- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Implementation of Fusion Primer-Driven Racket PCR Protocol for Genome Walking
Published: Vol 15, Iss 23, Dec 5, 2025 DOI: 10.21769/BioProtoc.5517 Views: 1129
Reviewed by: Shengze YaoAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2022
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Genome-walking protocols have been extensively used to clone unknown genomic sequences next to known DNAs. Existing genome-walking protocols need further improvement in methodological specificity or operation. Here, we describe a novel genome-walking protocol based on fusion primer–driven racket PCR (FPR-PCR). FPR-PCR involves four sequence-specific oligos (SSO), SSO1, SSO2, SSO3, and SSO4, which are sequentially chosen from known DNA in the direction 5’→3’. The fusion primer, mediating primary FPR-PCR, is generated by attaching SSO3 to the 5’ end of SSO1. The SSO3 encourages the target DNA of primary PCR to form a racket-like structure by mediating intra-strand annealing. SSO2 and SSO4 are directly used as sequence-specific primers (SSP) in secondary FPR-PCR, which selectively amplifies this racket-like DNA. This protocol was verified by cloning several unknown genomic sequences. Compared to traditional PCRs, FPR-PCR offers the advantages of higher specificity and fewer rounds, primarily attributed to the omission of arbitrary walking primers typically required in traditional methods.
Key features
• This FPR-PCR protocol builds upon the method constructed by Pei et al. [1].
• The FPR-PCR protocol relies on a multi-functional fusion primer (FP) that mediates the primary amplification and the formation of racket-like DNA.
• The FPR-PCR comprises only two rounds of amplification reactions.
Keywords: Genome-walking PCRGraphical overview
Background
Genome walking (GW) is a molecular technique for identifying unknown genomic sequences next to known DNAs [2–4]. GW has been extensively used to obtain regulatory sites of genes, amplify non-conserved regions based on conserved DNAs, identify T-DNA, discover new functional genes, or screen microbes [5–7]. Therefore, GW has made significant contributions to the development of life sciences [8–10].
To date, there are many protocols for genome walking, mainly including genomic library methods, ligation-based PCRs, and arbitrary PCRs [11–14]. The genomic library method has been abandoned due to its time-consuming nature [15]. The ligation-based PCRs are also showing a tendency to be phased out, as they require pretreatment of the genomic plate prior to PCR [16–19]. In contrast, arbitrary PCRs directly mediate walking by randomly annealing a walking primer to the unknown flank; in general, the target DNA is obtained after two to three nested amplifications [20–23]. Therefore, arbitrary PCRs are a more rapid and straightforward approach. However, existing arbitrary PCRs typically realize GW by the differential amplification between target DNA and non-target DNA. Obviously, non-target background arising from walking primer challenges these PCRs [24–26].
Herein, we propose an efficient but specific genome-walking protocol, fusion primer–driven racket PCR (FPR-PCR). This method utilizes a fusion primer (FP) to mediate the target amplicon of primary FPR-PCR to form a racket-like structure; then, a secondary PCR, driven by a sequence-specific primer (SSP) pair, selectively enriches this racket-like DNA. As a result, non-target amplification is basically overcome in FPR-PCR. The FPR-PCR was validated by successfully acquiring several unknown flanking genomic DNAs [27–29].
Materials and reagents
Biological materials
1. Genome of Levilactobacillus brevis [30–35], isolated with the TIANamp Bacteria DNA kit
Reagents
1. TIANamp Bacteria DNA kit (TIANGEN, catalog number: 4992448)
2. 1× TE buffer (Sangon, catalog number: B548106)
3. LA Taq polymerase (Takara, catalog number: RR02MA)
4. 6× Loading buffer (Takara, catalog number: 9156)
5. DiaSpin column DNA Gel Extraction kit (Sangon, catalog number: B110092)
6. DL 5,000 DNA marker (Takara, catalog number: 3428Q)
7. Agarose (Sangon, catalog number: A620014)
8. 0.5 M EDTA (Solarbio, catalog number: B540625)
9. GoldView I nucleic acid staining agent (10,000 ×) (Solarbio, catalog number: G8140)
10. Tris (Solarbio, catalog number: T8060)
11. Boric acid (Solarbio, catalog number: B8110)
12. Oligos (Sangon)
gadC-FPα: TGTTTTCTTCTTGCTCT|ATGGTTATTCTCTGGGG
gadC-FPβ: TGTTTTCTTCTTGCTCT|TCTCTGGGGATTGATTG
gadC-SSP2: TTGGGCGTTATAATTCCTGTTTTCTTCTTG
gadC-SSP4: GGAGCGGTAGTGTGTTAGTTGGGTT
Note: The two parts (SSO1 and SSO3) in an FP are separated by a vertical line. The left part is SSO3, and the right part is SSO1.
Solutions
1. 100 μM primer (see Recipes)
2. 10 μM primer (see Recipes)
3. 2.5× TBE buffer (see Recipes)
4. 0.5× TBE buffer (see Recipes)
5. 1.5% agarose gel (see Recipes)
Recipes
1. 100 μM primer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Primer powder | 100 μM | n/a |
| 1× TE buffer | 1× | Volume (μL) specified by the supplier |
| Total | n/a | Volume (μL) specified by the supplier |
2. 10 μM primer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 100 μM primer | 10 μM | 10 μL |
| Ultrapure water | n/a | 90 μL |
| Total | n/a | 100 μL |
3. 2.5× TBE buffer (pH 8.3)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris | 225 mM | 27 g |
| Boric acid | 225 mM | 13.75 g |
| 0.5 M EDTA | 5 mM | 10 mL |
| Ultrapure water | n/a | 950 mL |
| Total | n/a | 1,000 mL |
4. 0.5× TBE buffer (pH 8.3)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 2.5 × TBE buffer | 0.5× | 200 mL |
| Ultrapure water | n/a | 800 mL |
| Total | n/a | 1,000 mL |
5. 1.5% agarose gel
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Agarose powder | 1.5% | 1.5 g |
| 0.5× TBE buffer | 0.5× | 100 mL |
| GoldView I nucleic acid staining agent (10,000×) | 1× | 10 μL |
| Total | n/a | 100 mL |
Laboratory supplies
1. 0.2 mL PCR tubes (Kirgen, catalog number: KG2311)
2. 10 μL pipette tips (Sangon, catalog number: F600215)
3. 0.2 mL pipette tips (Sangon, catalog number: F600227)
4. 1 mL pipette tips (Sangon, catalog number: F630101)
5. 1.5 mL tubes (Labselect, catalog number: MCT-001-150)
Equipment
1. PCR cycler (Analtytikjena, model: Biometra TOne 96G PCR)
2. Electrophoresis apparatus (Beijing Liuyi, model: DYY-6C)
3. Gel imaging system (Bio-Rad, model: ChemiDoc XRS+)
4. Microcentrifuge (Tiangen, model: TGear)
Software and datasets
1. Oligo 7 software (Molecular Biology Insights, Inc., USA)
2. DNASTAR Lasergene software (DNASTAR, Inc., USA)
3. All data are available at https://www.frontiersin.org/articles/10.3389/fgene.2022.969840/full#supplementary-material (access date, 10/18/2022)
Procedure
A. Primer design
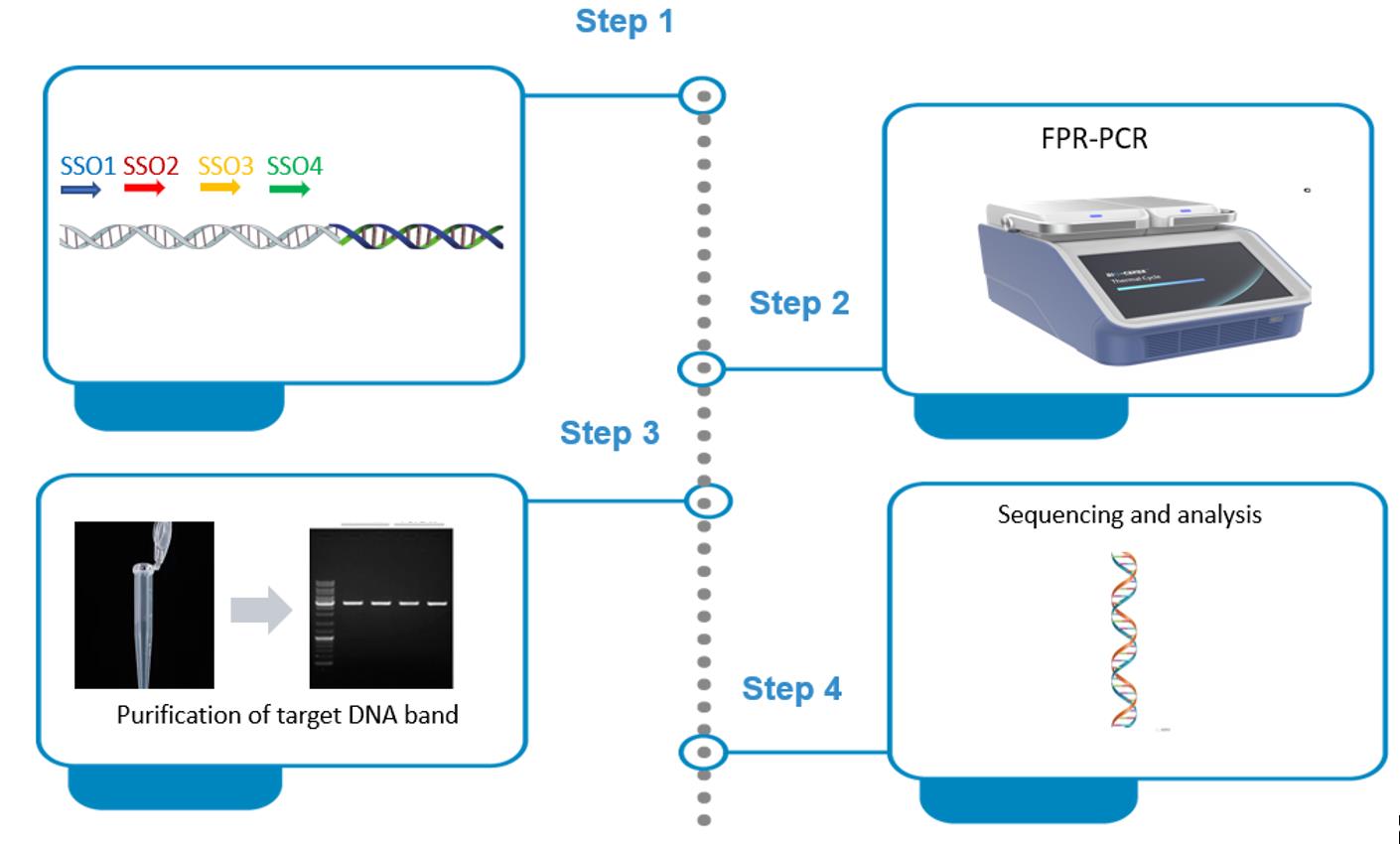
1. Sequentially pick a set of four SSOs—SSO1, SSO2, SSO3, and SSO4—from known DNA in the direction 5’→3′ (Figure 1).

Figure 1. Picking sequence-specific oligos (SSOs). The four SSOs are from known DNA. The fusion primer (FP) is made by attaching SSO3 to the 5’ end of SSO1. FP performs primary FPR-PCR, while SSO2 and SSO4 are directly used as sequence-specific primers (SSPs) to perform secondary FPR-PCR.
Note: We suggest picking more than one SSO1 (SSO1α, SSO1β, …), one SSO2, one SSO3, and one SSO4. Then, attach SSO3 to SSO1s to obtain FPs, so as to perform parallel FPR-PCRs for a walking experiment. In this study, two SSO1s (SSO1α and SSO1β) were picked from each gene to make FPα and FPβ, respectively.
Critical: SSO1s can partially overlap or be completely different. For the former, the difference at the 3′ ends must not be less than 3 nt. SSO1s are 17–21 nt with a melting temperature (Tm) of 50–55 °C. SSO3 is 17–18 nt with a Tm of 45–55 °C. SSO2 or SSO4 is 25–30 nt with a Tm of 60–66 °C.
2. Open the Oligo 7 software and click File and Open to enter a known DNA sequence (Figure 2A).

Figure 2. Screenshots exhibiting how to design a sequence-specific oligo (SSO). (A) How to enter known sequence. (B) How to define the length of SSO.
3. Click Change and Current Oligo Length to define SSO length (Figure 2B).
4. Click Analyze, Duplex Formation, and Current Oligo (Figure 3A) to assess SSO dimer (Figure 3B).
5. Click Analyze, Hairpin Formation, and Current Oligo (Figure 4A) to assess the SSO hairpin (Figure 4B).
Note: If the current SSO forms an obvious dimer(s) or hairpin(s) with a Tm ≥ 40 °C (Figure 4C), pick a new SSO and then assess it by repeating steps A3–A5 until a satisfactory one is obtained.
6. Attach SSO3 to the 5’ end of SSO1 to form FP and assess the FP by repeating steps A3–A5.
Note: If the current FP is unsatisfactory, redesign SSO3 or SSO1 to make a new one until a satisfactory FP is obtained.

Figure 3. Screenshots exhibiting how to assess sequence-specific oligo (SSO) dimer. (A) How to find Duplex Formation and Current Oligo. (B) Output SSO dimer(s).

Figure 4. Screenshots exhibiting how to assess sequence-specific oligo (SSO) hairpin. (A) How to find Hairpin Formation and Current Oligo. (B) Output SSO hairpin(s). (C) An unsatisfactory SSO selection.
B. FPR-PCR amplification
The process of FPR-PCR is outlined in Figure 5.

Figure 5. Outline of fusion primer (FP)-driven racket PCR. The FP’s 3′-part is SSO1, while the 5′-part is SSO3. Primary PCR uses a single FP (FP = SSO3|SSO1), without involving a reverse primer, and the secondary PCR uses nested primers SSO2 and SSO4.
Notes:
1. In primary PCR, the 25 °C cycle promotes FP to partially annealing to the unknown flank. The annealed FP synthesizes a target DNA by extending toward the known region, thereby mediating walking.
2. One type of non-target DNA is also expected to be produced in primary FPR-PCR. In secondary FPR-PCR, however, only the target DNA can be amplified because only this DNA has the binding sites for both SSO2 and SSO4.
1. Primary FPR-PCR
a. Mix primary FPR-PCR components into a PCR tube (Table 1).
Table 1. Composition of primary FPR-PCR
| Reagent | Final concentration | Volume (μL) |
|---|---|---|
| Genomic template | Microbe, 0.2–2 ng/μL; Oryza sativa, 2–20 ng/μL | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| FP (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (containing Mg2+) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| Ultrapure water | n/a | 34.5 |
| Total | n/a | 50 |
b. Fully mix the components using a pipette.
c. Centrifuge at 3,000× g for 10 s to gather the mixture.
d. Run PCR amplification (Table 2).
Table 2. Primary FPR-PCR cycling program
| Step | Temperature (°C) | Duration (min) | Cycle |
| Initial denaturation | 94 | 2 | 1 |
| Denaturation | 94 | 0.5 | 5 |
| Annealing | 55 | 0.5 | |
| Extension | 72 | 3 | |
| Denaturation | 94 | 0.5 | 1 |
| Annealing | 25 | 0.5 | |
| Extension | 72 | 3 | |
| Denaturation | 94 | 0.5 | 30 |
| Annealing | 65 | 0.5 | |
| Extension | 72 | 3 | |
| Final extension | 72 | 10 | 1 |
| Hold | 4 | forever | 1 |
Note: The 25 °C annealing cycle enables PF to partially anneal to the unknown flank and then extend toward the known region, thus mediating genome walking.
e. Take 1 μL of this PCR product as the template of the secondary FPR-PCR.
f. Store the rest of the product at -20 °C.
2. Secondary FPR-PCR
a. Mix secondary FPR-PCR components into a PCR tube (Table 3).
Table 3. Composition of secondary FPR-PCR
| Reagent | Final concentration | Volume (μL) |
|---|---|---|
| Primary FPR-PCR product | n/a | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| SSP2 (10 μM) | 0.2 μM | 1 |
| SSP4 (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (containing Mg2+) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| Ultrapure water | n/a | 33.5 |
| Total | n/a | 50 |
b. Fully mix the components.
c. Centrifuge at 3,000× g for 10 s to gather the mixture.
d. Run PCR amplification (Table 4).
Table 4. Secondary FPR-PCR cycling program
| Step | Temperature (°C) | Duration (min) | Cycle |
| Initial denaturation | 94 | 2 | 1 |
| Denaturation | 94 | 0.5 | 25 |
| Annealing | 60 | 0.5 | |
| Extension | 72 | 3 | |
| Hold | 4 | forever | 1 |
C. Gel electrophoresis
1. Mix 5 μL of FPR-PCR product and 1 μL of 6× loading buffer.
2. Transfer the mixture into a 1.5% agarose gel with 1× GoldView I nucleic acid staining agent.
3. Electrophorese at a voltage of 5 V/cm for 30 min.
4. Check the gel using the ChemiDoc XRS+ imaging system (Figure 6).

Figure 6. Mining the unknown flanking region of gadC. FPα and FPβ denote the two parallel sets of FPR-PCRs. The bands indicated by white arrowheads are the secondary FPR-PCR products. Lane P, primary PCR; lane S, secondary PCR; lane M, TaKaRa DL5000 DNA marker. All four bands marked with arrows were sequenced and verified to be correct.
D. Purification of PCR product
1. Mix 40 μL of secondary FPR-PCR product and 8 μL of 6× loading buffer.
2. Transfer the mixture into a 1.5% agarose gel with 1× GoldView I nucleic acid staining agent.
3. Electrophorese at a voltage of 5 V/cm for 30 min.
4. Check the gel using the ChemiDoc XRS+ imaging system (Figure 6) and cut out target DNA band(s) with a knife.
5. Purify the DNA band(s) from the cut gel using the DiaSpin column DNA Gel Extraction kit.
E. DNA Sequencing
1. Mail the purified product(s) to Sangon Biotech Co., Ltd for sequencing.
Data analysis
1. Analyze sequencing data using the MegAlign software. Open the software and then click File and Enter Sequences (Figure 7A) to input DNA sequences to be analyzed (Figure 7B).

Figure 7. Screenshots exhibiting how to input DNA sequences. (A) How to find Enter Sequences. (B) Input sequences.
2. Click Align and By Clustal W Method (Figure 8A) to get the result (Figure 8B).
Note: The experiment is considered successful if the SSO4-sided segment of the FPR-PCR product overlaps known DNA (Figure 8B).

Figure 8. Screenshots exhibiting how to analyze the input sequences. (A) How to find By Clustal W. (B) Final outcome.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Pei et al. [1]. Fusion primer driven racket PCR: A novel tool for genome walking. Frontiers in Genetics (Figure 6).
General notes and troubleshooting
General notes
1. FPR-PCR is a universal genome-walking protocol.
2. FPR-PCR relies on the formation of racket-like DNA mediated by SSO3.
3. Unlike other arbitrary PCRs that require three rounds of nested amplification, FPR-PCR only requires two amplifications because its secondary amplification is driven by an SSP pair.
4. Like other PCR genome-walking protocols, FPR-PCR also has the issue of multiple bands, but only the largest product needs to be considered.
5. Although the walking length is unpredictable, experimental data indicate that the typical amplicon size range observed in the current FPR-PCR is 0.3–1.6 kb.
6. The band patterns of FPR-PCR are unpredictable, and there is no necessary connection between the band patterns between primary and secondary PCRs. However, secondary PCR generally produces 1–2 DNA bands.
7. Running parallel FPR-PCR sets increases the success rate and efficiency of a walking cycle.
Troubleshooting
Problem 1: No distinct amplicon(s) appear in secondary FPR-PCR.
Possible causes: (i) A weak target amplification or a strong non-target amplification occurs in primary FPR-PCR; or (ii) the SSO set may not be suitable.
Solutions: Appropriately dilute the primary FPR-PCR product, then use 1 μL of the dilution as the template of secondary FPR-PCR. If no distinct amplicon(s) appear yet, redesign an SSO set.
Problem 2: A clear DNA band(s) is the non-target product.
Possible cause: The genome has sites homologous to the SSP(s) in other region(s).
Solution: Redesign an SSP set.
Problem 3: Direct sequencing of the FPR-PCR product is difficult.
Possible cause: There exists interference from non-target background.
Solution: Clone the FPR-PCR product and then sequence.
Problem 4: Smear DNA is observed.
Solution: Reduce cycle numbers if smear DNA appears.
Acknowledgments
This work was funded by the National Natural Science Foundation of China (grant No. 32160014), China; and the Jiangxi Provincial Department of Science and Technology (grant No. 20225BCJ22023), China. This FPR-PCR protocol has been originally described and validated in Frontiers in Genetics [1].
Competing interests
The authors declare no competing interests.
References
- Pei, J., Sun, T., Wang, L., Pan, Z., Guo, X. and Li, H. (2022). Fusion primer driven racket PCR: A novel tool for genome walking. Front Genet. 13: e969840. https://doi.org/10.3389/fgene.2022.969840
- Alquezar‐Planas, D. E., Löber, U., Cui, P., Quedenau, C., Chen, W. and Greenwood, A. D. (2020). DNA sonication inverse PCR for genome scale analysis of uncharacterized flanking sequences. Methods Ecol Evol. 12(1): 182–195. https://doi.org/10.1111/2041-210x.13497
- Leoni, C., Volpicella, M., Placido, A., Gallerani, R. and Ceci, L. R. (2010). Application of a genome walking method for the study of the spinach Lhcb1 multigene family. J Plant Physiol. 167(2): 138–143. https://doi.org/10.1016/j.jplph.2009.06.020
- Gao, D., Pan, Z., Pan, H., Gu, Y. and Li, H. (2025). Center degenerated walking-primer PCR: A novel and universal genome-walking method. Curr Issues Mol Biol. 47(8): 602. https://doi.org/10.3390/cimb47080602
- Grivet, D., Heinze, B., Vendramin, G. G. and Petit, R. J. (2001). Genome walking with consensus primers: application to the large single copy region of chloroplast DNA. Mol Ecol Notes. 1(4): 345–349. https://doi.org/10.1046/j.1471-8278.2001.00107.x
- Leoni, C., Volpicella, M., De Leo, F., Gallerani, R. and Ceci, L. R. (2011). Genome walking in eukaryotes. FEBS J. 278(21): 3953–3977. https://doi.org/10.1111/j.1742-4658.2011.08307.x
- Chen, H., Tian, B., Wang, R., Pan, Z., Gao, D. and Li, H. (2025). Uracil walking primer PCR: An accurate and efficient genome-walking tool. J Genet Eng Biotechnol. 23(2): 100478. https://doi.org/10.1016/j.jgeb.2025.100478
- Yu, Z., Wang, D., Lin, Z. and Li, H. (2025). Protocol to mine unknown flanking DNA using PER-PCR for genome walking. Bio-protocol. 15(1365): e5188. https://doi.org/10.21769/bioprotoc.5188
- Wu, H., Pan, H. and Li, H. (2025). Protocol to retrieve unknown flanking DNA using fork PCR for genome walking. Bio-protocol. 15(1363): e5161. https://doi.org/10.21769/bioprotoc.5161
- Trinh, Q., Shi, H., Xu, W., Hao, J., Luo, Y. and Huang, K. (2012). Loop‐linker PCR: An advanced PCR technique for genome walking. IUBMB Life. 64(10): 841–845. https://doi.org/10.1002/iub.1069
- Jia, M., Ding, D., Liu, X. and Li, H. (2025). Protocol to identify unknown flanking DNA using partially overlapping primer-based PCR for genome walking. Bio-protocol. 15(1364): e5172. https://doi.org/10.21769/bioprotoc.5172
- Ashoub, A. and Abdalla, K. S. (2006). A primer-based approach to genome walking. Plant Mol Biol Rep. 24(2): 237–243. https://doi.org/10.1007/bf02914062
- Chang, K., Wang, Q., Shi, X., Wang, S., Wu, H., Nie, L. and Li, H. (2018). Stepwise partially overlapping primer-based PCR for genome walking. AMB Express. 8(1): 77. https://doi.org/10.1186/s13568-018-0610-7
- Spalinskas, R., Van den Bulcke, M., Van den Eede, G. and Milcamps, A. (2012). LT-RADE: An efficient user-friendly genome walking method applied to the molecular characterization of the insertion site of genetically modified maize MON810 and rice LLRICE62. Food Anal Methods. 6(2): 705–713. https://doi.org/10.1007/s12161-012-9438-y
- Li, H., Ding, D., Cao, Y., Yu, B., Guo, L. and Liu, X. (2015). Partially overlapping primer-based PCR for genome walking. PLoS One. 10(3): e0120139. https://doi.org/10.1371/journal.pone.0120139
- Wang, R., Gu, Y., Chen, H., Tian, B. and Li, H. (2025). Uracil base PCR implemented for reliable DNA walking. Anal Biochem. 696: 115697. https://doi.org/10.1016/j.ab.2024.115697
- Li, H., Wei, C., Pan, Z. and Liu, X. (2024). Arbitrarily suffixed sequence-specific primer PCR for reliable genome walking: Taking genome walkings of Levilactobacillus brevis and rice as examples. Iran J Biotechnol. 22(4): 106–111. https://doi.org/10.30498/ijb.2024.449960.3896
- Tian, B., Wu, H., Wang, R., Chen, H. and Li, H. (2024). N7-ended walker PCR: An efficient genome-walking tool. Biochem Genet. 63(4): 3758–3772. https://doi.org/10.1007/s10528-024-10896-1
- Li, F., Fu, C. and Li, Q. (2019). A simple genome walking strategy to isolate unknown genomic regions using long primer and RAPD primer. Iran J Biotechnol. 17(2): 89–93. https://doi.org/10.21859/ijb.2183
- Guo, X., Zhu, Y., Pan, Z., Pan, H. and Li, H. (2024). Single primer site-specific nested PCR for accurate and rapid genome-walking. J Microbiol Methods. 220: 106926. https://doi.org/10.1016/j.mimet.2024.106926
- Li, H., Lin, Z., Guo, X., Pan, Z., Pan, H. and Wang, D. (2024). Primer extension refractory PCR: An efficient and reliable genome walking method. Mol Genet Genomics. 299(1): 27. https://doi.org/10.1007/s00438-024-02126-5
- Liu, Y. G. and Whittier, R. F. (1995). Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics. 25(3): 674–681. https://doi.org/10.1016/0888-7543(95)80010-j
- Chen, H., Wei, C., Lin, Z., Pei, J., Pan, H. and Li, H. (2024). Protocol to retrieve unknown flanking DNA sequences using semi-site-specific PCR-based genome walking. STAR Protoc. 5(1): 102864. https://doi.org/10.1016/j.xpro.2024.102864
- Pan, H., Guo, X., Pan, Z., Wang, R., Tian, B. and Li, H. (2023). Fork PCR: a universal and efficient genome-walking tool. Front Microbiol. 14: e1265580. https://doi.org/10.3389/fmicb.2023.1265580
- Wang, L., Jia, M., Li, Z., Liu, X., Sun, T., Pei, J., Wei, C., Lin, Z. and Li, H. (2023). Protocol to access unknown flanking DNA sequences using Wristwatch-PCR for genome-walking. STAR Protoc. 4(1): 102037. https://doi.org/10.1016/j.xpro.2022.102037
- Wei, C., Lin, Z., Pei, J., Pan, H. and Li, H. (2023). Semi-site-specific primer PCR: A simple but reliable genome-walking tool. Curr Issues Mol Biol. 45(1): 512–523. https://doi.org/10.3390/cimb45010034
- Lin, Z., Wei, C., Pei, J. and Li, H. (2023). Bridging PCR: An efficient and reliable scheme implemented for genome-walking. Curr Issues Mol Biol. 45(1): 501–511. https://doi.org/10.3390/cimb45010033
- Sun, T., Jia, M., Wang, L., Li, Z., Lin, Z., Wei, C., Pei, J. and Li, H. (2022). DAR-PCR: A new tool for efficient retrieval of unknown flanking genomic DNA. AMB Express. 12(1): 131. https://doi.org/10.1186/s13568-022-01471-1
- Wang, L., Jia, M., Li, Z., Liu, X., Sun, T., Pei, J., Wei, C., Lin, Z. and Li, H. (2022). Wristwatch PCR: A versatile and efficient genome walking strategy. Front Bioeng Biotechnol. 10: e792848. https://doi.org/10.3389/fbioe.2022.792848
- Wang, L., Jia, M., Gao, D. and Li, H. (2024). Hybrid substrate-based pH autobuffering GABA fermentation by Levilactobacillus brevis CD0817. Bioprocess Biosyst Eng. 47(12): 2101–2110. https://doi.org/10.1007/s00449-024-03088-z
- Li, H., Pei, J., Wei, C., Lin, Z., Pan, H., Pan, Z., Guo, X. and Yu, Z. (2023). Sodium-ion-free fermentative production of GABA with Levilactobacillus brevis CD0817. Metabolites. 13(5): 608. https://doi.org/10.3390/metabo13050608
- Li, H., Wang, L., Nie, L., Liu, X. and Fu, J. (2023). Sensitivity intensified ninhydrin-based chromogenic system by ethanol-ethyl acetate: Application to relative quantitation of GABA. Metabolites. 13(2): 283. https://doi.org/10.3390/metabo13020283
- Li, H., Sun, T., Jia, M., Wang, L., Wei, C., Pei, J., Lin, Z. and Wang, S. (2022). Production of gamma-aminobutyric acid by Levilactobacillus brevis CD0817 by coupling fermentation with self-buffered whole-cell catalysis. Fermentation. 8(7): 321. https://doi.org/10.3390/fermentation8070321
- Jia, M., Zhu, Y., Wang, L., Sun, T., Pan, H. and Li, H. (2022). pH auto-sustain-based fermentation supports efficient gamma-aminobutyric acid production by Lactobacillus brevis CD0817. Fermentation. 8(5): 208. https://doi.org/10.3390/fermentation8050208
- Gao, D., Chang, K., Ding, G., Wu, H., Chen, Y., Jia, M., Liu, X., Wang, S., Jin, Y., Pan, H., et al. (2019). Genomic insights into a robust gamma-aminobutyric acid-producer Lactobacillus brevis CD0817. AMB Express. 9(1): 72. https://doi.org/10.1186/s13568-019-0799-0
Article Information
Publication history
Received: Sep 22, 2025
Accepted: Oct 24, 2025
Available online: Nov 3, 2025
Published: Dec 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Gu, Y., Pei, J., Li, M., Tang, Q. and Li, H. (2025). Implementation of Fusion Primer-Driven Racket PCR Protocol for Genome Walking. Bio-protocol 15(23): e5517. DOI: 10.21769/BioProtoc.5517.
Category
Molecular Biology > DNA > Genome walking
Microbiology > Microbial genetics > DNA
Molecular Biology > DNA > PCR
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.