of culture, cellular debris was markedly reduced, and clear stratification was observed. The adherent layer consisted predominantly of astrocytes and fibroblasts, whereas the upper layer was composed of morphologically uniform round or oval-shaped microglia. The culture medium was replaced on day 8.
2. On day 9, seal the T25 flasks and place them on an orbital shaker at 200 rpm for 2 h at 37 °C. Collect the resulting suspended cells and reseed them into fresh T25 flasks to obtain isolated microglia. Replenish the original culture flasks with 5 mL of DMEM/F12 supplemented with 20% FBS and maintain for an additional 5 days; then, harvest a second batch of microglia using the same procedure (Figure 2A, B).
Notes:
1. According to quantitative analysis, approximately 1.6 × 106 primary microglial cells can be isolated from the cerebral cortex and hippocampal regions of a single mouse.
2. If the yield of suspended microglia on day 9 is insufficient, supplementation with M-CSF (10–50 ng/mL) may be applied to promote microglial expansion.
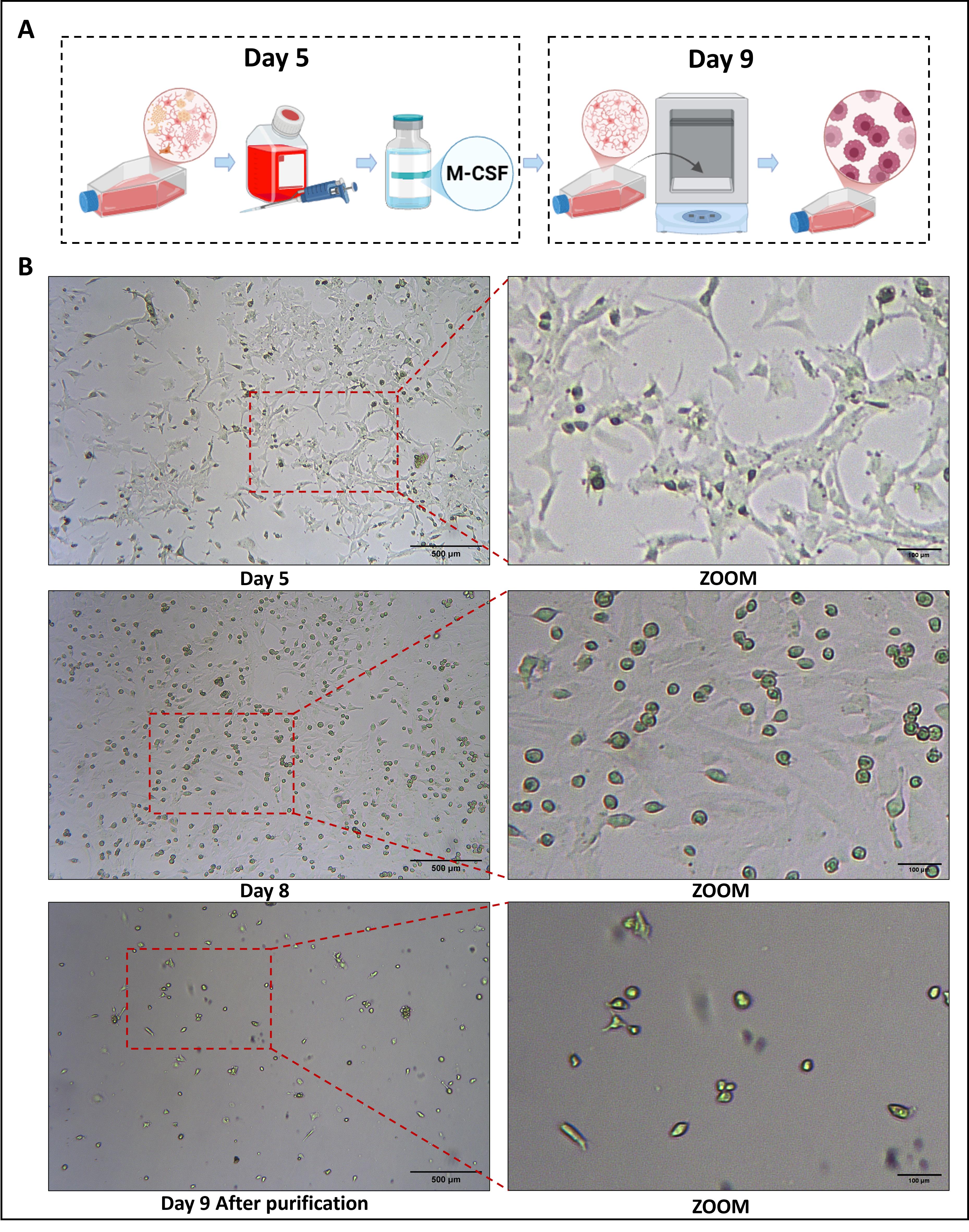
Figure 2. Morphological characteristics observed under a microscope on day 5 and day 8 of maintenance culture after medium change, and on day 9 after harvesting primary microglia. (A) Days 5–9: Schematic workflow of primary cell culture; (B) Days 5, 8, and 9: Cell morphological changes under light microscopy.
C. Identification of primary microglia
1. Cell preparation: Harvest cells by trypsin digestion, resuspend in complete DMEM/F12 supplemented with 20% FBS, and seed at a density of 2 × 105 cells/mL into 6-cm dishes and 12-well plates containing sterile glass coverslips. Incubate overnight at 37 °C in a humidified atmosphere with 5% CO2.
2. After adherence, remove the medium, rinse cells once with PBS, and subsequently fix with 4% paraformaldehyde at 4 °C overnight.
3. Subject cells grown on coverslips to immunofluorescence staining using the microglial marker Iba1 and the astrocytic marker GFAP (Figure 3A).
4. In parallel, the purity of primary microglia is further verified by flow cytometry using the microglia-specific surface marker CD11b (Figure 3B).
5. To evaluate functional activity, treat primary microglia with PBS or lipopolysaccharide (LPS) (1 μg/mL) for 24 h. Subsequently, analyze CD40 surface expression by flow cytometry to assess microglial activation markers (Figure 3C). Then, extract total RNA for qPCR analysis to determine the expression levels of pro-inflammatory cytokines (IL-6, IL-1β, TNF-α) and the anti-inflammatory factor TGF-β (Figure 3D).
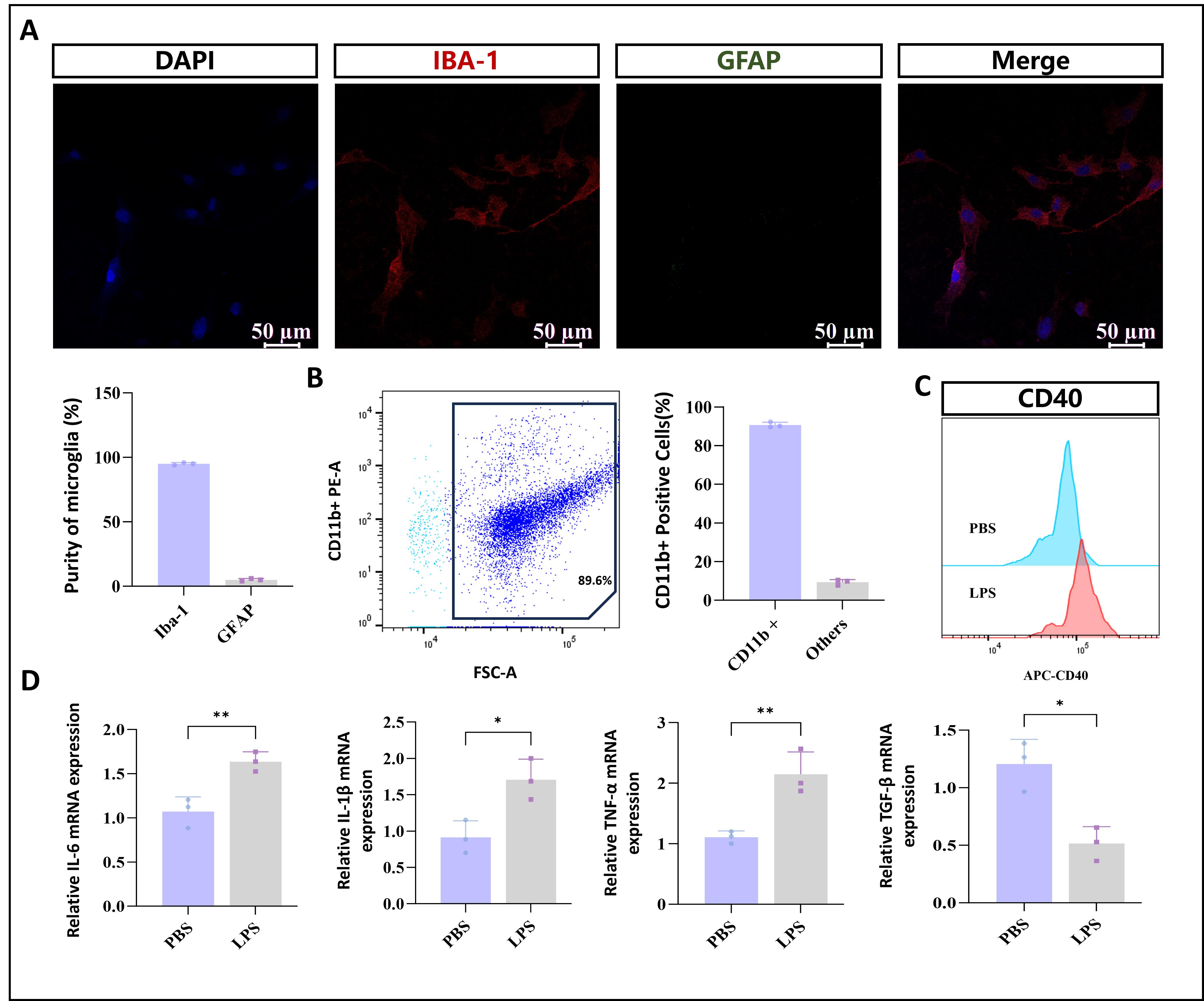
Figure 3. Identification of primary microglia. (A) Immunofluorescence staining of isolated cells with the microglial marker Iba1 and the astrocytic marker GFAP. (B) Flow cytometric analysis of primary microglia proportion using direct CD11b-PE labeling. (C) Flow cytometry analysis showing changes in CD40 surface expression in primary microglial cells from the two groups. (D) Functional validation of the model by stimulating isolated primary microglia with LPS (1 μg/mL). Data are presented as mean ± SD; n = 3 independent biological replicates per group. One-way ANOVA followed by Tukey’s post hoc test was used for multiple comparisons (*p < 0.05, **p < 0.01, ***p < 0.001). Analyses were performed in GraphPad Prism 9.
Data analysis
Flow cytometry data were analyzed using FlowJo software, and statistical analyses were performed with GraphPad Prism. Quantitative data are presented as mean ± standard deviation (mean ± SD). Comparisons between two groups were conducted using Student’s t-test, while differences among multiple groups were assessed by one-way analysis of variance (one-way ANOVA), according to the experimental design. A P-value < 0.05 was considered statistically significant.
Validation of protocol
1. The experimental protocol was reproducibly validated, and all data were analyzed using FlowJo for flow cytometry and GraphPad Prism for statistical evaluation.
2. This protocol has been used and validated in the following research article:
• Liu et al. [17]. LRRK2 Mediates α-Synuclein-Induced Neuroinflammation and Ferroptosis through the p62-Keap1-Nrf2 Pathway in Parkinson’s Disease. Inflammation.: e1007/s10753–025–02291–8. https://doi.org/10.1007/s10753-025-02291-8 (Figure 13).
General notes and troubleshooting
General notes
1. This experimental protocol is applicable to C57BL/6-Sncatm2.1(SNCA*A53T)Mdk/J transgenic mice, standard C57BL/6 mice, and transgenic mice on a C57BL/6 background.
2. According to quantitative analysis, approximately 1.6 × 106 primary microglial cells can be isolated from the cerebral cortex and hippocampal regions of a single mouse.
Troubleshooting
Problem 1: Insufficient yield of primary microglia.
Possible cause: Reduced viability of primary microglia.
Solution: If the number of microglia obtained after initial extraction and transfer into T25 culture flasks (day 9) is insufficient, supplementation with 20 ng/mL M-CSF during medium replacement is recommended. This treatment helps maintain normal dendritic or pseudopod-like morphology and promotes the proliferation of primary microglia.
Acknowledgments
Jianwei Li, Menglin Zhang, Xinran Zhang, Xiao Wu: Conceptualization, Methodology, Writing—Original Draft, Supervision, Formal Analysis. Zijian Zheng, Cheng Xue, Lan Shen: Investigation, Data Curation, Writing—Review & Editing. Guohui Lu: Review and proofreading. Jianwei Li, Zijian Zheng, and Menglin Zhang made the primary contributions to this work and are designated as first authors. All authors contributed to the literature search, read, and approved the final manuscript. All authors confirm their accountability for the research presented in this manuscript, and no further changes to the authorship will be made.
Funding: This study was supported by the National Natural Science Foundation of China (82460265), The Health and Wellness Innovation Talent Project of Jiangxi Province's "Gan po Talent Program" (gpyc20240210), Key Project of Scientific and Technological Innovation of Jiangxi Provincial Health Commission (2025ZD005), and the Yang Fan Project of the First Affiliated Hospital of Nanchang University (RSC-0036).
This protocol was used in [17].
Competing interests
The authors declare that they have no competing interests.
Ethical considerations
All experimental procedures involving animals were conducted in strict adherence to the guidelines outlined in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996). The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the First Affiliated Hospital of Nanchang Capital [Ethics Approval No. (2023) CDYFYYLK (05–017)]. It should be noted that this study did not involve human subjects, and therefore, human ethics approval was not applicable.
References
- Colonna, M. and Butovsky, O. (2017). Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 35(1): 441–468. https://doi.org/10.1146/annurev-immunol-051116-052358
- Hickman, S., Izzy, S., Sen, P., Morsett, L. and El Khoury, J. (2018). Microglia in neurodegeneration. Nat Neurosci. 21(10): 1359–1369. https://doi.org/10.1038/s41593-018-0242-x
- Nayak, D., Roth, T. L. and McGavern, D. B. (2014). Microglia Development and Function. Annu Rev Immunol. 32(1): 367–402. https://doi.org/10.1146/annurev-immunol-032713-120240
- Prinz, M., Jung, S. and Priller, J. (2019). Microglia Biology: One Century of Evolving Concepts. Cell. 179(2): 292–311. https://doi.org/10.1016/j.cell.2019.08.053
- Henn, A., Lund, S., Hedtjärn, M., Schrattenholz, A., Pörzgen, P., and Leist, M. (2009). The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex. 26: 83–94. https://doi.org/10.14573/altex.2009.2.83
- He, Y., Yao, X., Taylor, N., Bai, Y., Lovenberg, T. and Bhattacharya, A. (2018). RNA sequencing analysis reveals quiescent microglia isolation methods from postnatal mouse brains and limitations of BV2 cells. J Neuroinflammation. 15(1): 153. https://doi.org/10.1186/s12974-018-1195-4
- Yip, P. K., Kaan, T. K., Fenesan, D. and Malcangio, M. (2009). Rapid isolation and culture of primary microglia from adult mouse spinal cord. J Neurosci Methods. 183(2): 223–237. https://doi.org/10.1016/j.jneumeth.2009.07.002
- Scott, N., Witt, K. and Schober, J. M. (2022). A Simplified Procedure for Isolation of Primary Murine Microglia. Biotechniques. 73(6): 273–279. https://doi.org/10.2144/btn-2022-0054
- Bohlen, C. J., Bennett, F. C. and Bennett, M. L. (2018). Isolation and Culture of Microglia. Curr Protoc Immunol. 125(1): e70. https://doi.org/10.1002/cpim.70
- Berglund, C. M. D., Aarum, J., Budd Haeberlein, S. L., Nyengaard, J. R., Hökfelt, T., Sandberg, K., Näslund, J. and Persson, M. A. (2004). Characterization of long‐term mouse brain aggregating cultures: Evidence for maintenance of neural precursor cells. J Comp Neurol. 474(2): 246–260. https://doi.org/10.1002/cne.20153
- Chen, Y., Stevens, B., Chang, J., Milbrandt, J., Barres, B. A. and Hell, J. W. (2008). NS21: Re-defined and modified supplement B27 for neuronal cultures. J Neurosci Methods. 171(2): 239–247. https://doi.org/10.1016/j.jneumeth.2008.03.013
- Smith, A. M., Gibbons, H. M., Oldfield, R. L., Bergin, P. M., Mee, E. W., Curtis, M. A., Faull, R. L. M. and Dragunow, M. (2013). M-CSF increases proliferation and phagocytosis while modulating receptor and transcription factor expression in adult human microglia. J Neuroinflammation. 10(1): 85. https://doi.org/10.1186/1742-2094-10-85
- Giulian, D. and Ingeman, J. (1988). Colony-stimulating factors as promoters of ameboid microglia. J Neurosci. 8(12): 4707–4717. https://doi.org/10.1523/jneurosci.08-12-04707.1988
- Nakamura, Y., Si, Q. and Kataoka, K. (1999). Lipopolysaccharide-induced microglial activation in culture: temporal profiles of morphological change and release of cytokines and nitric oxide. Neurosci Res. 35(2): 95–100. https://doi.org/10.1016/s0168-0102(99)00071-1
- Vahsen, B. F., Gray, E., Candalija, A., Cramb, K. M. L., Scaber, J., Dafinca, R., Katsikoudi, A., Xu, Y., Farrimond, L., Wade-Martins, R., et al. (2022). Human iPSC co-culture model to investigate the interaction between microglia and motor neurons. Sci Rep. 12(1): 12606. https://doi.org/10.1038/s41598-022-16896-8
- Hedegaard, A., Stodolak, S., James, W. S. and Cowley, S. A. (2020). Honing the Double-Edged Sword: Improving Human iPSC-Microglia Models. Front Immunol. 11: e614972. https://doi.org/10.3389/fimmu.2020.614972
- Liu, X., Zheng, Z., Xue, C., Wang, X., Li, J., Liu, Z., Xin, W., Xu, X., Zhou, D., Yao, L., et al. (2025). LRRK2 Mediates α-Synuclein-Induced Neuroinflammation and Ferroptosis through the p62-Keap1-Nrf2 Pathway in Parkinson’s Disease. Inflammation.: e1007/s10753–025–02291–8. https://doi.org/10.1007/s10753-025-02291-8
Article Information
Publication history
Received: Aug 25, 2025
Accepted: Oct 19, 2025
Available online: Nov 19, 2025
Published: Dec 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Li, J., Zheng, Z., Zhang, M., Xue, C., Zhang, X. and Lu, G. (2025). Revisiting Primary Microglia Isolation Protocol: An Improved Method for Microglia Extraction. Bio-protocol 15(23): e5530. DOI: 10.21769/BioProtoc.5530.
Category
Neuroscience > Nervous system disorders > Parkinson's disease
Cell Biology > Cell isolation and culture > Cell isolation
Neuroscience > Basic technology
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.