- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Colocalizing Telomeres With PML or γH2AX Foci by IF-FISH in Mouse Brain Neurons
Published: Vol 15, Iss 21, Nov 5, 2025 DOI: 10.21769/BioProtoc.5485 Views: 1739
Reviewed by: Elena A. OstrakhovitchShubham GargAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Oct 2025
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Telomere length maintenance is strongly linked to cellular aging, as telomeres progressively shorten with each cell division. This phenomenon is well-documented in mitotic, or dividing, cells. However, neurons are post-mitotic and do not undergo mitosis, meaning they lack the classical mechanisms through which telomere shortening occurs. Despite this, neurons retain telomeres that protect chromosomal ends. The role of telomeres in neurons has gained interest, particularly in the context of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), where aging is a major risk factor. This has sparked interest in investigating telomere maintenance mechanisms in post-mitotic neurons. Nevertheless, most existing telomere analysis techniques were developed for and optimized using mitotic cells, posing challenges for studying telomeres in non-dividing neuronal cells. Thus, this protocol adapts an already established technique, the combined immunofluorescence and telomere fluorescent in situ hybridization (IF-FISH) on mitotic cells to study the processes occurring at telomeres in cortical neurons of the mouse ALS transgenic model, TDP-43 rNLS. Specifically, it determines the occurrence of DNA damage and the alternative lengthening of telomeres (ALT) mechanism through simultaneous labeling of the DNA damage marker, γH2AX, or the ALT marker, promyelocytic leukemia (PML) protein, together with telomeres. Therefore, the protocol enables the visualization of DNA damage (γH2AX) or the ALT marker (PML) concurrently with telomeres. This technique can be successfully applied to brain tissue and enables the investigation of telomeres specifically in cortical neurons, rather than in bulk tissue, offering a significant advantage over Southern blot or qPCR-based techniques.
Key features
• This protocol enables the labeling of telomeres in mouse brain tissue prepared from paraffin-embedded brain sections.
• This method facilitates concurrent labeling of proteins that are colocalized at telomere sites.
Keywords: IF-FISHGraphical overview
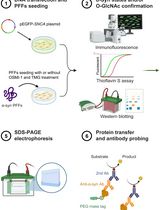
Approximate timeline for conducting immunofluorescence and telomere fluorescent in situ hybridization (IF-FISH)
Background
This protocol has the potential to advance the study of telomere length maintenance mechanisms, particularly in neurons of transgenic mouse models of neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS). As post-mitotic cells, neurons do not undergo cell division [1]; therefore, telomere homeostasis is likely regulated differently in these cells compared to mitotic cells, in which telomeres progressively shorten with each round of replication [2]. Telomere length maintenance represents an emerging and compelling area of research, especially in the context of pathological conditions such as ALS that affect the nervous system [3,4].
Established methods for studying telomeres include Southern blot analysis, which enables measurement of terminal restriction fragment (TRF) length [5], and quantitative PCR (qPCR)-based techniques [6], which estimate relative telomere length by comparing telomeric repeat copy number to that of a single-copy gene. While these methods are useful, they often lack cell-type specificity and are not well-suited for analyzing telomere dynamics in heterogeneous tissues such as the brain. ALS affects specific neuronal subtypes in the brain and spinal cord [7]; therefore, a method that enables telomere analysis within the desired cell type is particularly valuable for improving the understanding of the disease mechanism. Moreover, these techniques do not allow for the investigation of molecular events at telomeres, e.g., telomere dysfunction-induced foci (TIFs), and the occurrence of DNA damage or the recruitment of telomere-associated proteins.
In contrast, the described technique for immunofluorescence combined with fluorescence in situ hybridization (IF-FISH) enables the visualization and analysis of telomeric or telomere-associated proteins at telomeric sites, such as TRF2 or Rif1, thereby providing deeper insights into telomere-related molecular processes. This method is particularly valuable in studies requiring cell-type specificity and spatial resolution, making it especially suitable for complex tissues like the brain. While IF-FISH is less suitable for precise telomere length quantification compared to methods such as Southern blot or qPCR, it nonetheless allows for the evaluation of relative telomere length based on fluorescence intensity. Thus, although this approach does not yield exact telomere length measurements, it is still valuable for detecting and comparing telomere length differences between experimental groups. Furthermore, IF-FISH provides spatial information at the single-cell level. When combined with quantitative data from methods such as qPCR or Southern blotting, it enhances the overall interpretation by offering critical context regarding cell-to-cell heterogeneity and the spatial organization of telomeres. This integrative approach enables a more comprehensive and multidimensional analysis of telomere dynamics under various experimental conditions. However, the limitation of the IF-FISH method is its relatively time-consuming protocol, particularly when compared to qPCR-based approaches.
Materials and reagents
Biological materials
1. Mouse brain paraffin sections, 10 µm thick (derived from transgenic bigenic rNLS mouse or monogenic control mouse [8])
Reagents
1. Trisodium citrate·2H2O (Merck, catalog number: 1064480500)
2. Tween-20 (Merck, catalog number: 11332465001)
3. Normal donkey serum (NDS) (Jackson ImmunoResearch, catalog number: 017-000-121)
4. Histone H2AX [p Ser139] antibody (Novus Biologicals, catalog number: NB100-384)
5. Anti-PML antibody, mouse monoclonal (Merck-Millipore, catalog number: P6746-200UL)
6. Tris base (Merck, catalog number: 252859-500G)
7. Sodium chloride (Merck, catalog number: S9625-1KG)
8. Potassium chloride (Merck, catalog number: P3911-500G)
9. Sodium phosphate dibasic (Merck, catalog number: S9763-500G)
10. Potassium phosphate monobasic (Merck, catalog number: P0662-500G)
11. Xylene (histological grade), (Merck, catalog number: 534056-500ML)
12. Alexa FluorTM 488, anti-mouse (Invitrogen, catalog number: A-21202)
13. Alexa FluorTM 488, anti-rabbit (Invitrogen, catalog number: A-21206)
14. Paraformaldehyde, 16% (Thermo Fisher Scientific, catalog number: 043368.9M)
15. Texas Red-conjugated C-strand telomere PNA probe (stock solution of 0.3 μL/mL in Mili-Q water) (Panagene, custom fluorescent dye)
16. Blocking reagent (Merck, catalog number: 11096176001)
17. Formamide (Merck, catalog number: F7503-100ML)
18. Hydrochloric acid (Merck, catalog number: 258148-25ML)
19. DAPI (Merck, catalogue number: D9542-1MG)
Solutions
1. 10 mM citrate buffer containing 0.1% Tween-2, pH 6.0 (see Recipes)
2. 1× PBS, pH 7.4 (see Recipes)
3. 1× PBS, pH 7.4 with 0.1% Triton X-100 (see Recipes)
4. 5% NDS (see Recipes)
5. Histone H2AX [p Ser139] antibody 1:200 dilution (see Recipes)
6. Anti-PML antibody 1:200 dilution (see Recipes)
7. 90% ethanol (see Recipes)
8. 70% ethanol (see Recipes)
9. 10 mM Tris Cl, pH 7.5 (see Recipes)
10. 50 mM Tris Cl, pH 7.5 (see Recipes)
11. PNA wash buffer A (70% v/v formamide, 10 mM Tris Cl, pH 7.5) (see Recipes)
12. PNA wash buffer B (50 mM Tris Cl, pH 7.5, 150 mM NaCl, 0.8% v/v Tween 20) (see Recipes)
13. 2% paraformaldehyde in 1× PBS (see Recipes)
14. PNA hybridization solution (see Recipes)
15. PNA probe hybridization working solution (see Recipes)
Recipes
1. 10 mM citrate buffer containing 0.1% Tween-20, pH 6.0
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Trisodium citrate·2H2O (MW = 294.10 g/mol) | 10 mM | 2.94 g |
| Milli-Q water | - | To 1,000 mL total volume |
| Tween-20 | 0.1% v/v | 1.0 mL |
| Total | 1,000 mL |
If necessary, adjust to pH 6.0 with HCl.
2. 1× PBS, pH 7.4
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Sodium chloride (MW = 58.44 g/mol) | 137 mM | 8 g |
| Potassium chloride (MW = 74.55 g/mol) | 2.7 mM | 0.2 g |
| Sodium phosphate dibasic (MW = 141.96 g/mol) | 10 mM | 1.44 g |
| Potassium phosphate monobasic (MW = 136.09 g/mol) | 1.8 mM | 0.245 g |
| Milli-Q water | - | To 1,000 mL total volume |
| Total volume | 1,000 mL |
Adjust to pH 7.4 with HCl.
3. 1× PBS, pH 7.4 with 0.1% Triton X-100
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Sodium chloride (MW = 58.44 g/mol) | 137 mM | 8 g |
| Potassium chloride (MW = 74.55 g/mol) | 2.7 mM | 0.2 g |
| Sodium phosphate dibasic (MW = 141.96 g/mol) | 10 mM | 1.44 g |
| Potassium phosphate monobasic (MW = 136.09 g/mol) | 1.8 mM | 0.245 g |
| Milli-Q water | - | To 1,000 mL total volume |
| Triton X-100 | 0.1% v/v | 1.0 mL |
| Total | 1,000 mL |
Adjust to pH = 7.4 with HCl.
4. 5% NDS
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Normal donkey serum | 5% | 0.5 mL |
| 1× PBS with 0.1% Triton X-100 | - | 9.5 mL |
| Total | 10 mL |
5. Histone H2AX [p Ser139] antibody 1:200 dilution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 5% NDS | 5% | 199 μL |
| Histone H2AX [p Ser139] antibody | 5 μg/mL | 1 μL |
| Total volume per one slide | 200 μL |
6. Anti-PML antibody 1:200 dilution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 5% NDS | 5% | 199 μL |
| Anti-PML antibody | 10–12.5 μg/mL | 1 μL |
| Total volume per one slide | 200 μL |
7. 90% ethanol
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Absolute ethanol (100%) | 90% | 225 mL |
| Milli-Q water | - | 25 mL |
| Total | 250 mL |
8. 70% ethanol
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Absolute ethanol (100%) | 70% | 175 mL |
| Milli-Q water | - | 75 mL |
| Total | 250 mL |
9. 10 mM Tris Cl, pH 7.5
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris base (MW = 121.14 g/mol) | 10 mM | 1.21 g |
| Milli-Q water | - | up to 1,000 mL |
| Total | 1,000 mL |
Adjust to pH 7.5 with HCl.
10. 50 mM Tris Cl, pH 7.5
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris base (MW = 121.14 g/mol) | 10 mM | 6.06 g |
| Milli-Q water | - | up to 1,000 ml |
| Total | 1,000 mL |
Adjust to pH 7.5 with HCl.
11. PNA wash buffer A (70% v/v formamide, 10 mM Tris Cl, pH 7.5)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 10 mM Tris Cl, pH 7.5 | - | 150 mL |
| Formamide | 70% | 350 mL |
| Total | 500 mL |
12. PNA wash buffer B (50 mM Tris Cl, pH 7.5, 150 mM NaCl, 0.8% v/v Tween 20)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 50 mM Tris Cl, pH 7.5 | 50 mM | 490 mL |
| Sodium chloride (58.44 g/mol) | 150 mM | 4.39 g |
| Tween-20 | 0.8% v/v | 4 mL |
| 50 mM Tris Cl, pH 7.5 | 50 mM | To 500 mL total volume |
| Total | 500 mL |
13. 2% paraformaldehyde in 1× PBS
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 16% paraformaldehyde | 2% | 1.25 mL |
| 1× PBS, pH 7.4 | 8.75 mL | |
| Total | 10 mL |
14. PNA hybridization solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 10 mM Tris Cl, pH 7.5 | - | 2.975 mL |
| Formamide | 70% | 7 mL |
| Blocking reagent | 0.25% | 25 μL |
| Total | 10 mL |
15. PNA probe hybridization working solution
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| PNA hybridization solution | - | 999 μL |
| 0.3 μg/mL C-strand telomere PNA probe | 0.3 ng/mL | 1 μL |
| Total | 1,000 μL |
Laboratory supplies
1. Parafilm® M sealing film (Merck, catalog number: HS234526B-1EA)
2. Cover glasses (Merck, catalog number: C9802)
3. Coplin jars, e.g., BRAND® plastic staining trough, Hellendahl pattern (Merck, catalog number: BR474400)
4. Dark humidity chamber for slides, e.g., StainTray slide staining system (Merck, catalog number: Z670146-1EA)
5. ProLongTM Gold Antifade Mountant (ThermoFisher scientific, catalog number: P36930)
6. Deionized water
Equipment
1. Hot plate, e.g., benchmark hotplate (Merck, catalog number: Z742547)
2. Drying oven, e.g., binder multifunction drying oven FED series (Merck, catalog number: Z604011)
3. Water bath with heating, e.g., MyBathTM mini water bath (Merck, catalog number: Z741427)
Procedure
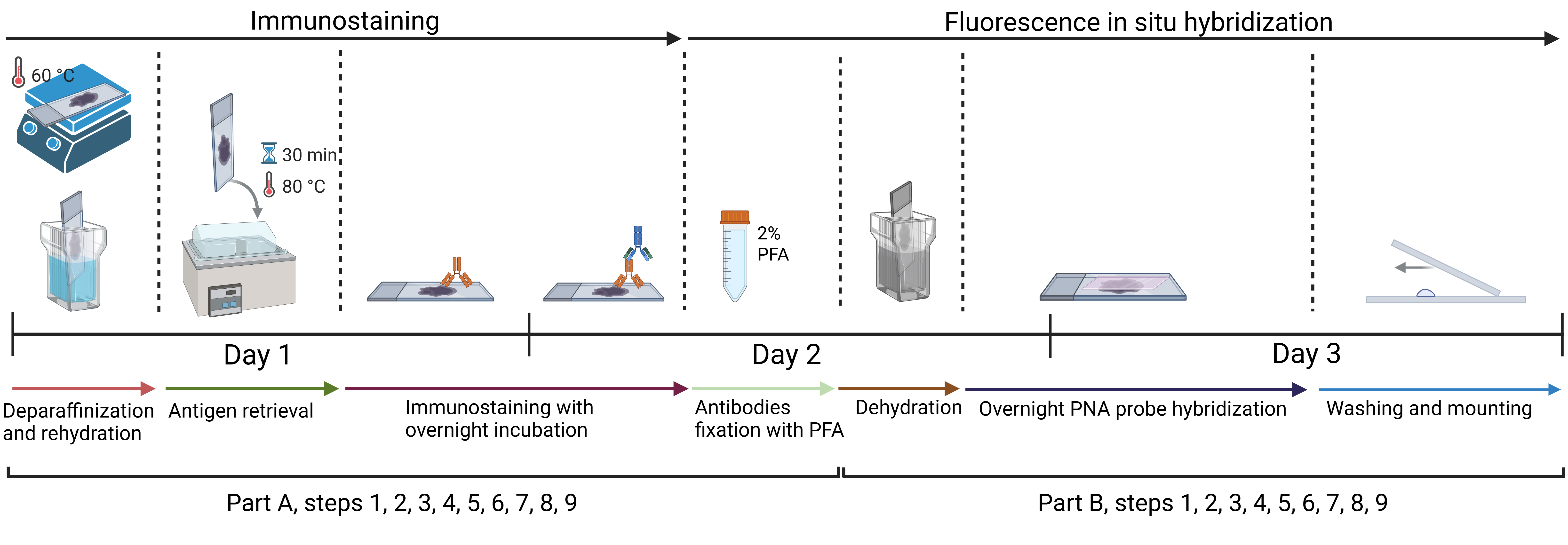
A. Immunofluorescence protocol on paraffin-embedded brain sections
Note: Turn on the water bath and set it to heat to 80 °C. Allow the bath to reach and stabilize at that temperature before placing slides inside later in the protocol.
1. Remove paraffin and rehydrate sections
a. Heat slides for 45 min at 60 °C on a hot plate; alternatively, a drying oven can be used.
Note: The following steps are performed at room temperature.
b. Immerse slides directly into pure xylene for 5 min, then repeat for a second 5 min.
c. Then, rehydrate slides with:
100% ethanol (2×, each 3 min)
90% ethanol (3 min)
70% ethanol (3 min)
d. Rinse slides in distilled water for 3 min.
2. Perform antigen retrieval
a. Incubate the slides in 10 mM citrate buffer containing 0.1% Tween-20, pH 6.0, at 80 °C in a water bath for 30 min, then leave them at RT to cool to room temperature.
Note: It is recommended to prepare the buffer fresh for each use; alternatively, it can be stored at 4 °C for up to two weeks.
b. Rinse in 1× PBS.
3. Block nonspecific binding
Note: Place the slides directly into a humidified, dark chamber. Although the dark chamber is not strictly required at this stage, starting in it removes the need to switch chambers later in the protocol.
a. Incubate slides for 30 min at room temperature in 200 μL of 5% NDS in 1× PBS 0.1% Triton per slide to reduce background staining.
Note: Optionally, draw a hydrophobic barrier around the tissue using a PAP pen. This step is not strictly required because the reagent drop usually remains stable.
4. Apply primary antibody
a. Dilute primary antibody (anti-γH2AX 1:200, 5 μg/mL or PML 1:200, 10–12.5 μg/mL) in 5% NDS to obtain 200 μL of solution per slide.
b. Incubate overnight at 4 °C in a humidified chamber.
5. Wash slides
a. Wash slides three times in 1× PBS with 0.1% Triton for 5 min. If a standard 5-slide Coplin jar is used for the washing step, 50 mL of buffer is required per wash.
6. Apply secondary antibody
a. Dilute Alexa Fluor488conjugated secondary antibody (1:200), 200 μL per slide.
b. Apply the solution on the slides and incubate for 2 h at RT in a dark, humidified chamber.
7. Wash slides
a. Wash slides two times in 1× PBS with 0.1% Triton for 5 min and once with 1× PBS for 5 min. If a standard 5-slide Coplin jar is used for the washing step, 50 mL of buffer is required per wash.
8. Fix antibody complexes
a. Fix slides in 2% paraformaldehyde for 10 min at room temperature, applying 200 μL of solution to cover the tissue.
9. Final wash
a. Rinse slides three times in deionized water in a Coplin jar. If a standard 5-slide Coplin jar is used for the washing step, 50 mL of buffer is required per wash.
B. PNA probe hybridization (fluorescent in situ hybridization)
Note: Place the hot plate in a dark or dimly lit room. Turn on the hot plate and set it to heat to 80 °C.
1. Dehydrate the slides through a graded ethanol series
Note: The following steps are performed at room temperature.
a. 70% for 5 min
b. 90% for 5 min
c. 100% for 5 min
d. Air-dry the slides, taking care not to over-dry them.
2. Apply 20–50 μL of the PNA probe hybridization working solution (0.3 ng/mL) and cover with a coverslip.
Note: The easiest way to do this is to apply the solution onto the coverslip first (placed on the bench) and then attach it to the microscope slide. Please see Figure 1.

Figure 1. Demonstration of PNA probe application on a glass slide
3. Place the slides on a pre-heated surface at 80 °C for 5 min, keeping them in the dark. Then, immediately transfer them to a humidified chamber in the dark and allow hybridization to proceed overnight at room temperature.
4. Remove the coverslip.
Note: Gently tap the edge of the disposable container with the hand holding the slide. The coverslip usually detaches easily. Please see Figure 2.
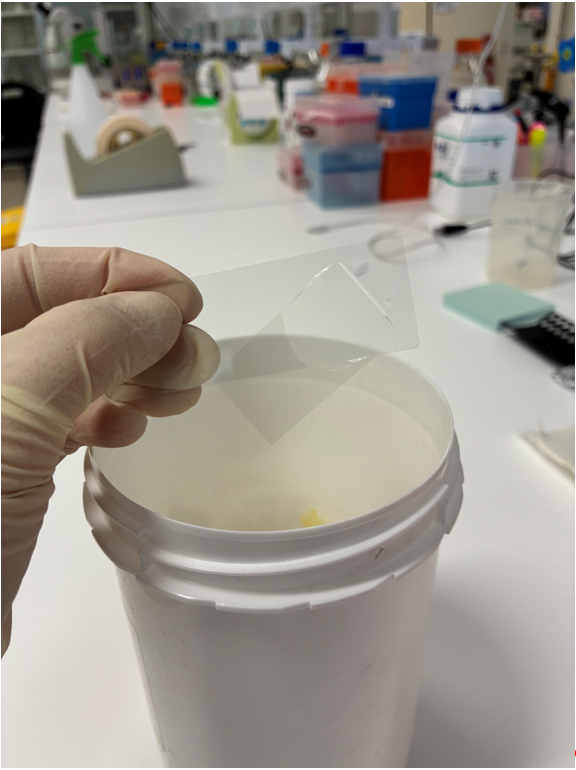
Figure 2. Demonstration of the removal of a coverslip
5. Wash the slides in PNA wash buffer A at room temperature, with shaking, 2 times for 10 min each.
Critical: Always prepare PNA wash buffer A fresh, immediately before each use. It is recommended to perform the wash in a Coplin jar, such as a standard 5-slide Coplin jar, which requires 50 mL of PNA wash buffer A per wash.
6. Wash the slides in PNA wash buffer B at room temperature, with shaking, 5 times for 5 min each.
Critical: Always prepare PNA wash buffer B fresh, immediately before each use. It is recommended to perform the wash in a Coplin jar, such as a standard 5-slide Coplin jar, which requires 50 mL of PNA wash buffer B per wash.
Note: Optionally stain with DAPI during the fourth wash if needed.
7. Rinse the slides 3 times in deionized water.
Critical: A proper rinse with deionized water helps shield the tissue from salt-induced damage during the subsequent dehydration step.
8. Again, dehydrate the slides through a graded ethanol series:
70% for 5 min
90% for 5 min
100% for 5 min.
9. Air-dry the slides and mount them in mounting medium. Figure 3 shows an example of the IF-FISH to detect PML and telomere in the cortical neurons of the mouse brain. The images were acquired using a Zeiss LSM 880 confocal microscope equipped with a Plan-Apochromat 63×/1.4 Oil DIC M27 objective.
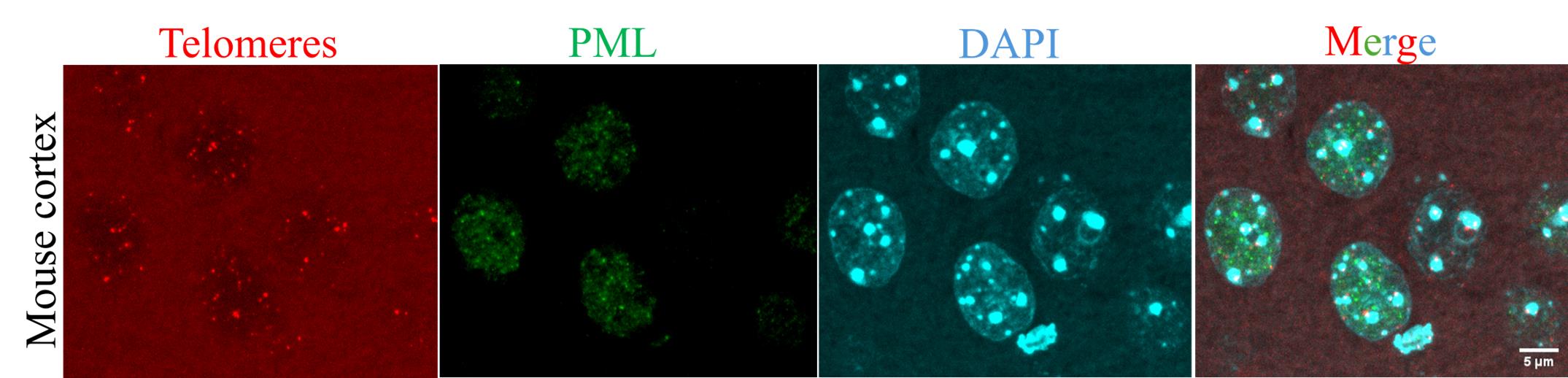
Figure 3. Example of immunofluorescence combined with fluorescence in situ hybridization (IF-FISH) performed on mouse brain tissue. Telomeres (red, by FISH), PML (green, by immunostaining), and nuclei (cyan, by DAPI). Scale bar: 5 μm.
Data analysis
The data analysis was performed as described in the original article [9]. Colocalization between PML bodies or γH2AX foci and telomeres was assessed using Mander’s coefficient via the JaCoP plugin [10] in Fiji (ImageJ) software [11]. Single optical slices from confocal z-stacks were used for the analysis. The individual cells were cropped from merged images containing the two relevant fluorescence channels (PML or γH2AX and telomeres). These were then split into separate channels, and after adjusting thresholds, Mander’s coefficient was calculated to quantify colocalization. Figure 4 illustrates an example of the workflow.
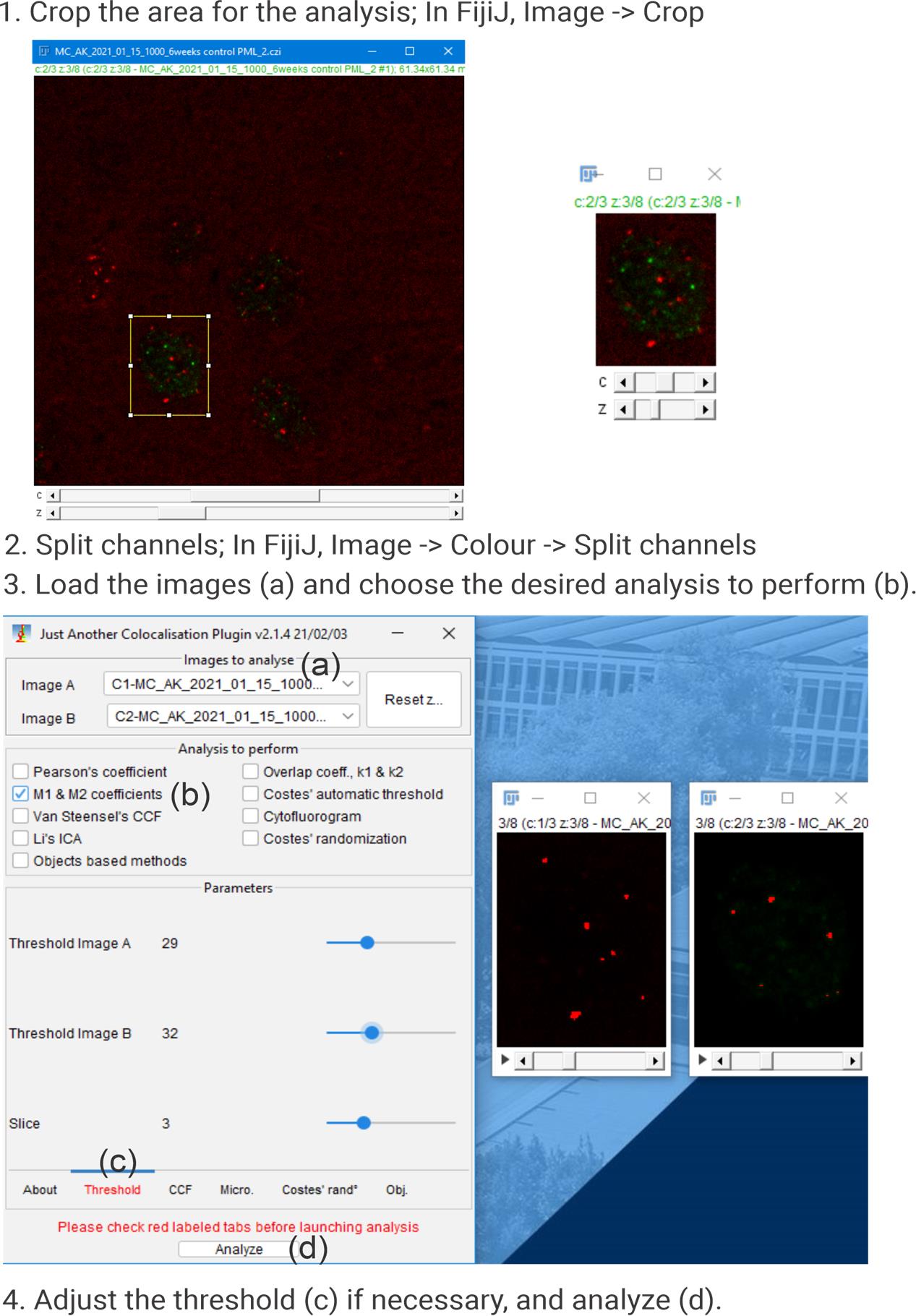
Figure 4. Colocalization analysis using the JaCoP plugin in Fiji (ImageJ) software
Validation of protocol
This protocol has been used and validated in the following research article:
Konopka et al [9]. Pathological forms of TDP-43 in amyotrophic lateral sclerosis (ALS) promote aberrant telomere elongation. Biochim Biophys Acta Mol Basis Dis (Figure 4A, B; Figure 5A, B).
General notes and troubleshooting
General notes
The protocol was optimized and validated using mouse tissue. However, applying it to human tissue did not yield satisfactory staining quality. A potential reason for this could be the quality of preserved brain section morphology. In this case, a neurodegenerative disease (ALS) was investigated, and the specimens were collected at the terminal stage, when extensive neurodegeneration is typically observed.
To strengthen the rigor of the study, the inclusion of the following negative controls is proposed: (1) omission of the primary antibody for immunostaining, (2) omission of the PNA probe, and/or (3) use of a scrambled PNA probe with a meaningless sequence.
Troubleshooting
Problem 1: Weak immunofluorescence signal.
Possible cause: The formamide treatment during PNA probe hybridization.
Solution: It is possible to perform staining after hybridization. If staining is to be performed after PNA hybridization, then the steps involving incubation with the primary and secondary antibodies should be carried out only after the wash step using PNA wash buffer B at step B6. Please note that antigen retrieval should be performed in the original order.
Acknowledgments
A.K. contributed to conceptualization, investigation, writing, review, and editing. The protocol was described and validated in [9] and modified based on [12].
Graphical overview was created in BioRender. Konopka, A. (2025) https://BioRender.com/2vmu4lg
Competing interests
The authors declare no conflicts of interest.
Ethical considerations
All experimental protocols, including animal care and handling, were conducted under the approval of the Internal Biosafety Committee of Macquarie University (NLRD 5974–52019597412350) and the Animal Ethics Committee of Macquarie University, New South Wales, Australia (ARA 2015/042, ARA 2017/020‑5).
References
- Herrup, K. and Yang, Y. (2007). Cell cycle regulation in the postmitotic neuron: oxymoron or new biology?. Nat Rev Neurosci. 8(5): 368–378. https://doi.org/10.1038/nrn2124
- Levy, M. Z., Allsopp, R. C., Futcher, A., Greider, C. W. and Harley, C. B. (1992). Telomere end-replication problem and cell aging. J Mol Biol. 225(4): 951–960. https://doi.org/10.1016/0022-2836(92)90096-3
- Liu, M. Y., Nemes, A. and Zhou, Q. G. (2018). The Emerging Roles for Telomerase in the Central Nervous System. Front Mol Neurosci. 11: e00160. https://doi.org/10.3389/fnmol.2018.00160
- Saretzki, G. (2022). Telomerase and neurons: an unusual relationship. Neural Regener Res. 17(11): 2364. https://doi.org/10.4103/1673-5374.336133
- Kimura, M., Stone, R. C., Hunt, S. C., Skurnick, J., Lu, X., Cao, X., Harley, C. B. and Aviv, A. (2010). Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat Protoc. 5(9): 1596–1607. https://doi.org/10.1038/nprot.2010.124
- O'Callaghan, N. J. and Fenech, M. (2011). A quantitative PCR method for measuring absolute telomere length. Biol Proced Online. 13(1): 3. https://doi.org/10.1186/1480-9222-13-3
- Rojas, P., Ramírez, A. I., Fernández-Albarral, J. A., López-Cuenca, I., Salobrar-García, E., Cadena, M., Elvira-Hurtado, L., Salazar, J. J., de Hoz, R., Ramírez, J. M., et al. (2020). Amyotrophic Lateral Sclerosis: A Neurodegenerative Motor Neuron Disease With Ocular Involvement. Front Neurosci. 14: e566858. https://doi.org/10.3389/fnins.2020.566858
- Walker, A. K., Spiller, K. J., Ge, G., Zheng, A., Xu, Y., Zhou, M., Tripathy, K., Kwong, L. K., Trojanowski, J. Q., Lee, V. Y., et al. (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 130(5): 643–660. https://doi.org/10.1007/s00401-015-1460-x
- Konopka, A., Jamali, M. S., Fowler, M., Mehta, P., Parakh, S., Takalloo, Z., Farzana, F., Mumtaz, N., Hunter, J., Shadfar, S., et al. (2025). Pathological forms of TDP-43 in amyotrophic lateral sclerosis (ALS) promote aberrant telomere elongation. Biochim Biophys Acta Mol Basis Dis. 1871(7): 167906. https://doi.org/10.1016/j.bbadis.2025.167906
- Bolte, S. and Cordelieres, F. P. (2006). A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 224(3): 213–232. https://doi.org/10.1111/j.1365-2818.2006.01706.x
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods. 9(7): 676–682. https://doi.org/10.1038/nmeth.2019
- Cesare, A. J., Heaphy, C. M. and O'Sullivan, R. J. (2015). Visualization of Telomere Integrity and Function In Vitro and In Vivo Using Immunofluorescence Techniques. Curr Protoc Cytom. 73(1): ecy1240s73. https://doi.org/10.1002/0471142956.cy1240s73
Article Information
Publication history
Received: Aug 9, 2025
Accepted: Sep 25, 2025
Available online: Oct 9, 2025
Published: Nov 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Konopka, A. (2025). Colocalizing Telomeres With PML or γH2AX Foci by IF-FISH in Mouse Brain Neurons. Bio-protocol 15(21): e5485. DOI: 10.21769/BioProtoc.5485.
Category
Neuroscience > Nervous system disorders > Neurodegeneration
Molecular Biology > DNA > DNA damage and repair
Neuroscience > Basic technology > Chromatin biology
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.