- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Practical CRISPR-Based Method for Rapid Genome Editing in Caulobacter crescentus
Published: Vol 15, Iss 21, Nov 5, 2025 DOI: 10.21769/BioProtoc.5490 Views: 2075
Reviewed by: Alessandro DidonnaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2025
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The RNA-guided Cas enzyme specifically cuts chromosomes and introduces a targeted double-strand break, facilitating multiple kinds of genome editing, including gene deletion, insertion, and replacement. Caulobacter crescentus and its relatives, such as Agrobacterium fabrum and Sinorhizobium meliloti, have been widely studied for industrial, agricultural, and biomedical applications; however, their genetic manipulations are usually characterized as time-consuming and labor-intensive. C. crescentus and its relatives are known to be CRISPR/Cas-recalcitrant organisms due to intrinsic limitations of SpCas9 expression and possible CRISPR escapes. By fusing a reporting gene to the C terminus of SpCas9M and precisely manipulating the expression of SpCas9M, we developed a CRISPR/SpCas9M-reporting system and achieved efficient genome editing in C. crescentus and relatives. Here, we describe a protocol for rapid, marker-less, and convenient gene deletion by using the CRISPR/SpCas9M-reporting system in C. crescentus, as an example.
Key features
• CRISPR-SpCas9M-reporting system overcomes the limitation of CRISPR escape and achieves a high apparent editing efficiency.
• The method enables multiple kinds of genome editing, generating in-frame and marker-less chromosomal modifications.
• The method completes a cycle of genome editing within one week.
• The method could be readily applied for genome editing in C. crescentus, A. fabrum, and S. meliloti.
Keywords: CRISPRGraphical overview
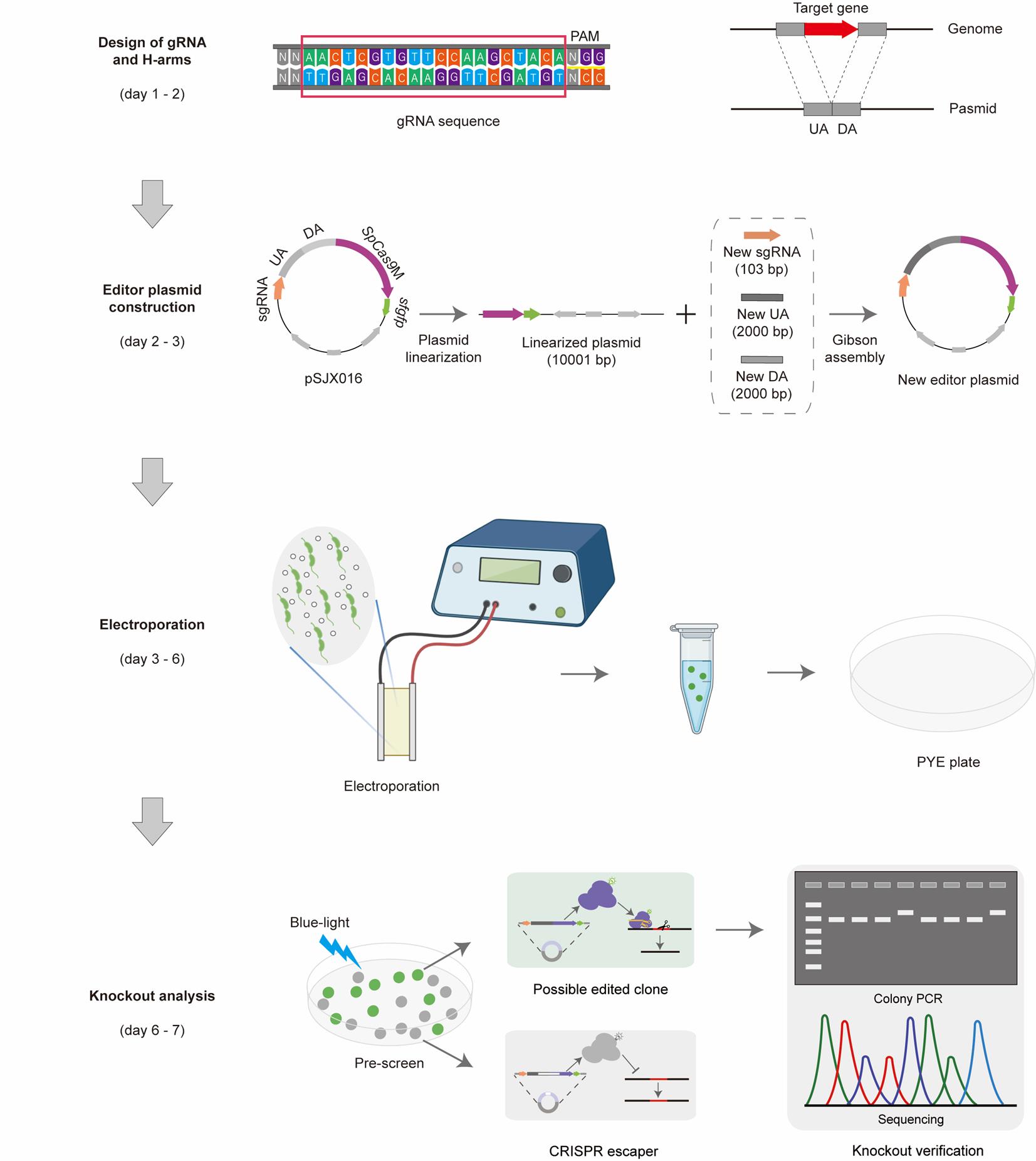
Workflow of gene deletion using the CRISPR/SpCas9M-reporting system. Step 1: Design of gRNA and H-arms. All the gRNA sequences were designed in CHOPCHOP [1]. H-arms consist of the upstream arm (UA, 2,000 bp) and the downstream arm (DA, 2,000 bp) of the targeted gene. Step 2: Editor plasmid construction. The starting plasmid pSJX016 was used as a starting point by replacing the sgRNA and H-arms in just one round of Gibson assembly. The corrected editor plasmids were checked by PCR and sequencing. Step 3: Electroporation. The new editor plasmid was transferred into the competent cells of C. crescentus through electroporation, and the possible edited cells were first selected on the PYE plates with the corresponding resistance. Step 4: Knockout analysis. The clones normally expressing SpCas9M were prescreened by the indication of fluorescent sfGFP under a blue-light lamp, and edited clones were finally confirmed by colony PCR and sequencing.
Background
Caulobacter crescentus, a member of Caulobacterales within α-proteobacteria, is a free-living bacterium widely distributed in freshwater environments [2,3]. Due to its tightly regulated cell cycle and asymmetric cell division, C. crescentus serves as an excellent model for studying cell polarity and cell differentiation [4–6]. The current genetic tool for chromosomal deletions and insertions in C. crescentus was first established in 1991 by Ely et al., utilizing sacB-based counterselection and allelic exchange via homologous recombination (HR) [7]. However, this method is labor-intensive and exhibits a relatively low editing efficiency [8]. In 2006, Martin Thanbichler and Lucy Shapiro et al. reported a single-crossover integration strategy that enables efficient gene insertion in C. crescentus [9,10]. Despite its greatly improved efficiency, this approach integrates the entire editing plasmid (including the selection marker) into the genome and is unsuitable for gene knockout.
CRISPR technology enables precise genome editing by utilizing a vector encoding the Cas enzyme and single-guide RNA (sgRNA). Guided by the sgRNA, the Cas enzyme induces a double-strand break (DSB) at the target sequence [11,12]. The corresponding cells then repair the DSB through either non-homologous end joining (NHEJ) or homology-directed repair (HDR), facilitating targeted gene knockout or knock-in [13]. Unlike the first-generation zinc finger nucleases (ZFNs) and the second-generation transcription activator-like effector nucleases (TALENs), CRISPR relies on the base pairing between gRNA and target DNA rather than protein–DNA binding interactions [14]. This enables CRISPR to exhibit distinct advantages, including both accuracy (editing specificity) and practicality (speed and cost-effectiveness).
For decades, scientists have attempted to develop efficient genome-editing tools for C. crescentus and its relatives via HR-based approaches. However, these efforts have largely been unsuccessful [15–17]. To address the challenges, we developed a CRISPR/Cas-assisted HR method for C. crescentus using an all-in-one editor plasmid, named CRISPR/SpCas9M-reporting system [18]. The reporting system facilitates the identification of CRISPR escape events and increases the apparent editing efficiency in C. crescentus. This method enables targeted gene deletion and insertion within less than a week, producing in-frame and marker-less edits on the chromosomes. Moreover, it has been successfully applied to two C. crescentus relatives, Agrobacterium fabrum and Sinorhizobium meliloti, establishing it as a general editing strategy [18]. We anticipate that this practical tool could be applicable to other difficult-to-edit organisms, facilitating both basic and applied research in α-proteobacteria.
Materials and reagents
Biological materials
1. C. crescentus NA1000 (Lucy Shapiro lab, Department of Developmental Biology, Stanford University School of Medicine)
2. Trans5α chemically competent cell (Transgen Biotech, catalog number: CD201-01)
3. pSJX016 plasmid (our lab, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology)
Reagents
1. PCR primers (Sangon Biotech), standard 25–50 nmol scale
2. 2× Phanta Flash Master Mix (Vazyme, catalog number: P520-01/02/03)
3. KOD OneTM PCR Master Mix (TOYOBO, catalog number: 450800)
4. Agarose (Baygene Biotech, catalog number: 232475)
5. BactoTM peptone (BD, catalog number: 211677)
6. BactoTM yeast extract (BD, catalog number: 212750)
7. BactoTM agar (BD, catalog number: 214010)
8. Magnesium sulfate (MgSO4) (Sigma, catalog number: 10034-99-8)
9. SYBRTM Safe DNA gel stain (Invitrogen, catalog number: 2978645)
10. Ethanol absolute (Macklin, catalog number: 64-17-5)
11. 50× TAE (Sangon Biotech, catalog number: B548101-0500)
12. LB broth (Huankai Microbial, catalog number: 028324)
13. LB agar (Huankai Microbial, catalog number: 028334)
14. Gel Extraction kit (Omega, catalog number: D2500020000E20Y090)
15. EasyPure Plasmid MiniPrep kit (TransGen Biotech, catalog number: EM101-02)
16. ClonExpress MultiS One Step Cloning kit (Vazyme, catalog number: C113-01/02)
17. FastDigest DpnI (Thermo Scientific, catalog number: FD1704)
18. 10× FastDigest buffer (Thermo Scientific, catalog number: FD1704)
19. DNA marker (Accurate Biology, catalog number: AG11909)
20. Kanamycin (Sangon Biotech, catalog number: A600286-0025)
21. ddH2O
22. Vanillic acid (Sigma, catalog number: 34770-10G)
23. MgSO4 (Sigma, catalog number: 10043-52-4)
24. CaCl2 (Sigma, catalog number: 10034-99-8)
Solutions
1. Kanamycin stock solution (50 mg/mL) (see Recipes)
2. LB agar medium (see Recipes)
3. LB liquid medium (see Recipes)
4. Peptone yeast extract (PYE) agar medium (see Recipes)
5. PYE liquid medium (see Recipes)
Recipes
1. Kanamycin stock solution (50 mg/mL)
a. Dissolve 0.5 g of kanamycin (powder) in 10 mL of distilled water at room temperature and filter the solution through a 0.2 μm filter. Then, aliquot the solution into 1 mL fractions and store in a -20 °C freezer.
b. When necessary, supplement media with kanamycin at the following concentrations: 5/25 and 50/50 (liquid/solid media for C. crescentus and for E. coli, respectively, in μg/mL).
2. LB agar medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Bacto tryptone | 1% (w/v) | 10 g |
| Yeast extract | 0.5% (w/v) | 5 g |
| Agar | 1.5% (w/v) | 15 g |
| NaCl | 0.5% (w/v) | 5 g |
| H2O | n/a | Fill up to 1 L |
| Total | n/a | 1 L |
3. LB liquid medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Bacto tryptone | 1% (w/v) | 10 g |
| Yeast extract | 0.5% (w/v) | 5 g |
| NaCl | 0.5% (w/v) | 5 g |
| H2O | n/a | Fill up to 1 L |
| Total | n/a | 1 L |
4. PYE agar medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Peptone | 0.2% (w/v) | 2 g |
| Yeast extract | 0.1% (w/v) | 1 g |
| Bacto agar | 1.5% (w/v) | 15 g |
| 1 M MgSO4 | 1 mM | 1 mL |
| 1 M CaCl2 | 0.5 mM | 0.5 mL |
| H2O | n/a | Fill up to 1 L |
| Total | n/a | 1 L |
5. PYE liquid medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Peptone | 0.2% (w/v) | 2 g |
| Yeast extract | 0.1% (w/v) | 1 g |
| 1 M MgSO4 | 1 mM | 1 mL |
| 1 M CaCl2 | 0.5 mM | 0.5 mL |
| H2O | n/a | Fill up to 1 L |
| Total | n/a | 1 L |
Laboratory supplies
1. Petri dishes (Fisher Scientific, catalog number: FB0875712)
2. 0.22 μm Millipore filter (Millipore, catalog number: R7MA69670)
3. 0.2 mL PCR tube (Corning Life Sciences, catalog number: PCR-0208-C)
4. 1.5 mL microtube (Axygen, catalog number: MCT-150-C)
5. Electroporation cuvette (Bio Rad, catalog number:1652082)
6. Polypropylene round-bottom tube (Falcon, catalog number: 352059)
7. Disposable cell spreader (BKMAMLAB, catalog number: 110305003)
8. Erlenmeyer flask (HEQI GLASS, catalog number: B-000204)
9. High-temperature resistant tissue sealing film (BKMAMLAB, catalog number: 110104005)
10. Pipette tips (KIRGEN, catalog number: KG1333)
11. Cuvettes (Loikaw, catalog number: B-002016)
12. Spectrophotometer (SHST, Model: SH-NanoOne)
Equipment
1. Pipettes (any source)
2. PCR thermocycler (Thermo Fisher Scientific, model: ProFlexTM Base)
3. DNA electrophoresis apparatus (Guangzhou Dao Yi Science and Technology Co., Ltd, model: EPHC 400)
4. Gel image analysis system (Tanon, model: MINI SPACE 960)
5. Nanodrop spectrophotometer (Shenhua Science Technology, model: SH-NnoOne)
6. Centrifuge (Eppendorf, model: 5425)
7. Bacterial incubator (Shanghai Bluepard Instruments)
8. Shaker (Shanghai Chuzhi Biotechnology, model: ZQLY-180ES)
9. Water bath (Shanghai Bluepard Instruments, model: DK-8D)
10. Electroporator (Bio-Rad, model: MicroPulser)
11. Blue-light lamp (MIULAB, model: DUT-48)
Software and datasets
1. Snapgene®4.3.6 (GSL Biotech)
2. Adobe Illustrator, 2021
3. GraphPad Prism 8
4. CHOPCHOP (https://chopchop.cbu.uib.no/)
5. TargetFinder algorithm (https://github.com/ ECBCgit/targetFinder)
Procedure
A. gRNA design
1. Design gRNA in CHOPCHOP sgRNA designer (https://chopchop.cbu.uib.no/) [1] using C. crescentus NA1000 as the target organism.
2. Search for the gene of interest in the genome of C. crescentus NA1000. Here, we selected spmX as the gene of interest (Figure 1).
3. Click the Target box and paste the gene sequence of spmX in FASTA format.
4. Click the In box and select Caulobacter crescentus (NA1000).
5. Click the Using box and select CRISPR/Cas9.
6. Click the Using box and select knockout.
7. Click the button Find Target Sites! (All the parameters in the options are default parameters).
8. The length of the gRNA sequence is 20 bp, and the protospacer adjacent motif (PAM) for Streptococcus pyogenes Cas9 (SpCas9) is 5’-NGG-3’ (Table 1).
9. The spacer sequence may be located on the non-transcribed strand or the transcribed strand.
10. Select gRNAs based on computational predictions of high on-target editing efficiency (efficiency score > 60), while excluding sequences with potential self-complementarity or off-target genomic binding.
11. The three most promising gRNAs should be selected.
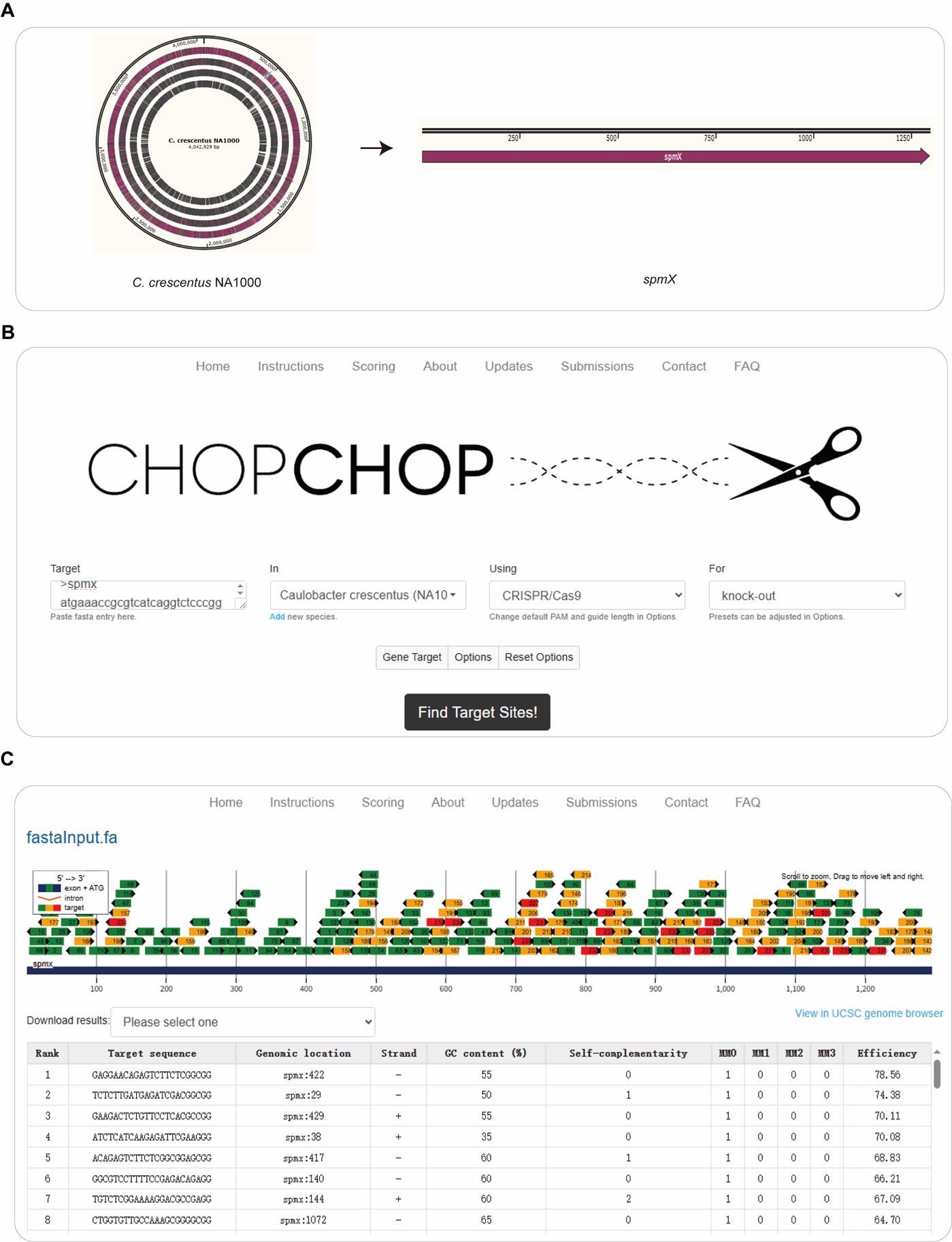
Figure 1. Design gRNAs in CHOPCHOP. (A) C. crescentus NA1000 was selected as the target organism, and spmX was selected as the gene of interest. (B, C) The gene sequence of spmX was pasted in the target box in fasta format on the CHOPCHOP web interface (https://chopchop.cbu.uib.no/). The following parameters were used: “Caulobacter crescentus (NA1000)” was selected in the In field, “CRISPR/Cas9” was chosen in the Using field, and “Knockout” was specified in the For field. After clicking the Find Target Sites! button, a list of all potential gRNA sequences was generated within spmX. From these output results, the most promising gRNAs with relatively high predicted on-target editing efficiencies (>60) were selected for further use.
Table 1. gRNAs for spmX editing, designed by CHOPCHOP
| Target sequence | Self-complementarity | MM0 | MM1 | MM2 | MM3 | Efficiency |
|---|---|---|---|---|---|---|
| GAGGAACAGAGTCTTCTCGGCGG | 0 | 1 | 0 | 0 | 0 | 78.56 |
| GAAGACTCTGTTCCTCACGCCGG | 0 | 1 | 0 | 0 | 0 | 70.11 |
| ATCTCATCAAGAGATTCGAAGGG | 0 | 1 | 0 | 0 | 0 | 70.08 |
MM0 = 0 mismatches, MM1 = 1 mismatches, MM2 = 2 mismatches, MM3 = 3 mismatches. The regions in bold font represent the PAM sequences of the target DNA.
B. Editor plasmid construction
1. Design H-arms according to the target gene. H-arms consist of upstream (UA) and downstream arms (DA). Select the upstream sequence (2,000 bp) of the start codon as the UA and the downstream sequence (2,000 bp) of the stop codon as DA.
2. Preparation of linearized vector, H-arms, and sgRNA fragments: The primers used to amplify the linearized fragments are listed in Table 2.
Table 2. Primers used in editor plasmid construction
| Fragment | Forward primer | Reverse primer | Size |
|---|---|---|---|
| Vector | tccaagctggcgcgcccagcggcgcgga | ccgagaagactctgttcctcactagtattatacctaggactgagctagctg | 11,011 bp |
| sgRNA | gaggaacagagtcttctcgggttttagagctagaaatagcaagttaaaataaggctagtc | gggtgattgtaaaaaaagcaccgactcggtg | 113 bp |
| UA | accgagtcggtgctttttttacaatcacccgatcgccc | ggcaagaaaccgcctagattgacccagtttgag | 2,020 bp |
| DA | aatctaggcggtttcttgcctcgccacc | gctgggcgcgccagcttggaccgatcgt | 2,020 bp |
a. Amplify the linearized vector via PCR using pSJX016 (Figure 2) as the template (Figure 3).
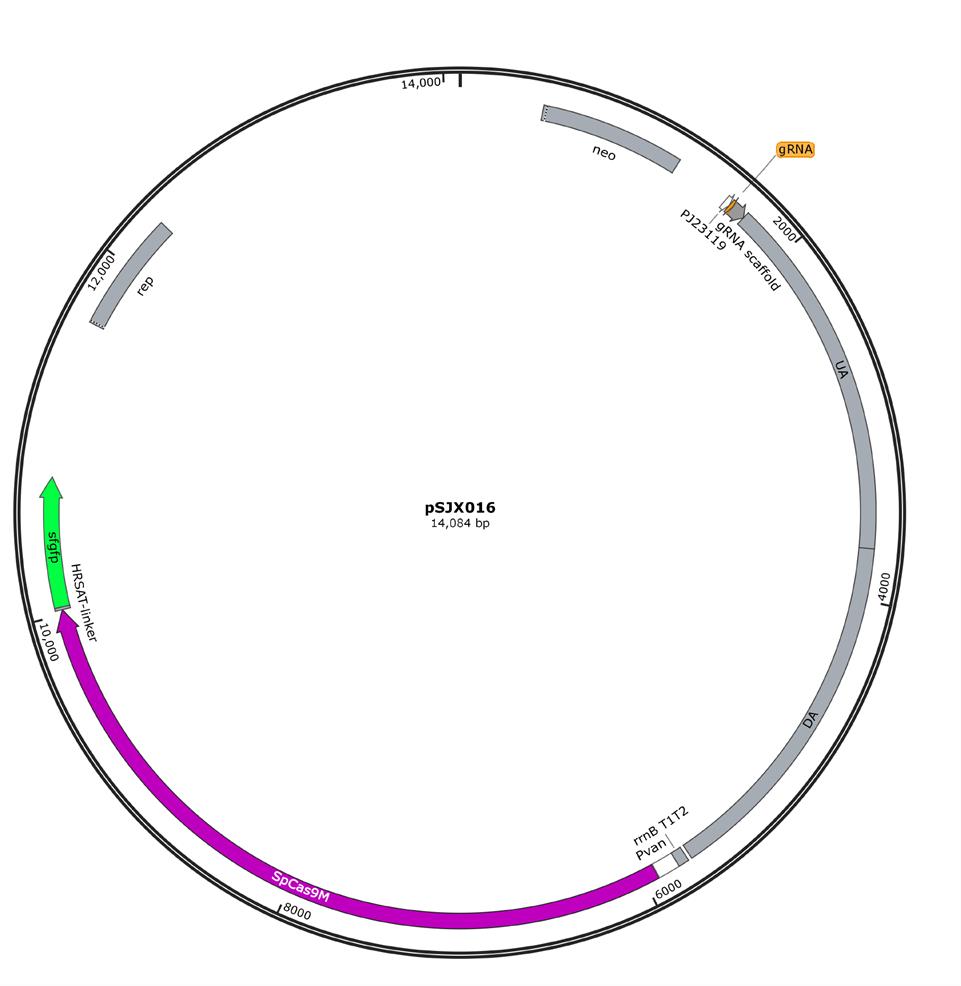
Figure 2. Map of starting editor plasmid pSJX016. pSJX016 is used for genome editing in C. crescentus and confers kanamycin resistance in the host cells. It contains the codon-optimized SpCas9M that is driven by an inducible promoter (Pvan, working concentration: 50 µM vanillic acid), the reporter gene, sfgfp, which is fused to the C terminus of SpCas9M via a flexible HRSAT linker to prescreen the clones normally expressing SpCas9M, the sgRNA that is driven by a constitutive promoter (PJ23119), and the H-arms, which are used for homologous recombination. To generate new knockout constructs, both gRNA and H-arms can be efficiently replaced using Gibson assembly.
b. Amplify the linearized UA and DA fragments via PCR using C. crescentus NA1000 genomic DNA as the template (Figure 3).
c. Amplify the linearized sgRNA fragment via PCR using pSJX016 (Figure 2) as the template. Add gRNA sequence as the homologous region to the upstream of the gRNA scaffold (Figures 2 and 3).
d. Conduct the PCR in a 40 μL reaction mixture consisting of 20 μL of KOD OneTM PCR Master Mix, 2 μL of forward primer (10 μM), 2 μL of reverse primer (10 μM), 1 μL of template DNA, and 15 μL of H2O. Amplification is conducted under the following PCR program: 98 °C for 5 min; 28 cycles of 98 °C for 10 s, 60 °C for 5 s, and 68 °C for 1–6 min (2 kb/min); and a final 68 °C for 5 min.
3. Construct the editor plasmids through the Gibson assembly method [19] (Figure 3) in a 20 μL reaction mixture: 0.03 pmol linearized vector, 0.03 pmol linearized sgRNA, 0.03 pmol linearized UA and DA, 4 μL of 5× CE MultiS Buffer, 2 μL of Exnase MultiS, and H2O up to 20 μL. Then, incubate the 20 μL mixture at 37 °C for 50 min.
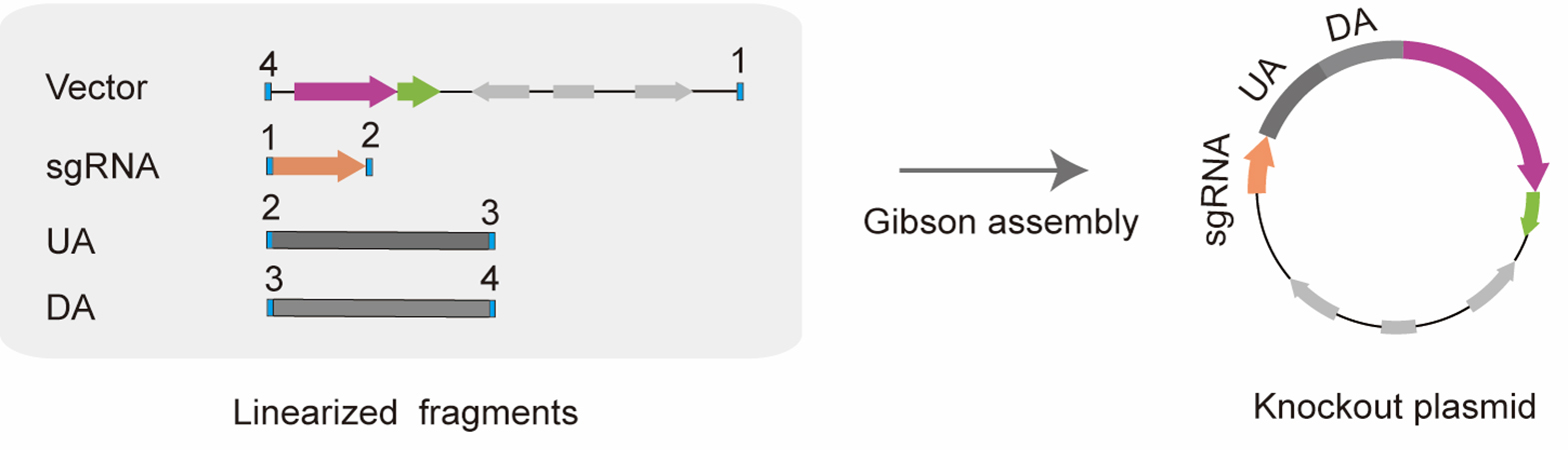
Figure 3. Diagram of constructing editor plasmid by Gibson assembly. By introducing homologous sequences at the 5' end of specific primers, linearized fragments with homologous regions at both ends could be amplified via PCR. Using Gibson assembly, the four linearized fragments could be assembled into a new editor plasmid by sequential homologous recombination with one another. The box filled with blue represents homologous regions (20 bp), and the homologous regions between adjacent fragments are represented by the same number.
C. Competent cells preparation
1. Inoculate 5 μL of bacterial culture of C. crescentus NA1000 onto a PYE plate at 30 °C for 2–3 days.
2. Pick the mono-clone, inoculate it in 3 mL of PYE liquid medium, and grow overnight at 30 °C, 200 rpm.
3. Transfer 1 mL of culture into 100 mL of PYE liquid medium and culture it to a final OD600 of 0.7–1.0 at 30 °C.
4. Collect the culture into a sterilized ice-cold 50 mL centrifuge tube and centrifuge it at 6,000× g for 10 min at 4 °C.
5. Discard the supernatant and resuspend the pellets in 40 mL of cold distilled water; then, centrifuge it at 6,000× g for 10 min at 4 °C.
6. After washing three times, resuspend the pellets in 1 mL of cold distilled water and then separate it into 100 μL aliquots.
D. Electroporation
1. Mix 1 μg of editor plasmid into 100 μL of competent cells for 20 min on ice.
2. Transfer the mixture to an ice-cold Bio-Rad Gene Pulser cuvette and subject it to a 2.5 kV electric shock.
3. Set the capacitor to 25 µF and the resistance on the pulse controller to 200 ohms.
4. After the electric shock, transfer the cells from the cuvette to a 1.5 mL tube containing 1 mL of PYE liquid medium for recovery (30 °C). During the recovery period, add the inducer of Pvan, vanillic acid, to the PYE liquid medium. The working concentration of vanillic acid is 50 µM.
5. After a 3-h recovery period at 30 °C, plate the C. crescentus cells onto the appropriate selective PYE plates.
E. Genome editing analysis
1. Pick a single clone from plates and suspend it in 20 μL of reaction mixture, containing 10 μL of 2× Phanta Flash Master Mix, 1 μM primer, and 8 μL of H2O.
2. Design the primer pairs for genome editing analysis (Figure 4B). The forward primer was located within the UA region, and the reverse primer was located downstream of the DA region.
3. Separate the PCR products by 1% agarose gels and stain with SYBR Safe. Negative controls (usually wild-type cells) and experimental samples were analyzed in parallel to identify nonspecific bands.
4. Verify the PCR products by Sanger sequencing to further confirm the knockout results.
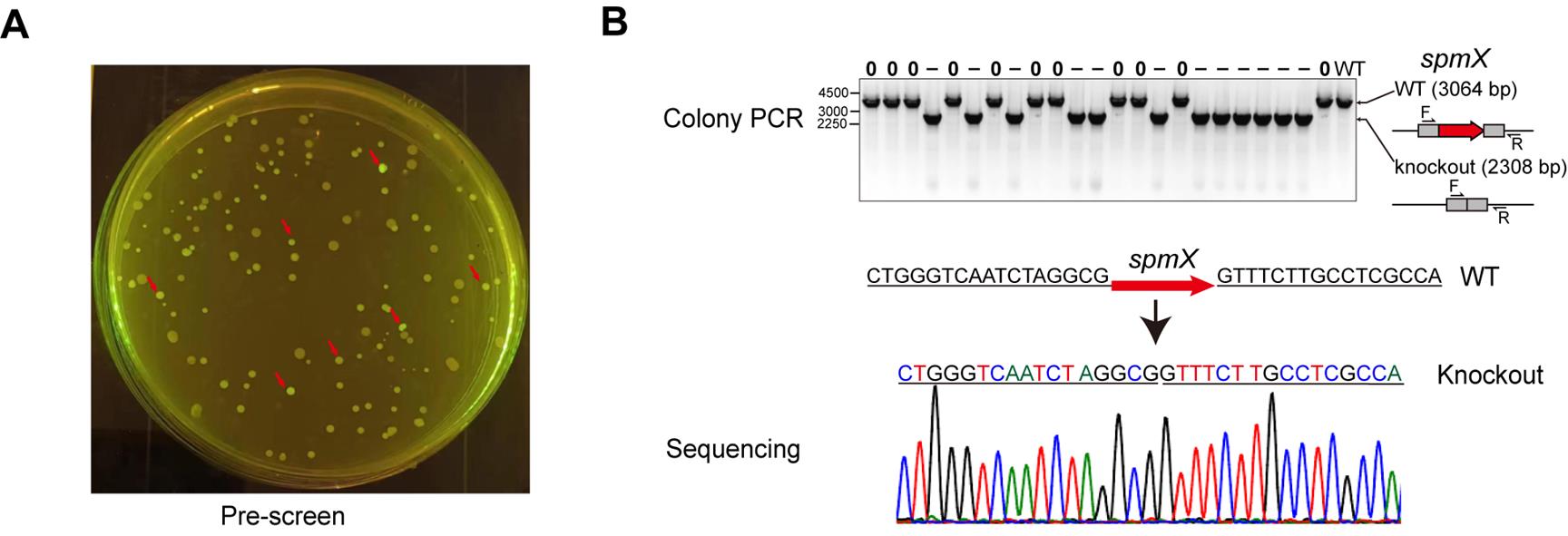
Figure 4. Analysis of spmX knockout. (A) The clones normally expressing SpCas9M (indicated by red arrows) were prescreened by the indication of fluorescent sfGFP under a blue-light lamp. (B) The edited clones were finally confirmed by colony PCR and Sanger sequencing.
F. Editor plasmid curing
1. Culture the correct knockout clone of C. crescentus in 5 mL of PYE liquid medium (without any antibiotics) at 30 °C, 200 rpm.
2. After 15–18 h, collect the cells and dilute into 1:1,000 in fresh PYE liquid medium.
3. Spread 50 μL of diluted culture on PYE plates without antibiotics and then incubate at 30 °C for about 2 days.
4. Pick the single clones and replica-spot them on PYE plates with no antibiotics and on PYE plates with antibiotics.
5. Select the clones that grew only on the antibiotic-free plates as the cured strains.
Validation of protocol
This protocol has been used and validated in the following research article:
Sun et al. [18]. A CRISPR/SpCas9M-reporting system for efficient and rapid genome editing in Caulobacter crescentus. Nucleic acids research (Figure 1, Figure 2, and Figure 3).
General notes and troubleshooting
General notes
1. Editing efficiency varies from 10% to 80% depending on the target, with some genes being far more difficult to edit than others. For essential genes, the conventional knockout is not possible because their loss results in non-viable cells.
2. Due to the significant variation in editing efficiency among different gRNAs, three or more gRNAs need to be selected to ensure successful knockout.
3. To minimize CRISPR escape events, clones exhibiting brighter green fluorescence could be prioritized for colony PCR detection. This pre-selection strategy can improve apparent editing efficiency.
4. The method is still effective for large gene deletions, including a gene as large as 7.7 kb, which was successfully knocked out in this study.
5. Curing efficiencies were usually high, up to 100% within one round.
6. Several common pitfalls, such as low electroporation efficiency, weak GFP expression, and PCR false positives, also influence the results of genome editing, and the corresponding troubleshooting advice is provided in Table 3.
Troubleshooting
Table 3. Troubleshooting advice for common pitfalls
| No. | Step | Problem | Suggestion |
|---|---|---|---|
| 1 | Electroporation | Low electroporation efficiency | Increase the concentration of competent cells and prepare the editor plasmid with high quality. |
| 2 | Electroporation | Weak GFP expression | Increase the concentration of Pvan inducer appropriately in PYE liquid medium. |
| 3 | Genome editing analysis | PCR false positives | Confirm the results by PCR again after curing the editor plasmid or change the checking primers. |
Acknowledgments
We acknowledge the National Natural Science Foundation of China (U22A20540, 32471494), the Guangdong Basic and Applied Basic Research Foundation (2023A1515030069), and the Shenzhen Science and Technology Plan Platform and Carrier Special Project (No. ZDSYS20220303153551001). This protocol was adapted from previous work [18].
Competing interests
The authors declare no competing financial interests.
References
- Labun, K., Montague, T. G., Krause, M., Torres Cleuren, Y. N., Tjeldnes, H. and Valen, E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47: W171–W174. https://doi.org/10.1093/nar/gkz365
- Hentchel, K. L., Reyes Ruiz, L. M., Curtis, P. D., Fiebig, A., Coleman, M. L. and Crosson, S. (2018). Genome-scale fitness profile of Caulobacter crescentus grown in natural freshwater. ISME J. 13(2): 523–536. https://doi.org/10.1038/s41396-018-0295-6
- Tan, W., Cheng, S., Li, Y., Li, X. Y., Lu, N., Sun, J., Tang, G., Yang, Y., Cai, K., Li, X., et al. (2022). Phase separation modulates the assembly and dynamics of a polarity-related scaffold-signaling hub. Nat Commun. 13(1): 7181. https://doi.org/10.1038/s41467-022-35000-2
- Tsokos, C. G. and Laub, M. T. (2012). Polarity and cell fate asymmetry in Caulobacter crescentus. Curr Opin Microbiol. 15(6): 744–750. https://doi.org/10.1016/j.mib.2012.10.011
- Govers, S. K. and Jacobs-Wagner, C. (2020). Caulobacter crescentus: model system extraordinaire. Curr Biol. 30(19): R1151–R1158. https://doi.org/10.1016/j.cub.2020.07.033
- Lu, N., Duvall, S. W., Zhao, G., Kowallis, K. A., Zhang, C., Tan, W., Sun, J., Petitjean, H. N., Tomares, D. T., Zhao, G. P., et al. (2023). Scaffold-Scaffold Interaction Facilitates Cell Polarity Development in Caulobacter crescentus. mBio. 14(2): e03218–22. https://doi.org/10.1128/mbio.03218-22
- Ely, B. (1991). Genetics of Caulobacter crescentus. Methods Enzymol. 204:372–384. https://doi.org/10.1016/0076-6879(91)04019-k
- Ebersbach, G., Briegel, A., Jensen, G. J. and Jacobs-Wagner, C. (2008). A Self-Associating Protein Critical for Chromosome Attachment, Division, and Polar Organization in Caulobacter. Cell. 134(6): 956–968. https://doi.org/10.1016/j.cell.2008.07.016
- Wang, S. C. E., West, L. and Shapiro, L. (2006). The Bifunctional FtsK Protein Mediates Chromosome Partitioning and Cell Division in Caulobacter. J Bacteriol. 188(4): 1497–1508. https://doi.org/10.1128/jb.188.4.1497-1508.2006
- Thanbichler, M., Iniesta, A. A. and Shapiro, L. (2007). A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 35(20): e137–e137. https://doi.org/10.1093/nar/gkm818
- Barrangou, R., Fremaux, C., Deveau, H., Richards, M., Boyaval, P., Moineau, S., Romero, D. A. and Horvath, P. (2007). CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science. 315(5819): 1709–1712. https://doi.org/10.1126/science.1138140
- Horvath, P. and Barrangou, R. (2010). CRISPR/Cas, the Immune System of Bacteria and Archaea. Science. 327(5962): 167–170. https://doi.org/10.1126/science.1179555
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., et al. (2013). Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 339(6121): 819–823. https://doi.org/10.1126/science.1231143
- Liu, X. S., Wu, H., Ji, X., Stelzer, Y., Wu, X., Czauderna, S., Shu, J., Dadon, D., Young, R. A., Jaenisch, R., et al. (2016). Editing DNA Methylation in the Mammalian Genome. Cell. 167(1): 233–247.e17. https://doi.org/10.1016/j.cell.2016.08.056
- Rodrigues, S. D., Karimi, M., Impens, L., Van Lerberge, E., Coussens, G., Aesaert, S., Rombaut, D., Holtappels, D., Ibrahim, H. M. M., Van Montagu, M., et al. (2020). Efficient CRISPR-mediated base editing in Agrobacterium spp.. Proc Natl Acad Sci USA. 118(2): e2013338118. https://doi.org/10.1073/pnas.2013338118
- Cui, Y., Dong, H., Tong, B., Wang, H., Chen, X., Liu, G. and Zhang, D. (2022). A versatile Cas12k-based genetic engineering toolkit (C12KGET) for metabolic engineering in genetic manipulation-deprived strains. Nucleic Acids Res. 50(15): 8961–8973. https://doi.org/10.1093/nar/gkac655
- Trujillo Rodríguez, L., Ellington, A. J., Reisch, C. R. and Chevrette, M. G. (2023). CRISPR-Associated Transposase for Targeted Mutagenesis in Diverse Proteobacteria. ACS Synth Biol. 12(7): 1989–2003. https://doi.org/10.1021/acssynbio.3c00065
- Sun, J., Yu, X., Tang, G., Chen, M., Zheng, Y., Hu, Y., Li, Q., Li, X., Li, N., Li, Z., et al. (2025). A CRISPR/SpCas9M-reporting system for efficient and rapid genome editing in Caulobacter crescentus. Nucleic Acids Res. 53(8): gkaf353. https://doi.org/10.1093/nar/gkaf353
- Bordat, A., Houvenaghel, M. C. and German-Retana, S. (2015). Gibson assembly: an easy way to clone potyviral full-length infectious cDNA clones expressing an ectopic VPg. Virol J. 12(1): 89. https://doi.org/10.1186/s12985-015-0315-3
Article Information
Publication history
Received: Aug 9, 2025
Accepted: Sep 27, 2025
Available online: Oct 14, 2025
Published: Nov 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Yuan, X., Yu, X., Zhao, W. and Sun, J. (2025). A Practical CRISPR-Based Method for Rapid Genome Editing in Caulobacter crescentus. Bio-protocol 15(21): e5490. DOI: 10.21769/BioProtoc.5490.
Category
Microbiology > Microbial genetics > Genome editing
Microbiology > Microbial genetics > CRISPR-Cas9
Molecular Biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.