- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Ribozyme-Mediated Knockdown of lncRNA Gene Expression in Drosophila
Published: Vol 15, Iss 20, Oct 20, 2025 DOI: 10.21769/BioProtoc.5477 Views: 2128
Reviewed by: Jibin SadasivanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2024

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Long noncoding RNAs (lncRNAs) are increasingly understood to play important roles in cell biology, development, and disease, though the vast majority of annotated lncRNAs have yet to be functionally characterized. Disrupting lncRNAs is often challenging owing to their tolerance for mutations (e.g., single-nucleotide polymorphisms and short indels) along with the limitations of other genetic knockdown strategies such as RNA interference (RNAi). Here, we describe a protocol to achieve robust knockdown of lncRNAs in the fruit fly Drosophila using a self-cleaving ribozyme. The 111-bp ribozyme cassette, which consists of the N79 hammerhead ribozyme flanked by flexible linker sequences, is inserted into transcript regions of lncRNA genes using CRISPR/Cas9-mediated homology-directed repair (HDR). The fluorescent eye transformation marker is then removed using a piggyBac transposase, leaving no other modifications at the lncRNA locus save the ribozyme cassette insertion. When transcribed as part of the lncRNA, the ribozyme folds and catalyzes its own self-cleavage, resulting in two RNA cleavage fragments. The efficacy of lncRNA knockdown is then evaluated using reverse transcription quantitative PCR (RT-qPCR) and single-molecule RNA fluorescence in situ hybridization (smFISH). This approach has resulted in efficient knockdown of both nuclear and cytoplasmic lncRNAs in Drosophila, with knockdown of steady-state RNA levels in 3' cleavage fragments typically exceeding 90% and no evidence of off-target effects. The method can also be applied to protein-coding genes in order to knock down specific mRNA isoforms. Thus, self-cleaving ribozymes are a valuable addition to the genetic toolkit in Drosophila.
Key features
• The ribozyme has the potential to knock down any RNA but is particularly useful for long noncoding RNAs, which can be resistant to mutations.
• The ribozyme is useful for the knockdown of nuclear-localized RNAs as well as RNAs that overlap with other genes in the genome.
• Ribozyme knockdown is typically stronger than RNAi and occurs in every cell.
• Insertion of the ribozyme cassette in the 5' region of a transcript ensures that most of the transcript is degraded.
Keywords: Long noncoding RNAGraphical overview
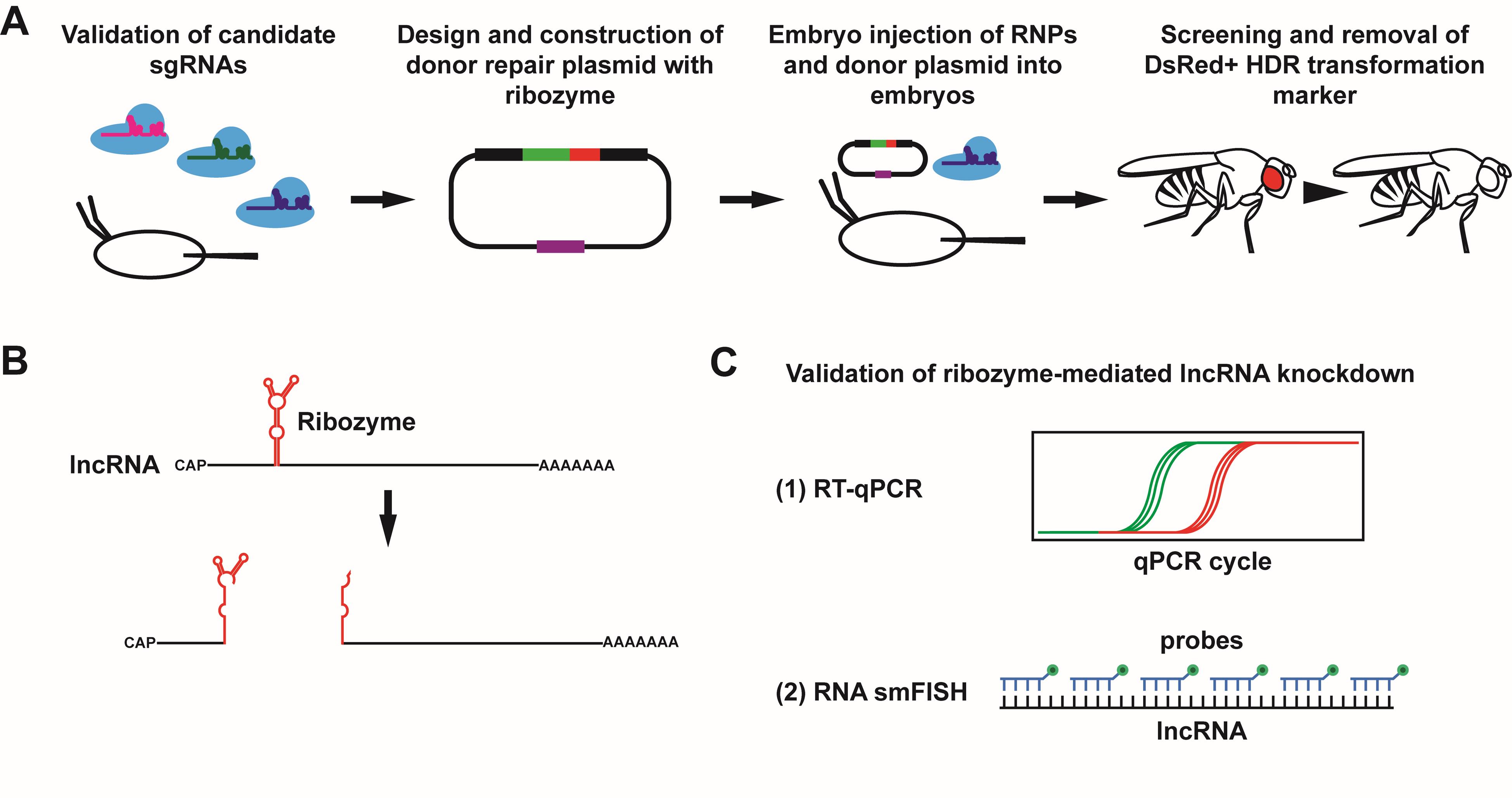
Overview of protocol to achieve and validate ribozyme-mediated knockdown of lncRNA gene expression in Drosophila. (A) To achieve knockdown of a target long noncoding RNA (lncRNA) using a self-cleaving ribozyme, the ribozyme cassette is integrated into the target lncRNA gene. Multiple candidate single-guide RNAs (sgRNAs) are first tested for cleavage efficiency in Drosophila embryos. Once a suitable sgRNA is identified, a donor repair plasmid is designed and constructed containing the ribozyme cassette, a selectable DsRed cassette, and two homology arms. See Figure 5 for a more detailed schematic of the donor repair plasmid. The Cas9/sgRNA RNPs and donor repair plasmid are then injected into Drosophila embryos to integrate the ribozyme cassette via homology-directed repair (HDR), and the progeny of injected flies are screened for the DsRed+ transformation marker. Once identified, the DsRed cassette is removed using a piggyBac transposase, resulting in the scarless insertion of the ribozyme cassette. (B) Upon transcription of the lncRNA, the ribozyme folds and cleaves itself, resulting in two RNA fragments with unprotected ends. (C) Successful knockdown of the lncRNA target can be validated using reverse transcription quantitative polymerase chain reaction (RT-qPCR) and single-molecule RNA fluorescent in situ hybridization (RNA smFISH).
Background
Long noncoding RNAs (lncRNAs) are RNA transcripts longer than 200 nucleotides that do not code for proteins. They are extensively transcribed in eukaryotic genomes and increasingly understood to play important roles in cell biology, development, and disease [1,2]. For example, the lncRNAs Xist [3,4] and roX [5–7] have been shown to play important roles in sex chromosome dosage compensation in mammals and Drosophila, respectively. The lncRNA Malat1 [8–10] has been implicated in tumorigenesis in various types of cancers. Despite striking examples such as these, however, relatively few of the thousands of annotated lncRNAs have been functionally characterized. In part, this is due to the inherent challenges in disrupting lncRNAs [11,12]. lncRNAs tend to be more tolerant of the types of mutations (i.e., single-nucleotide polymorphisms and short indels) that are often used to disrupt open reading frames in protein-coding genes. While large deletions of lncRNA genes are feasible, doing so may also remove DNA regulatory elements, leaving the interpretation of any resulting phenotypes murky. Further, many lncRNAs overlap with other genes in the genome, precluding their complete removal. Although RNA interference (RNAi) knockdown is feasible, RNAi has variable knockdown efficiency [13], is known to produce off-target effects [14,15], and, since the RNAi machinery is primarily localized to the cytoplasm, it may have limited efficacy against nuclear-localized lncRNAs.
Self-cleaving ribozymes have been used to achieve RNA knockdown in cis in mammalian cells [16–19], zebrafish [20], the nematode worm Caenorhabditis elegans [21], and Drosophila melanogaster [22,23], including knockdown of lncRNAs [16,17,23]. When inserted into a transcription unit within the genome, the ribozyme is transcribed along with its target gene, folds, and then cleaves itself, thus resulting, at the very least, in two RNA cleavage fragments. These cleavage fragments may then be susceptible to degradation by the endogenous exonucleases that attack unprotected RNAs. Because these self-cleaving ribozymes operate in cis, off-target effects are expected to be rare.
Here, we detail the protocol we used to knock down several lncRNAs in Drosophila using a self-cleaving ribozyme, as previously published [23]. We used a 111-bp ribozyme cassette that consists of the N79 hammerhead ribozyme [18], a variant engineered from the naturally occurring Sm1 hammerhead ribozyme found in the parasitic fluke Schistosoma mansoni. The ribozyme sequence is surrounded by flexible linker sequences to insulate it from surrounding RNA sequences when folding [24] (Figure 1). The N79 ribozyme cassette was then inserted into lncRNA transcription units within the Drosophila genome using CRISPR/Cas9-mediated homology-directed repair (HDR) from donor DNA as previously described [25–28]. In addition to the N79 ribozyme cassette, the donor DNA contains a selection marker (DsRed) for detection of integration events and a counterselection marker (eya RNAi) for detection of homologous integration events [27]. The selection marker is then precisely excised by PiggyBac transposition to yield a genome with only the N79 cassette inserted [25,28]. Using this method, we were able to achieve a strong knockdown in four lncRNAs, as verified with reverse transcription quantitative PCR (RT-qPCR) and single-molecule RNA fluorescence in situ hybridization (RNA smFISH). These lncRNAs included both cytoplasmic and nuclear lncRNAs. RNA levels after ribozyme-mediated knockdown were typically diminished by 90% or more for the 3' cleavage fragments, higher than the knockdown typically achieved by RNAi [13]. However, knockdown of the 5' cleavage fragment was more variable. Further, we did not detect any off-target effects using ribozyme-mediated knockdown, even when inserted near essential protein-coding genes. Thus, self-cleaving ribozymes offer great potential for characterization of lncRNAs in eukaryotes and are a valuable addition to the genetic toolkit for Drosophila.
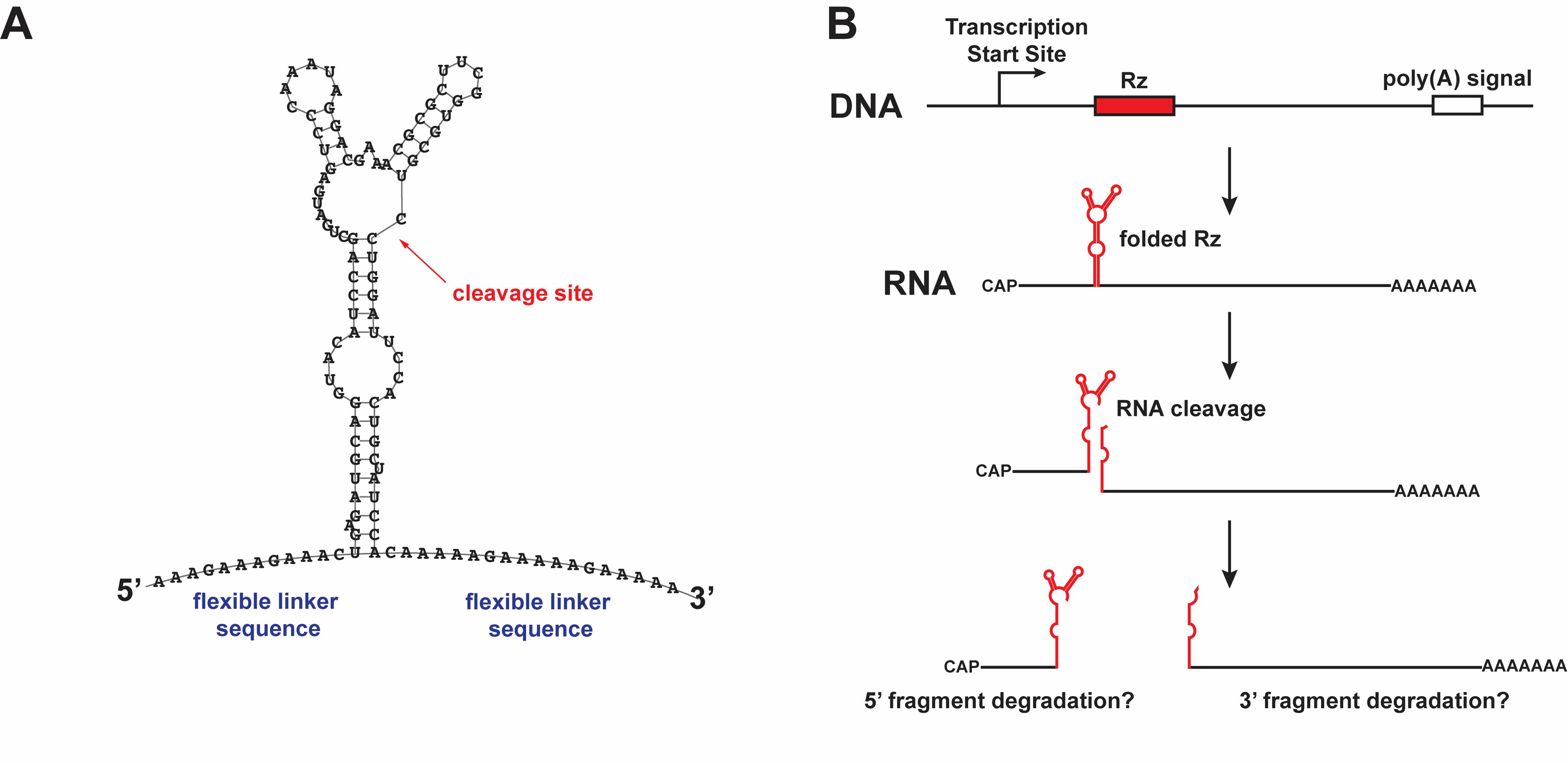
Figure 1. RNA knockdown using a self-cleaving ribozyme. (A) The sequence and predicted secondary structure of the N79 ribozyme cassette, as predicted by RNAfold [29], with the cleavage site indicated. (B) Model of ribozyme-mediated cleavage in cis, producing two cleavage fragments that may be susceptible to degradation by exonucleases. This figure is modified from Nyberg et al. [23] with permission from Oxford University Press (license 6070880155798).
Materials and reagents
Biological materials
1. Drosophila stock for CRISPR/Cas9 injections, preferably a stock with the white allele w[1118] (Bloomington Drosophila Stock Center, catalog number: 6326)
2. X chromosome Drosophila balancer stock (e.g., l(1)*[*], w[1118]/FM7c, Bloomington Drosophila Stock Center, catalog number: 7756)
3. 2nd chromosome Drosophila balancer stock (e.g., y[1] w[*]; nub[2] Adc[b-1] sna[Sco] pr[1] cn[1]/CyO, Bloomington Drosophila Stock Center, catalog number: 3628)
4. 3rd chromosome Drosophila balancer stock (e.g., y[1] w[*];; TM3, Sb[1]/TM6B, Tb[+], Bloomington Drosophila Stock Center, catalog number: 3720)
5. piggyBac transposase Drosophila stock w[1118]; Herm[3xP3-eCFP, alphatub-piggyBacK10]M6 (Bloomington Drosophila Stock Center, catalog number: 32070)
6. Electrocompetent DH5α E. coli (ThermoFisher Scientific, catalog number: 11319019)
Reagents
Chemicals
1. Nuclease-free water (100 mL) (New England Biolabs, catalog number: B1500L)
2. Agarose (100 g) (ThermoFisher Scientific, catalog number: 16500-100)
3. GreenGlo Safe DNA Dye (500 μL at 20,000× in water) (Thomas Scientific, catalog number: C788T73)
4. Tris base (1 kg) (Fisher Scientific, catalog number: BP152-1, product format: resuspend in double-distilled water to 1 M, adjust pH to 8.0 or 7.4, and sterilize by autoclave)
5. Glacial acetic acid (2.5 L) (Fisher Scientific, catalog number: A490-212)
6. Ethylenediaminetetraacetic acid disodium salt dihydrate (EDTA) (500 g) (Millipore Sigma, catalog number: E5134-500G, product format: resuspend in double-distilled water to 0.5 M, adjust pH to 8.0, and sterilize by autoclave)
7. Absolute ethanol (200 proof) molecular biology grade (500 mL) (Fisher Scientific, catalog number: BP2818500)
8. Sodium dodecyl sulfate (SDS) (500 g) [Fisher Scientific, catalog number: BP166-500, product format: resuspend in double-distilled water to 0.5% (w/v) in a spray bottle]
9. Sodium chloride (NaCl) (10 kg) (Fisher Scientific, catalog number: BP358-10, product format: resuspend in double-distilled water to 5 M and sterilize by autoclave)
10. Bacto yeast extract (500 g) (ThermoFisher Scientific, catalog number: 212750)
11. Tryptone (500 g) (Fisher Scientific, catalog number: BP1421-500)
12. Agar (80–100 mesh) (500 g) (Fisher Scientific, catalog number: BP2641-500)
13. Carbenicillin, disodium salt (5 g) (Research Products International, catalog number: C46000-5.0, product format: resuspend to 100 mg/mL in double-distilled water and sterilize using a 0.2 μm filter with syringe)
14. Potassium chloride (KCl) (500 g) (Millipore Sigma, catalog number: P3911-500G, product format: resuspend to 2 M in double-distilled water and filter-sterilize)
15. Halocarbon oil 700 (50 mL) (Millipore Sigma, catalog number: H8898-50ML)
16. Halocarbon oil 27 (100 mL) (Millipore Sigma, catalog number: H8773-100ML)
17. Boric acid (1 kg) (Fisher Scientific, catalog number: BP168-1)
18. Ethidium bromide (10 mL at 10 mg/mL) (ThermoFisher Scientific, catalog number: 15585011)
19. Isopropanol (1 L) (ThermoFisher Scientific, catalog number: 327270010)
20. Sodium acetate (NaOAc) (500 g) (Fisher Scientific, catalog number: S210-500)
21. Dimethyl sulfoxide (DMSO) (50 mL) (Millipore Sigma, catalog number: D2438-50ML)
22. TRIzol reagent (100 mL) (ThermoFisher Scientific, catalog number: 15596026)
23. Sodium bicarbonate (NaHCO3) (500 g) (Fisher Scientific, catalog number: S233-500, product format: resuspend in double-distilled water to 1 M and adjust pH to 8.3)
24. Atto 633 NHS ester (1 mg) (Millipore Sigma, catalog number: 01464-1MG-F, product format: resuspend in DMSO to 33.4 mM)
25. Amino-11-ddUTP (200 nmol) (Lumiprobe, catalog number: A5040, product format: resuspend in nuclease-free water to 10 mM)
26. Triethylammonium acetate buffer (TEAA buffer) (250 mL at 1 M) (Millipore Sigma, catalog number: 69372-250ML, product format: dilute to 100 mM in double-distilled water and sterilize by filtering using a 500 mL 0.2 μm filter unit)
27. Acetonitrile, HPLC grade (1 L) (Fisher Scientific, catalog number: A998SK-1)
28. Phosphate-buffered saline (PBS) (1 L at 10×) (Fisher Scientific, catalog number: BP399-1, product format: dilute to 1× in double-distilled water)
29. Tween 20 (50 mL) (Millipore Sigma, catalog number: P9416-50ML)
30. Paraformaldehyde (500 g) (Millipore Sigma, catalog number: P6148-500G)
31. Diethyl pyrocarbonate (DEPC) (Millipore Sigma, catalog number: D5758-25ML)
32. Methanol (4 L) (Fisher Scientific, catalog number: A412SK-4)
33. Saline sodium citrate (SSC) (500 mL at 20×) (ThermoFisher Scientific, catalog number: AM9770)
34. Bovine serum albumin (BSA) (10 g) (Millipore Sigma, catalog number: A2153-10G, product format: resuspend to 100 mg/mL in nuclease-free water and store at -20 °C)
35. Dextran sulfate sodium salt (100 g) (ThermoFisher Scientific, catalog number: 433241000)
36. Salmon sperm DNA solution (5 × 1 mL, 10 mg/mL) (ThermoFisher Scientific, catalog number: 15632011)
37. Vanadyl ribonucleoside complexes (10 mL at 200 mM) (New England Biolabs, catalog number: S1402S)
38. DAPI [4′,6-Diamidino-2-phenylindole dihydrochloride, 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride] (5 mg) (Millipore Sigma, catalog number: D9542-5MG, product format: resuspend to 5 mg/mL in nuclease-free water for stock solution)
39. Vectashield Plus antifade mounting medium (10 mL) (Vector Laboratories, catalog number: H-1900-10)
Enzymes and enzyme kits
1. Phusion High-Fidelity DNA Polymerase (100 units at 2,000 units/mL) (New England Biolabs, catalog number: M0530S)
2. MEGAscript T7 Transcription kit (40 reactions) (ThermoFisher Scientific, catalog number: AM1334)
3. RiboLock RNase Inhibitor (2,500 units at 40 units/μL) (ThermoFisher Scientific, catalog number: EO0381)
4. Alt-R S.p. Cas9 Nuclease V3 (500 μg at 10 μg/μL) (Integrated DNA Technologies, catalog number: 1081059)
5. Proteinase K (1 mL at 20 mg/mL) (ThermoFisher Scientific, catalog number: EO0491)
6. Taq DNA polymerase (500 reactions) (ThermoFisher Scientific, catalog number: 10342020)
7. T7 Endonuclease I (1,250 units at 10,000 units/mL) (New England Biolabs, catalog number: M0302L)
8. EcoRV-HF (4,000 units at 20,000 units/mL) (New England Biolabs, catalog number: R3195S)
8. NEBuilder HiFi DNA Assembly Master Mix (50 reactions) (New England Biolabs, catalog number: E2621L)
9. RQ1 RNase-free DNase (1,000 units at 1 unit/μL) (Promega, catalog number: M6101)
10. SuperScript III Reverse Transcriptase (50 reactions) (ThermoFisher Scientific, catalog number: 18080044)
11. RNaseOUT Ribonuclease Inhibitor (5,000 units at 40 units/μL) (ThermoFisher Scientific, catalog number: 10777019)
12. TB Green Premix Ex Taq II (Tli RNase H Plus), 1 × 5 mL (200 reactions) (Takara Bio USA, catalog number: RR820L)
13. Terminal deoxynucleotidyl transferase (TdT) (500 units at 20 units/μL) (ThermoFisher Scientific, catalog number: EP0161)
Nucleic acids
1. Set of dATP, dCTP, dGTP, dTTP (40 μmol each at 100 mM) (Promega, catalog number: U1330)
2. pU6-BbsI-chiRNA plasmid (Addgene, catalog number: 45946)
3. Oligo primers for single-guide RNA (sgRNA) amplification (Integrated DNA Technologies, custom order, product format: resuspend to 100 μM in TE buffer for stock solution; dilute to 10 μM in nuclease-free water for working solution)
sgRNA_F: 5'-TTAATACGACTCACTATAGG[sgRNA_target_sequence_noPAM]GTTTTAGAGCTAGAAATAG-3'
sgRNA_R: 5'-AAAAGCACCGACTCGGTGCC-3'
4. 1 kb DNA ladder (200 gel lanes) (New England Biolabs, catalog number: N3232S)
5. 100 bp DNA ladder (100 gel lanes) (New England Biolabs, catalog number: N3231S)
6. pBS-GMR-eya(shRNA) plasmid (Addgene, catalog number: 157991)
7. pScarlessHD-DsRed plasmid (Addgene, catalog number: 64703)
8. Random oligo primers (Integrated DNA Technologies custom oligo 5’-N1-N1-N1-N1-N1-N1-N1-N1-N-3’ where N1 is hand-mixed ACGT at a ratio of 1:1:1:1 and where N is machine-mixed ACGT. Order 5 μmol and dissolve in double-distilled water to 500 μM. Dilute this stock to 50 μM and store as aliquots at -20 °C)
9. Anchored oligo(dT) primers (Integrated DNA Technologies custom oligo 5’-TTT TTT TTT TTT TTT TTT TTT TT-N2-N-3’ where N2 is hand-mixed ACGT at a ratio of 1:1:1:0 and where N is machine-mixed ACGT. Order 1 μmol and dissolve in double-distilled water to 50 μM. Store as aliquots at -20 °C)
Commercial clean-up kits
1. Monarch Spin PCR & DNA Cleanup kit (5 μg) (250 preps) (New England Biolabs, catalog number: T1130L)
2. Monarch Spin Plasmid Miniprep kit (250 preps) (New England Biolabs, catalog number: T1110L)
3. Monarch Spin RNA Cleanup kit (50 μg) (100 preps) (New England Biolabs, catalog number: T2040L)
4. Monarch Spin DNA Gel Extraction kit (50 preps) (New England Biolabs, catalog number: T1120S)
5. HiSpeed Plasmid Midi kit (25 preps) (Qiagen, catalog number: 12643)
6. EndoFree Plasmid Mega kit (5 preps) (Qiagen, catalog number: 12381)
7. Microspin G-25 columns (50 columns) (Cytiva, catalog number: 27532501)
Drosophila husbandry
1. Active dry yeast (2 lbs) (Genesee Scientific, catalog number: 62–103, product format: mix 7 g of yeast with 10 mL of deionized water in a 50 mL beaker and mix until it forms a paste with the consistency of peanut butter. Store covered with parafilm at 4 °C for up to a week, adding water or yeast as necessary to maintain consistency)
2. Molasses (1 gal) (Genesee Scientific, catalog number: 62-117)
3. Ethyl acetate (500 mL) (Fisher Scientific, catalog number: E145-500)
4. Propionic acid (1 L) (ThermoFisher Scientific, catalog number: 149300010)
5. Tegosept (1 kg) [Genesee Scientific, catalog number: 20-258, product format: resuspend to 10.67% (w/v) by adding 16 g of Tegosept to 150 mL of 95% ethanol and store at room temperature]
Solutions
1. dNTP mix (see Recipes)
2. Tris-EDTA buffer (TE) (see Recipes)
3. 50× Tris-acetate-EDTA buffer (TAE) (see Recipes)
4. 5× Tris-borate-EDTA buffer (TBE) (see Recipes)
5. Luria broth (LB) (see Recipes)
6. Luria broth (LB) agar plates (see Recipes)
7. Embryo collection plates (see Recipes)
8. Halocarbon oil mix (see Recipes)
9. Squish buffer (see Recipes)
10. DEPC water (see Recipes)
11. PBT (see Recipes)
12. Paraformaldehyde (PFA) fixative (see Recipes)
13. RNA smFISH hybridization buffer (see Recipes)
14. RNA smFISH wash buffer (see Recipes)
Recipes
1. dNTP mix
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| dATP (100 mM) | 10 mM | 50 μL |
| dCTP (100 mM) | 10 mM | 50 μL |
| dGTP (100 mM) | 10 mM | 50 μL |
| dTTP (100 mM) | 10 mM | 50 μL |
| Nuclease-free water | 300 μL |
a. Thaw the individual dNTP stocks (100 mM each).
b. Briefly vortex them and spin them down.
c. Mix each dNTP as indicated below.
d. The dNTP mix can be stored at -20 °C.
2. Tris-EDTA buffer (TE)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 1 M Tris pH 8.0 | 10 mM | 500 μL |
| 0.5 M EDTA | 1 mM | 100 μL |
| Double-distilled water | 49.4 mL |
a. Mix the individual stock solutions as indicated below.
b. Sterilize by filtering with a Steriflip vacuum filter unit.
c. Store at room temperature.
3. 50× Tris-acetate-EDTA buffer (TAE)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris base (MW = 121.14 g/mol) | 2 M | 242 g |
| Glacial acetic acid | 1 M | 57.1 mL |
| 0.5 M EDTA | 50 mM | 100 mL |
| Double-distilled water | fill to 1,000 mL |
a. Mix the individual reagents as indicated below in ~650 mL of double-distilled water in a 1 L glass beaker with a stir bar until all reagents are in solution.
b. Bring volume up to 1,000 mL with double-distilled water and split between two 1 L glass bottles.
c. Sterilize by autoclave on a liquid cycle for at least 30 min.
d. Store at room temperature.
e. Dilute to 1× TAE in double-distilled water for standard agarose gel electrophoresis.
4. 5× Tris-borate-EDTA buffer (TBE)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris base (MW = 121.14 g/mol) | 450 mM | 54.51 g |
| Boric acid (MW = 61.83 g/mol) | 450 mM | 27.82 g |
| 0.5 M EDTA | 10 mM | 20 mL |
| Double-distilled water | fill to 1,000 mL |
a. Mix the individual reagents as indicated below in ~700 mL of double-distilled water in a 1 L glass beaker with a stir bar until all reagents are in solution.
b. Bring volume up to 1,000 mL with double-distilled water and split between two 1 L glass bottles.
c. Sterilize by autoclave in a liquid cycle for at least 30 min.
d. Store at room temperature.
e. Dilute to 0.5× TBE in double-distilled water for agarose gel electrophoresis of T7EI digestion products.
5. Luria broth (LB)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| NaCl | 5 g/L | 5 g |
| Tryptone | 10 g/L | 10 g |
| Bacto yeast extract | 5 g/L | 5 g |
| Double-distilled water | fill to 1,000 mL |
a. Mix the individual reagents as indicated below in ~700 mL of double-distilled water in a 1 L glass beaker with a stir bar until all reagents are in solution.
b. Bring volume up to 1,000 mL with double-distilled water and split between two 1 L glass bottles.
c. Sterilize by autoclave on a liquid cycle for at least 30 min.
d. Store LB without antibiotics at room temperature.
e. Add appropriate antibiotic (e.g., carbenicillin at a final concentration of 100 μg/mL) after LB is cooled down.
f. Store LB with antibiotics at 4 °C for up to 1 month.
6. Luria broth (LB) agar plates
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| NaCl | 5 g/L | 5 g |
| Tryptone | 10 g/L | 10 g |
| Bacto yeast extract | 5 g/L | 5 g |
| Double-distilled water | fill to 1,000 mL | |
| Agar | 15 g/L | 15 g |
a. Mix the individual reagents as indicated in ~700 mL of double-distilled water in a 2 L Erlenmeyer flask with a stir bar until all reagents are in solution.
b. Bring volume up to 1,000 mL with double-distilled water. Add agar, which will not go into solution until after autoclave sterilization.
c. Sterilize by autoclave on a liquid cycle for at least 30 min, leaving the stir bar in the flask.
d. Allow to cool to ~50 °C. Add appropriate antibiotics (e.g., carbenicillin at a final concentration of 100 μg/mL) and stir for 5 min.
e. Promptly pour the liquid mixture into 100 mm bacteriological Petri dishes with sterile technique and allow to cool at room temperature.
f. Once solidified, store LB agar plates with antibiotics at 4 °C for up to 1 month.
7. Embryo collection plates
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Agar | 33.4 g/L | 22 g |
| Molasses | 13.7% (v/v) | 90 mL |
| Double-distilled water | 560 mL | |
| Ethyl acetate | 0.76% (v/v) | 5 mL |
| Propionic acid | 0.49% (v/v) | 3.2 mL |
| 10.67% (w/v) Tegosept solution | 0.20% (w/v) | 125 μL |
a. Mix agar, molasses, and water in a 2 L Erlenmeyer flask with a stir bar.
b. Agar will not go into solution until after autoclave sterilization.
c. Sterilize by autoclave on a liquid cycle for at least 30 min., leaving the stir bar in the flask.
d. After autoclaving, stir on a stir plate until the temperature cools to ~50 °C. Add the three inhibitors (ethyl acetate, propionic acid, and Tegosept) and stir for 5 min.
e. Promptly pour liquid mixture into 60 mm disposable Petri dishes. Do not flame the lip of the flask when pouring plates, as the mixture is flammable.
f. Allow plates to cool at room temperature overnight so that condensation evaporates.
g. Store embryo collection plates at 4 °C for up to 3 months.
8. Halocarbon oil mix
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Halocarbon oil 700 | 75% (v/v) | 6 mL |
| Halocarbon oil 27 | 25% (v/v) | 2 mL |
a. Mix reagents as indicated below in a 15 mL conical tube.
b. Allow to mix thoroughly on a nutator.
c. Store at room temperature.
9. Squish buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| 1 M Tris pH 8.0 | 10 mM | 500 μL |
| 5 M NaCl | 25 mM | 250 μL |
| 0.5 M EDTA | 1 mM | 100 μL |
| Double-distilled water | fill to 49.5 mL | |
| Proteinase K (20 mg/mL) | 0.2 mg/mL | 1 μL per 100 μL squish buffer |
a. Mix Tris, NaCl, EDTA, and water in a 50 mL conical tube.
b. Sterilize by filtering with a Steriflip vacuum filter unit.
c. Store this squish buffer without Proteinase K at room temperature.
d. Immediately before use, add 1 μL of Proteinase K (20 mg/mL) to 99 μL of squish buffer, scaling up or down as needed.
e. Add squish buffer (with Proteinase K) directly to samples.
10. DEPC water
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Diethyl pyrocarbonate (DEPC) | 0.1% (v/v) | 1 mL |
| Double-distilled water | 1 L |
a. Add DEPC directly to double-distilled water.
b. Invert several times to mix and let it sit overnight.
c. Autoclave on a liquid cycle for at least 30 min to sterilize and decompose the DEPC.
d. Store at room temperature.
11. PBT
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| PBS, 10× | 1× | 5 mL |
| Tween 20 | 0.1% (v/v) | 50 μL |
| DEPC water | 45 mL |
a. Add 10× PBS, DEPC water, and Tween 20 to a 50 mL conical tube.
b. Vortex briefly and rock on a nutator for at least 20 min.
c. Store at room temperature.
12. Paraformaldehyde (PFA) fixative
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| PBS, 10× | 1× | 1 mL |
| DEPC water | 9 mL | |
| Paraformaldehyde | 4% (w/v) | 0.4 g |
| Tween 20 | 0.1% (v/v) | 10 μL |
a. Add 10× PBS and DEPC water to a 50 mL glass beaker.
b. Swirl briefly and then microwave for 10 s.
c. Using gloves, add a stir bar and then stir on a heated stir plate (~70 °C) in a chemical fume hood.
d. Add paraformaldehyde and stir until it goes into solution.
e. Transfer to a 15 mL conical tube and replace any volume that has evaporated with fresh DEPC water to 10 mL.
f. Add Tween 20, vortex briefly, and rock on a nutator for 20 min.
g. Store at room temperature until use. Prepare fresh daily.
13. RNA smFISH hybridization buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Dextran sulfate | 10% (w/v) | 0.5 g |
| Nuclease-free water | 3.8 mL | |
| BSA (100 mg/mL) | 0.2 mg/mL | 10 μL |
| Salmon sperm DNA solution (10 mg/mL) | 0.1 mg/mL | 50 μL |
| Vanadyl ribonucleoside complexes (200 mM) | 2 mM | 50 μL |
| Tween 20 | 0.1% | 5 μL |
a. Add dextran sulfate to nuclease-free water in a 15 mL conical tube and vortex vigorously until dextran sulfate is in solution.
b. Add BSA, salmon sperm solution, vanadyl ribonucleoside complexes, and Tween 20, and vortex.
c. Store at 4 °C shielded from light for up to a year.
14. RNA smFISH wash buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Saline sodium citrate (SSC) (20×) | 4× | 10 mL |
| Tween 20 | 0.1% | 50 μL |
| DEPC water | 40 mL |
a. Add 20× SSC, DEPC water, and Tween 20 to a 50 mL conical tube.
b. Vortex briefly and rock on a nutator for at least 20 min.
c. Store at room temperature.
Laboratory supplies
1. 1.5 mL low retention microcentrifuge tubes (nuclease-free) (ThermoFisher Scientific, catalog number: 3451)
2. 0.2 mL PCR 8-tube strips (USA Scientific, catalog number: 1402-2900)
3. 1,250 μL low retention universal pipette tips (VWR, catalog number: 76323-456)
4. 200 μL low retention universal pipette tips (VWR, catalog number: 76323-390)
5. 10 μL low retention universal pipette tips (VWR, catalog number: 76323-394)
6. 1,250 μL SHARP low retention barrier pipette tips (nuclease-free) (Thomas Scientific, catalog number: 1159M42)
7. 200 μL SHARP low retention barrier pipette tips (nuclease-free) (Thomas Scientific, catalog number: 1159M40)
8. 20 μL SHARP low retention barrier pipette tips (nuclease-free) (Thomas Scientific, catalog number: 1159M43)
9. 10 μL SHARP low retention barrier pipette tips (nuclease-free) (Thomas Scientific, catalog number: 1159M41)
10. 5 mL disposable polystyrene serological pipettes (Corning, catalog number: 4487)
11. 10 mL disposable polystyrene serological pipettes (Corning, catalog number: 4488)
12. 25 mL disposable polystyrene serological pipettes (Corning, catalog number: 4489)
13. 50 mL disposable polystyrene serological pipettes (Greiner, catalog number: 768180)
14. 100 mm × 15 mm bacteriological Petri dishes (Falcon, catalog number: 351029)
15. 60 mm × 15 mm disposable Petri dishes (for embryo collection plates) (VWR, catalog number: 25384-328)
16. 100 mL Tri-Pour graduated disposable beakers (for embryo collection cages) (United States Plastic Corp., catalog number: 76076, product format: poke small holes in the top of the cage to allow air flow)
17. Microloader pipette tips (Eppendorf, catalog number: 930001007)
18. 18 mm × 18 mm micro cover glass (VWR, catalog number: 48366-045)
19. Frosted microscope slides, 75 mm × 25 mm (Fisher Scientific, catalog number: 12-544-2)
20. Borosilicate capillary glass with filament (outer diameter = 1.0 mm, inner diameter = 0.5 mm, length = 10 cm, for microinjection needles) (Sutter Instrument, catalog number: BF100-50-10)
21. Masterstroke finest red sable paint brush, round, size 0 (Blick Art Materials, catalog number: 05457-1100)
22. Hypodermic needle, 26 gauge, 5/8 in. (Becton Dickinson, catalog number: 305115)
23. Razor blade, 0.22 mm (VWR, catalog number: 55411-050)
24. 50 mL polypropylene conical centrifuge tubes (Falcon, catalog number: 352098)
25. 15 mL polypropylene conical centrifuge tubes (Falcon, catalog number: 352099)
26. Kimwipes, 8.2 in. × 4.4 in. (Kimberly-Clark Professional, catalog number: 34120)
27. Saran wrap
28. RNase-free disposable pellet pestle (Fisher Scientific, catalog number: 12-141-364)
29. MicroAmp EnduraPlate optical 96-well clear reaction plate (for qPCR) (Applied Biosystems, catalog number: A36924)
30. MicroAmp optical adhesive film (for qPCR) (Applied Biosystems, catalog number: 4311971)
31. High performance cover glass, No. 1.5H, 22 mm × 22 mm (Zeiss, catalog number: 474030-9020-000)
32. Clear nail polish (Sally Hansen, catalog number: 45083)
33. Steriflip 0.22 μm vacuum filter unit, 50 mL (Millipore Sigma, catalog number: SCGP00525)
34. Disposable 0.2 μm filter unit, 500 mL (Fisher Scientific, catalog number: FB12566504)
35. Syringe filter, 0.2 μm (50 filters) (ThermoFisher Scientific, catalog number: 725-2520)
36. Syringe, 20 mL (50 syringes) (Fisher Scientific, catalog number: 14-817-111)
37. Magnetic stir bar kit (Fisher Scientific, catalog number: 14-513-82)
Equipment
General molecular biology
1. Thermocycler (Bio-Rad, model: T100)
2. Pipetman 4-pipet kit (P2, P20, P200, P1000) (Gilson, catalog number: F167360)
3. Owl EasyCast B2 mini gel electrophoresis system (ThermoFisher Scientific, catalog number: B2-BP)
4. Owl compact power supply (ThermoFisher Scientific, catalog number: EC300XL)
5. Ultraviolet gel imaging system (Azure Biosystems, model: c600)
6. Vortex mixer (Fisher Scientific, catalog number: 02-215-414)
7. Electronic controller for serological pipets (Gilson, catalog number: F110120)
8. MyFuge mini centrifuge (Braintree Scientific, catalog number: C1008)
9. Centrifuge with 24 × 2 mL rotor (Eppendorf, model: 5425, catalog number: 5405000441)
10. Refrigerated centrifuge with 24 × 2 mL rotor (Eppendorf, model: 5425R, catalog number: 5406000445)
11. NanoDrop Ultra UV-Vis spectrophotometer (ThermoFisher Scientific, catalog number: NDULTRAGL)
12. Qubit 4 fluorometer (ThermoFisher Scientific, catalog number: Q33238)
13. Heat block (Fisher Scientific, catalog number: 88-860-022)
14. Gene Pulser Xcell Microbial System (Bio-Rad, catalog number: 1652662)
15. Shaking incubator for microbiology (37 °C) (New Brunswick Scientific, model: Innova43)
16. Autoclave (Spire Integrated Solutions, model: PRIMUS)
17. Microwave (Sharp, model: SMC1441CW)
18. Magnetic hot/stir plate (VWR, catalog number: 76447-044)
19. Nalgene autoclave pan (ThermoFisher Scientific, catalog number: 6900-0020)
20. 1 L PYREX glass beaker (Corning, catalog number: 10031L)
21. 50 mL glass beaker (Fisher Scientific, catalog number: FB10050)
22. 2 L PYREX glass Erlenmeyer flask (Corning, catalog number: 49802L)
23. 1 L Wheaton glass bottle (DWK Life Sciences, catalog number: 219760)
24. Refrigerator (4 °C)
25. Freezer (-20 °C)
26. Ultra-low temperature freezer (-80 °C)
Drosophila husbandry
1. Stereomicroscope (Zeiss, model: Stemi 508)
2. Dual gooseneck fiber-optic illuminator (AmScope, catalog number: HL150-AY)
3. Drosophila anesthesia system (Genesee Scientific, catalog number: 59-122BC)
4. NightSea full system with royal blue illumination (Electron Microscopy Sciences, catalog number: SFA-RB)
5. NightSea add-on light and filter set, violet (Electron Microscopy Sciences, catalog number: SFA-LFS-VI)
6. NightSea add-on light and filter set, green (Electron Microscopy Sciences, catalog number: SFA-LFS-GR)
7. Drosophila incubator (Genesee Scientific, catalog number: 59-407L)
Embryo injections
1. Inverted phase contrast microscope (for microinjections) (Nikon, model: TMS)
2. Femtojet 4i microinjector (Calibre Scientific, catalog number: EPE-5252000021)
3. Mechanical micromanipulator (Leica, model: Micromanipulator)
4. Micropipette grinder (Narishige, model: EG-40)
5. Micropipette puller (Sutter Instrument, model: P-97)
RT-qPCR
1. Plate centrifuge (Eppendorf, model: 5810)
2. Real-time PCR system (Applied Biosystems, model: QuantStudio 3)
RNA smFISH
1. ZORBAX Eclipse Plus C18 Reverse Phase HPLC column (Agilent Technologies, catalog number: 959961-902)
2. High-pressure liquid chromatography (HPLC) system (Agilent Technologies, model: 1100)
3. Savant SpeedVac DNA 130 system (ThermoFisher Scientific, catalog number: DNA130-115)
4. ProBlot 12 hybridization oven (Labnet, catalog number: H1200A)
5. Nutating mixer (Corning, catalog number: 6720)
6. Laser scanning confocal microscope (Leica Microsystems, model: SP8)
Software and datasets
1. FlyBase (https://flybase.org)
2. flyCRISPR Optimal Target Finder (https://flycrispr.org/target-finder/)
3. Benchling (https://www.benchling.com)
4. NEBuilder Assembly Tool (https://nebuilder.neb.com/)
5. Primer3Plus (https://www.primer3plus.com)
6. Integrated DNA Technologies (IDT) OligoAnalyzer Tool (https://www.idtdna.com/calc/analyzer)
Procedure
The first goal is to find an sgRNA that efficiently targets the gene of interest for ribozyme knockdown (Figure 2). Several sgRNAs are tested for their efficiency at inducing nonhomologous end joining (NHEJ) events at the selected target site.
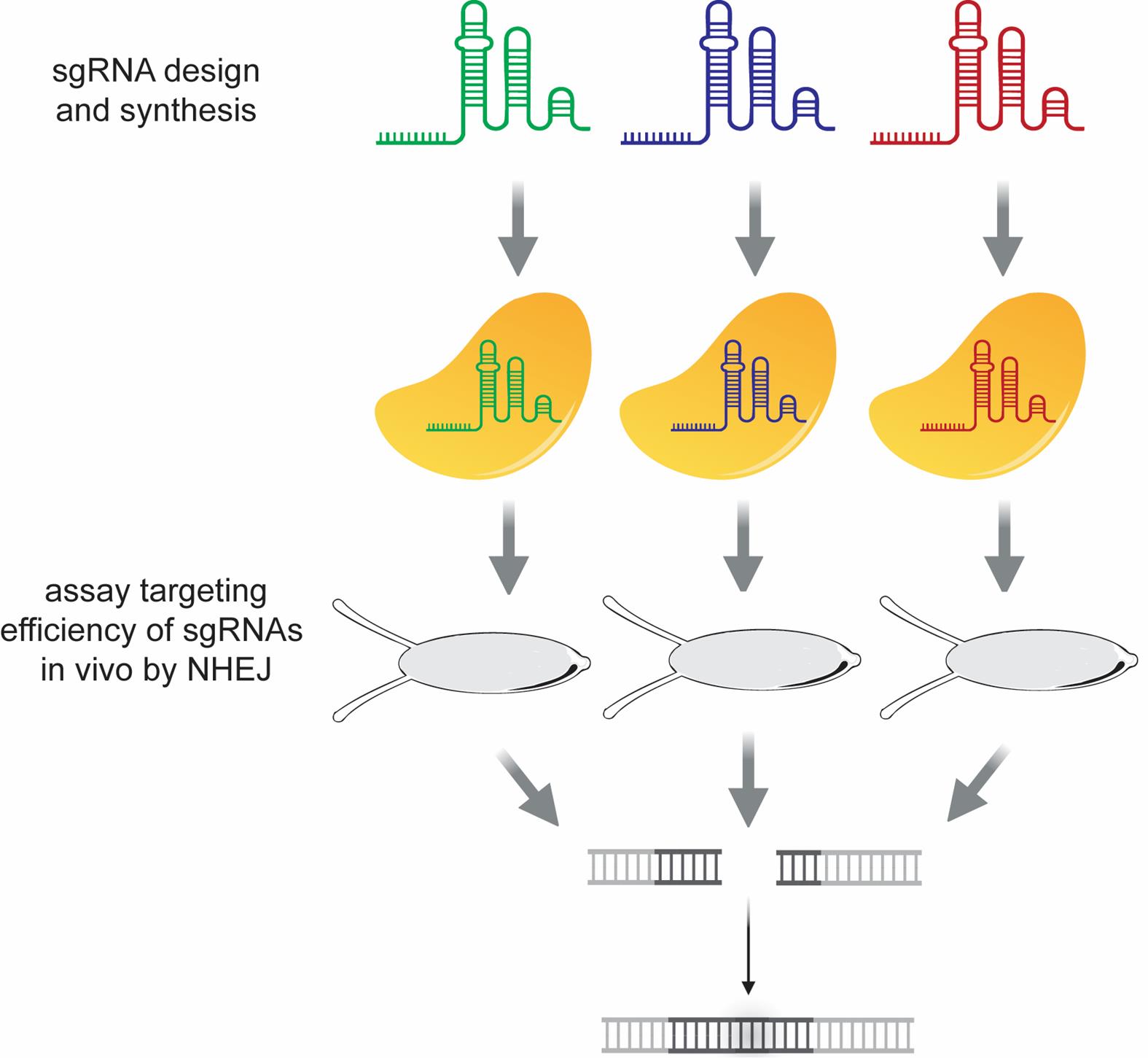
Figure 2. Workflow for the design and assessment of cleavage efficiencies in vivo of multiple sgRNAs targeting the lncRNA gene
A. Design considerations for selecting appropriate ribozyme-knockdown targets
The N79 ribozyme cassette used here resulted in efficient RNA cleavage and degradation of the 3' cleavage fragment in all lncRNA genes targeted thus far. The fate of the 5' cleavage fragment has been more variable. Thus, we strongly recommend inserting the ribozyme in the 5' region of transcripts that will be knocked down. Insertion of the ribozyme cassette just downstream (~85–100 bp) of the transcription start site works well.
In addition, consider the following when choosing where to insert the ribozyme cassette in a target gene:
1. The insertion site needs to be close to a naturally occurring PAM site (i.e., NGG). If possible, insert the ribozyme cassette within or immediately 5' to the PAM site. Doing so will disrupt the sgRNA target sequence in the donor repair plasmid, ensuring that Cas9-mediated cleavage will only occur in the genome.
2. To ensure scarless removal of the DsRed marker after integration, the ribozyme insertion site should be near a naturally occurring TTAA site (ideally within 30 bp). If a naturally occurring TTAA site is not present nearby, a similar sequence (e.g., TTAT, TAAA, TTCA) can be substituted with TTAA in the donor repair plasmid.
3. Insertion of the ribozyme cassette within an intron may result in knockdown of spliced RNA, though the size of the intron likely is a factor. We have previously shown that insertion of the ribozyme cassette at the 5' end of a ~17,500-bp intron in CR44833 could produce robust knockdown in Drosophila [23]. However, insertion of the N79 ribozyme into a small intron of ~500 bp failed to produce knockdown in mammalian cells [18].
B. In vitro transcription of candidate sgRNAs
1. To identify candidate sgRNAs that target the genomic region of interest, use flyCRISPR Optimal Target Finder (https://flycrispr.org/target-finder/).
a. Enter the genomic DNA sequence to be targeted. Follow the design considerations discussed in Section A.
b. Select the appropriate Drosophila melanogaster genome. If the Drosophila line that will be modified is not included here, choose the reference genome (r_6). The sequence of the genomic region of interest in the chosen Drosophila line will need to be verified using PCR and Sanger sequencing to ensure there are no polymorphisms that disrupt sgRNA target sites.
c. Select a 20 nt guide length for maximum specificity.
d. Select All CRISPR targets. A 5' G or 5' GG in the target sequence is not necessary for in vitro transcription of RNA using T7 RNA polymerase.
e. Select Find CRISPR targets.
f. Select High Stringency and NGG Only PAM sites.
g. Select Evaluate.
2. Select 2–3 highly ranked sgRNAs as candidates (see Section A for design criteria). Candidate sgRNAs having minimal predicted off-target sites (i.e., 0 or 1) are preferable.
3. Synthesize the candidate sgRNAs in vitro. Design and order the following PCR primers:
sgRNA_R: 5'-AAAAGCACCGACTCGGTGCC-3' (used for all sgRNAs)
sgRNA_F: 5'-TTAATACGACTCACTATAGG[sgRNA_target_sequence_noPAM]GTTTTAGAGCTAGAAATAG-3' (specific for each sgRNA)
Critical: Do not include the PAM site for each sgRNA target in the sgRNA_F primers.
4. For each candidate sgRNA, perform a 50 μL PCR reaction with the following conditions:
Reaction mix:
10 μL of 5× Phusion HF buffer
1 μL of 10 mM dNTP mix
2.5 μL of 10 μM sgRNA_F primer
2.5 μL of 10 μM sgRNA_R primer
10 ng of pU6-BbsI-chiRNA plasmid DNA
0.5 μL of Phusion High-Fidelity DNA polymerase (2,000 units/mL)
to 50 μL of nuclease-free water
Touchdown PCR thermocycling conditions:
(1) 98 °C for 2 min.
(2) 98 °C for 15 s.
(3) 60 °C - 0.5 °C/cycle for 15 s.
(4) 72 °C for 20 s.
(5) To step 2, 20 times
(6) 98 °C for 15 s.
(7) 50 °C for 15 s.
(8) 72 °C for 20 s.
(9) To step 6, 13 times
(10) 72 °C for 5 min.
Note: Other high-fidelity DNA polymerases should also work just as well as Phusion HF DNA polymerase.
5. Verify successful PCR amplification using 1% (w/v) agarose gel electrophoresis with 1× TAE buffer and 1× GreenGlo DNA dye. The PCR product should be ~120 bp in size.
6. Purify the PCR product using the Monarch Spin PCR & DNA Cleanup kit, eluting in 20 μL of nuclease-free water.
7. Measure the concentration of the purified PCR product using a NanoDrop spectrophotometer. The concentration should be 40 ng/μL or higher.
Pause point: Purified PCR products can be safely stored at -20 °C until ready to proceed.
8. For each candidate sgRNA, perform the following in vitro transcription reaction using the MEGAscript T7 transcription kit:
Reaction mix:
2 μL of 10× reaction buffer
2 μL of ATP solution
2 μL of CTP solution
2 μL of GTP solution
2 μL of UTP solution
300 ng of purified PCR product from above
2 μL of T7 RNA polymerase enzyme mix
0.5 μL of RiboLock RNase inhibitor (40 U/μL)
to 20 μL of nuclease-free water
Reaction incubation conditions: 37 °C overnight (up to 16 h).
Note: DNase treatment is not necessary after in vitro transcription.
9. Purify the in-vitro transcribed sgRNA product using the Monarch Spin RNA Cleanup kit (50 μg), eluting in 20 μL of nuclease-free water.
10. Measure the concentration of the purified sgRNA using a NanoDrop spectrophotometer. Successful in vitro transcription should yield concentrations of 2,000 ng/μL or higher. Purified RNA should have a 260/280 ratio of ~2.0 and a 260/230 ratio between 2.0 and 2.3.
Note: Successful in vitro transcription should also produce a bright, discrete band when running 0.5 μL on a 1% (w/v) agarose gel.
Pause point: Purified sgRNA can be safely stored at -80 °C until ready to proceed.
C. Empirical testing of cleavage efficiencies for candidate sgRNAs
To maximize the probability of successful CRISPR/Cas9-mediated insertion of the N79 cassette, empirical testing of cleavage efficiency for each candidate sgRNA should be performed (Figure 2). To do this, Cas9/sgRNA ribonucleoproteins (RNPs) will be assembled in vitro using purified Cas9 protein and the sgRNAs prepared in Section B. RNPs will be injected into non-dechorionated embryos (eggshells still intact) using the method of Gompel and Schröder (http://gompel.org/wp-content/uploads/2015/12/Drosophila-transformation-with-chorion.pdf). Indels created by RNP-mediated cleavage and NHEJ in genomic DNA from injected embryos will be detected in vitro after digestion with T7 endonuclease I (T7EI), which recognizes and cleaves mismatched regions in double-stranded DNA. RNP-mediated cleavage efficiency correlates with T7EI digestion efficiency.
1. Three days before embryo injections, set up 2–3 embryo collection cages with embryo collection plates containing a dab of yeast paste. Use ~120 females and ~60 males per cage of the Drosophila line that will be modified. Incubate in a 25 °C incubator and flip the cages onto fresh embryo collection plates with yeast paste at least daily until injection day.
2. On the day of injection, pre-clear any developing embryos that the females may be internally storing by flipping the flies onto fresh embryo collection plates with yeast paste and allowing them to lay for 60–90 min in a 25 °C incubator in the dark. Perform this at least twice, discarding the embryos each time.
Note: Flies lay embryos best either early morning or late afternoon. We typically aim to start our first embryo injections at 4 pm.
3. After at least two pre-clears, flip the cages again onto fresh embryo collection plates with yeast paste and allow the females to lay embryos for ~45 min in a 25 °C incubator in the dark. These embryos will be collected for injections.
4. While collecting embryos for injection, prepare the RNPs for each candidate sgRNA:
RNP mix:
1.19 μL of Alt-R S.p. Cas9 Nuclease V3 (10 μg/μL)
0.38 μL of 2 M KCl
2.36 μg of purified sgRNA from Section B
to 5 μL of nuclease-free water
Incubation conditions: Room temperature for 10 min.
5. Centrifuge the RNP mix at 16,000 rcf for 10 min. Transfer 4 μL of supernatant into a fresh tube to be loaded into the injection needles. Keep at room temperature until needle loading.
6. Load a microinjection needle with the RNP mix using a microloader pipette tip.
7. Adhere an 18 mm × 18 mm cover glass to a microscope slide using water. Then, use a size 0 red sable paint brush to transfer embryos from the collection plate to the cover glass and line up 40–50 embryos on the cover glass, with the posterior end of the embryo oriented to face the injection needle.
8. Gently apply a layer of halocarbon oil mix over the embryos.
9. Inject the RNP mix for a particular candidate sgRNA into the embryos. Skip any embryos that have cellularized or look to have developmental defects. A volume of ~100 pL should be injected into each embryo.
10. After injecting all suitable embryos on the slide, use a hypodermic needle to rupture any embryos that were skipped due to cellularization or developmental defects.
11. Remove as much of the halocarbon oil from the injected embryos as possible using Kimwipes and a razor blade.
Critical: Take care when using a razor blade to prevent cuts.
12. Gently remove the cover glass with embryos from the slide and place it on a fresh embryo collection plate with the embryos face up, being careful not to crush them. Place the lid on the collection plate and place the plate in a humid chamber (e.g., a beaker lined with damp paper towels and partially covered with Saran Wrap to retain moisture but also allow air).
13. Repeat steps C6–12 for each candidate sgRNA.
Note: We typically prepare slides from a fresh collection of embryos for each candidate sgRNA to ensure that the majority of embryos have not undergone cellularization.
14. For a negative control, perform one final collection of embryos. Place this plate of uninjected embryos in the humid chamber as well.
15. Incubate the embryos in the humid chamber at 25 °C overnight.
16. Collect genomic DNA from injected embryos. Approximately 24 h after the final injections, pick up individual L1 larvae (preferable) or late-stage embryos from each collection plate using a pipette tip. Place each individual into its own PCR tube that is pre-loaded with 20 μL of squish buffer plus Proteinase K. Place 8 individuals per sgRNA treatment, as well as 8 individuals from the negative control treatment.
17. Grind each larva or embryo sample with a pipette tip. Verify under a stereomicroscope that the larva or embryo is still in the tube and that it has ruptured. Briefly spin down the mixes.
Critical: It is very easy to lose a larva or embryo, so verify that the individual is still in the tube.
18. Incubate the tubes at 37 °C for 30 min and then 95 °C for 5 min to inactivate the Proteinase K. Store the genomic DNA preps at 4 °C.
Pause point: Genomic DNA preps can be stored up to a few weeks at 4 °C.
19. In preparation for the T7EI assay, design PCR primers that amplify the genomic region of interest containing the sgRNA cleavage sites. PCR amplicons should be between 700 and 1,200 bp long, with the sgRNA target sites located in the middle of the amplicons.
Note: T7EI cleaves duplex DNA at sites of sequence mismatch. If sgRNA RNPs generate NHEJ-induced mutations in vivo, the T7EI will digest the DNA amplicon at those sites. However, T7EI also cleaves non-B DNA structures such as cruciform DNA. Thus, even T7EI assays of unmodified DNA may produce cleavage products. Therefore, we recommend pre-testing multiple PCR primer pairs to ensure that the predicted cleavage products produced by sgRNA targeting do not overlap with any side-reaction cleavage products. Steps C20–23 describe this pre-testing procedure.
20. For each candidate PCR primer pair, amplify DNA from individual noninjected control genomic DNA. Perform 2–3 separate PCR reactions in 50 μL PCR reactions. For each reaction, use 3 μL of genomic DNA and standard Taq DNA polymerase.
21. After the PCR is complete, denature and reanneal 10 μL of the PCR products using the following protocol in a thermocycler:
(1) 95 °C for 3 min.
(2) 94 °C for 1 min.
(3) Repeat 90 times for 1 min each at a temperature, stepping down 1 °C for each cycle
(4) 6 °C for 10 s.
(5) 8 °C for 10 s.
(6) 10 °C for 10 s.
(7) 12 °C, hold.
22. Perform the following T7EI reaction using the denatured and reannealed PCR product:
Reaction mix:
10 μL of denatured and reannealed PCR product
2 μL of 10× NEBuffer 2
0.2 μL of T7 Endonuclease I (10,000 units/mL)
to 20 μL of nuclease-free water
Reaction incubation conditions: 37 °C for 1 h.
23. Run the T7EI-digested DNA side-by-side with 10 μL of undigested DNA as a control on a 2% (w/v) agarose gel. To increase sensitivity and detect faint cleavage products, use 0.5× TBE as the electrophoresis buffer and ethidium bromide (0.5 μg/mL) as the fluorescent dye.
24. From screening various primer pairs in steps C20–23, select a primer pair that both (1) produces a strong PCR product and (2) does not produce any digestion products that overlap in size with predicted fragments from sgRNA-targeted NHEJ events.
25. With the selected primer pair, repeat steps C20–23 (PCR amplification, denaturation and reannealing, T7EI digestion, and agarose gel electrophoresis) for all RNP-injected and uninjected control samples generated in steps C4–18.
26. Identify sgRNA-mediated cleavage products of the expected size from RNP-injected individuals that are not present in uninjected and undigested controls (Figure 3). sgRNAs that generate cleavage products in multiple injected individuals are highly likely to be successful and should be selected for subsequent HDR efforts.
Notes:
1. sgRNA-mediated cleavage products may be faint, so make sure exposure time is high enough to capture them when imaging the gel.
2. In our experience, sgRNAs that produce the expected cleavage fragments in at least 50% of embryos are very likely to result in successful integration of the ribozyme cassette via HDR.
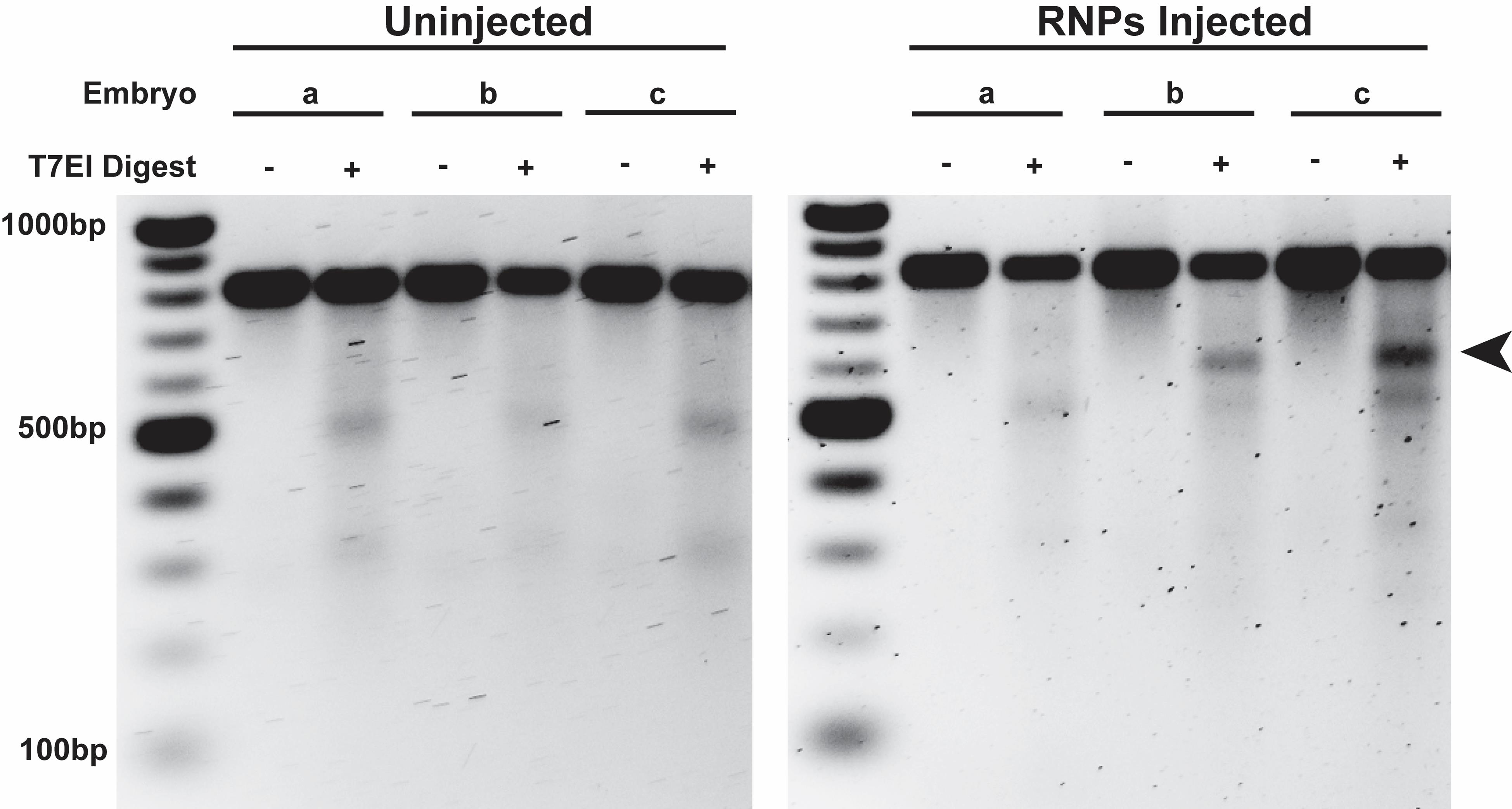
Figure 3. In vivo screening of sgRNA cleavage activity using the T7 endonuclease I assay. PCR products (826 bp) amplified from the lncRNA CR45715 gene in uninjected (left) and RNP-injected (right) embryos were subjected to T7 endonuclease I (T7EI) digestion. T7EI digestion of this PCR amplicon was predicted to produce cleavage products of 596 and 230 bp upon successful cleavage and NHEJ by the sgRNA/Cas9 RNP. The 596-bp cleavage product (marked by an arrow) is clearly visible in two of three RNP-injected flies, though the 230-bp product is too faint to visualize. Bands at ~500 and ~300 bp are produced in all embryos, including uninjected, after T7EI digestion and are not indicative of successful targeting by sgRNA/Cas9 RNP. This sgRNA was subsequently used to successfully insert the ribozyme cassette in the CR45715 locus [23]. The figure is reproduced from Nyberg et al. [26] with permission from Springer Nature (license 6070871136981).
Once an sgRNA is selected, a tailor-made donor plasmid is constructed and used to induce HDR-mediated ribozyme insertion at the target site (Figure 4). The resultant Drosophila lines carrying the ribozyme insertion are then characterized for knockdown of gene expression using RT-qPCR and smFISH, where appropriate.
Figure 4. Workflow for the integration of the ribozyme cassette into the Drosophila genome via homology-directed repair (HDR) and validation of lncRNA knockdown using RT-qPCR and RNA smFISH
D. Design of the donor plasmid containing the ribozyme cassette
After finding an effective sgRNA, computationally design the HDR donor plasmid for ribozyme insertion by using the bioinformatics tool Benchling (https://www.benchling.com) (Figure 5). The donor plasmid consists of five parts:
(1) the backbone plasmid containing a counterselection marker [pBS-GMR-eya(shRNA)]
(2) the left homology arm
(3) the right homology arm
(4) the N79 ribozyme cassette
(5) the integration marker 3xP3-DsRed
Critical: Computationally design the donor plasmid sequence first before worrying about how to construct it.
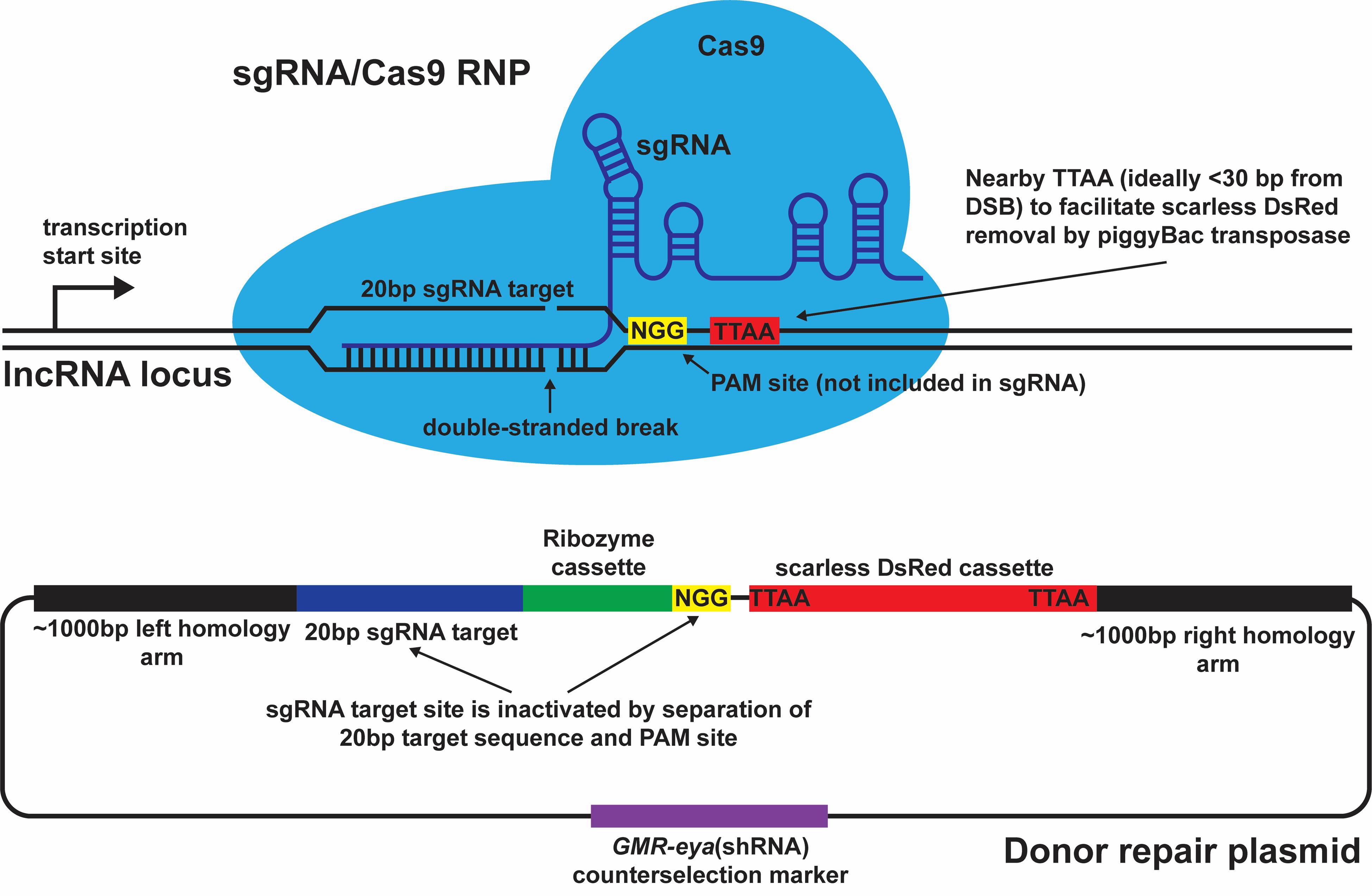
Figure 5. Design of the sgRNA and donor repair plasmid. Important design considerations are depicted at and around the sgRNA target site. The double-strand break (DSB) occurs 3 bp upstream of the NGG PAM site, which is adjacent to the sgRNA target site but not included in the sgRNA itself. The scarless DsRed cassette can be inserted at a nearby TTAA site, ideally within 30 bp of the DSB. The ribozyme cassette itself is inserted between the NGG PAM site and the sgRNA target site within the donor repair plasmid, thus preventing the sgRNA/Cas9 RNP from targeting and cleaving the donor plasmid during injections.
1. In Benchling, create a new sequence file containing the 3,845-bp pBS-GMR-eya(shRNA) plasmid, which will be used as the backbone of the donor plasmid. If integrated into Drosophila, it expresses a short hairpin RNA driven by an eye-specific enhancer that results in RNAi knockdown of the eya gene [27]. Thus, integration of the entire donor plasmid into the Drosophila genome via nonhomologous insertion results in adults with small, rough eya- eyes. However, successful homologous insertion of the ribozyme cassette via HDR results in adults with normal-sized eya+ eyes.
2. To generate homology arms of ~1,000 bp in length, copy the genome sequence that includes the predicted sgRNA target site plus ~1,000 bp on either side of this site. Paste the ~2,000-bp sequence into the EcoRV site of the pBS-GMR-eya(shRNA) sequence in Benchling. Ensure that the ends of both homology arms contain optimal sequences for Gibson assembly (moderate GC content and nonrepetitive sequence within the terminal 30 bp). Homology arm length can be slightly increased or decreased to obtain optimal end sequences.
3. In Benchling, insert the ribozyme cassette sequence, which consists of the N79 ribozyme flanked by flexible linker sequences on both sides. Insert the sequence within or immediately 5' to the PAM of the sgRNA target site. Doing so will disrupt the sgRNA target sequence in the donor plasmid and ensure that only the sgRNA target site in the genome is cleaved in vivo by Cas9. The 111 nt sequence of the ribozyme cassette is: 5'- AAAGAAAGAAACTGAGATGCAGGTACATCCAGCTGATGAGTCCCAAATAGGACGAAACGCGCTTCGGTGCGTCCTGGATTCCACTGCTATCCACAAAAAGAAAAAGAAAAA-3'
Critical: Make sure that the ribozyme cassette is inserted in the same 5' to 3' orientation as the lncRNA transcript that will be knocked down.
4. The 3xP3-DsRed marker gene is used to screen for integration of the ribozyme cassette into the genome. If inserted into the genome, the marker gene expresses DsRed in adult eyes, which is easily detected under a fluorescent stereomicroscope. Once the marker gene has been used to find successful insertions of the N79 cassette, the marker gene is precisely excised from the genome by transposition. The 3xP3-DsRed marker gene is flanked by piggyBac inverted terminal repeat sequences, with TTAA on both ends. Upon excision of the 3xP3-DsRed marker by the piggyBac transposase, the two TTAA sequences are reduced to a single TTAA sequence. Thus, the 3xP3-DsRed marker sequence should be inserted at a naturally occurring TTAA site in the genome. This will ultimately allow for insertion of the ribozyme cassette in the target lncRNA without any other modifications to the genome.
Notes:
1. 3xP3-DsRed fluorescence is easiest to see in white-eyed flies. Thus, use a white-eyed stock for the injections, if possible. In adults with wild-type red eye color, 3xP3-DsRed fluorescence is visible only in a small number of ommatidia in the pseudopupil, as well as the ocelli on the dorsal side of the head, making it difficult, though not impossible, to detect.
2. The 3xP3-DsRed marker can be placed on either side of the sgRNA/ribozyme, as it will ultimately be removed.
Critical: The TTAA site should be located within 30 bp of the double-strand break (DSB) made by the sgRNA to ensure that homologous recombination does not occur between 3xP3-DsRed and the DSB. If a naturally occurring TTAA site is not present nearby, substitute a similar sequence (e.g., TTAT, TAAA, TTCA) with TTAA.
E. Construction and purification of the donor plasmid containing the ribozyme cassette
Construction of the donor plasmid is done using Gibson assembly, using five DNA fragments:
(1) pBS-GMR-eya(shRNA) linearized with EcoRV
(2) a PCR amplicon containing the left homology arm
(3) a PCR amplicon containing the right homology arm
(4) a PCR amplicon containing the 3xP3-DsRed marker
(5) a synthetic double-stranded DNA fragment containing the N79 ribozyme cassette
1. The pBS-GMR-eya(shRNA) plasmid is linearized using EcoRV restriction enzyme cleavage at the plasmid’s multicloning site. Since EcoRV produces blunt-ended fragments, no nucleotides will be removed by the 5'–3' exonuclease activity of the Gibson assembly reaction when assembling the donor plasmid. The rest of the donor plasmid will be assembled directly at the EcoRV cut site.
2. Perform the following restriction digest reaction using pBS-GMR-eya(shRNA) plasmid DNA:
Reaction mix:
5 μg of pBS-GMR-eya(shRNA) DNA
25 μL of 10× rCutSmart Buffer
5 μL of EcoRV-HF (20,000 units/mL)
to 250 μL of nuclease-free water
Reaction incubating conditions: 37 °C for 15 min.
3. Run the entire reaction of digested plasmid on a 1% (w/v) agarose gel, using multiple lanes to accommodate the large volume. Quickly excise the linearized plasmid from the gel with a razor blade, minimizing the exposure of the DNA to long wavelength UV light. Purify the linearized plasmid using the Monarch Spin DNA Gel Extraction kit, eluting in a total of 30 μL of nuclease-free water. Store purified DNA at -20 °C until the Gibson Assembly reaction.
4. Design each of the PCR amplicons and synthetic fragments to share ~30 bp of overlapping sequence with adjacent fragments in the assembly. For the three PCR amplicons, add sequences to the 5' ends of the oligonucleotide primers to generate these 30-bp sequence overlaps. Verify that all oligo primers can bind in the computationally assembled donor plasmid sequence.
Notes:
1. Use the NEBuilder Assembly Tool (https://nebuilder.neb.com/) to aid in the design of overlap regions, using the following settings: Product/kit: NEBuilder HiFi DNA Assembly Master Mix; minimum overlap: 30 nt; circularize: yes; PCR Polymerase/kit: Phusion High-Fidelity DNA Polymerase (HF Buffer); PCR primer conc.: 500 nM; min. primer length: 18 nt.
2. Maximum oligo primer lengths are typically 60 nt when ordering from Integrated DNA Technologies.
Critical: The terminal 17 bp on either side of the 3xP3-DsRed marker gene are identical. Thus, only one of these ends should be used to form a Gibson overlap region. Because the synthetic fragment containing the ribozyme cassette is adjacent to the 3xP3-DsRed marker, we typically extend the synthetic fragment well into the 3xP3-DsRed marker. We have found that the following sequences within the 3xP3-DsRed sequence make good Gibson overlap sequences:
piggyBac left (5') region: 5'-GTCGTTATAGTTCAAAATCAGTGACACTTA-3'
piggyBac right (3') region: 5'-AGATAATCATGCGTAAAATTGACGCATGTG-3'
5. To generate the PCR amplicon containing the 3xP3-DsRed marker, perform a 50 μL touchdown PCR reaction using 30–50 ng of pScarlessHD-DsRed plasmid DNA as template. Otherwise, use the reaction mix and reaction conditions as indicated in step B4, adjusting the extension times accordingly to account for the length of the PCR amplicon. Store completed PCR reactions at 4 °C until gel purification. Because the reaction contains small amounts of plasmid template, PCR amplicons must be purified via gel electrophoresis. See step E7.
6. To generate PCR amplicons containing the homology arms, perform a 50 μL touchdown PCR reaction for each homology arm using as template 50 ng of genomic DNA from the Drosophila stock that will be used for injections. Use the reaction mix and reaction conditions as indicated in step B4, adjusting the extension times accordingly to account for the length of the PCR amplicons. Store completed PCR reactions at 4 °C until amplicon purification. If the PCR reaction generates a single strong band, it can be purified using a standard column purification kit (e.g., Monarch Spin PCR & DNA Cleanup kit). If the reaction produces multiple bands, the amplicon must be purified via gel electrophoresis. See step E7.
7. For PCR reactions that used plasmid DNA as the template or that generated multiple products, run the entire reaction on a 1% (w/v) agarose gel. Quickly excise the desired PCR amplicon from the gel with a razor blade, minimizing the exposure of the DNA to long-wavelength UV light. Purify the DNA using the Monarch Spin DNA Gel Extraction kit, eluting each in 20 μL of nuclease-free water. Store purified DNA at -20 °C until the Gibson Assembly reaction.
8. Design and order the synthetic duplex DNA fragment containing the ribozyme cassette using a commercial service such as Integrated DNA Technology's gBlocks. Since the ribozyme sequence is difficult to PCR amplify on account of its secondary structure, DNA synthesis is required. Resuspend the lyophilized DNA in nuclease-free water at a final concentration of 10 ng/μL. Incubate at 50 °C for 15 min to facilitate resuspension. Store purified DNA at -20 °C until the Gibson Assembly reaction.
9. Determine the concentration of all five purified DNA fragments using either a spectrophotometer (e.g., NanoDrop) or a fluorometer (e.g., Qubit).
10. Calculate the volumes needed for each fragment in the Gibson assembly reaction. When performing a 5-piece Gibson assembly using NEBuilder HiFi DNA Assembly Master Mix, the five fragments should be added in equimolar amounts, with the total DNA content of the reaction not exceeding 0.5 pmol. A quantity of 0.08–0.1 pmol of each fragment works well. The combined volume of all five fragments should be 10 μL or less.
11. Mix all five of the DNA fragments together, adding nuclease-free water to a final volume of 10 μL. Perform a negative control reaction in parallel by adding just the fragments that may contain trace amounts of uncut plasmid (i.e., the linearized backbone plasmid and the scarless 3xP3-DsRed PCR fragment).
12. Mix the 10 μL of DNA with 10 μL of NEBuilder HiFi DNA Assembly Master Mix. Mix well.
13. Incubate at 50 °C for 1 h in a thermocycler with a heated lid.
14. Transform 1 μL of Gibson assembly reaction into a strain of competent E. coli having high transformation efficiency (e.g., electrocompetent DH5α). Plate transformed bacteria on LB plus carbenicillin agar plates.
Note: Transformation efficiencies are typically higher with electrocompetent E. coli than chemically competent E. coli.
15. Pick several individual colonies and grow them overnight in 5 mL cultures of LB broth plus carbenicillin. Purify plasmid samples using a Monarch Spin Plasmid Miniprep kit, eluting in 30 μL of nuclease-free water. Sequence the whole plasmid DNA from each sample using a commercial Oxford Nanopore Sequencing (ONT) service. Ensure that all five fragments are assembled correctly.
Note: Ensure that there are no disabling mutations in coding regions of the homology arms or in the 3xP3-DsRed and ribozyme cassettes. Polymorphisms in noncoding regions of the homology arms are not uncommon and are generally benign.
16. Purify the verified donor plasmid DNA for microinjection using the Qiagen HiSpeed Plasmid Midi kit, following a modified protocol to remove endotoxins using two reagents from the Qiagen EndoFree Plasmid Mega kit (buffer ER and buffer QN).
a. Before beginning, pre-chill buffer P3 at 4 °C and make fresh isopropanol and 70% ethanol solutions.
b. Centrifuge the entirety of a 50 mL overnight LB culture at 6,000 rcf for 15 min at 4 °C.
c. Decant the supernatant and resuspend the pellet by vortexing vigorously in 6 mL of buffer P1 with added RNase A.
d. Add 6 mL of buffer P2 and mix well by inverting 4–6 times. Incubate at room temperature for 5 min.
Critical: Do not vortex after the addition of buffer P2.
e. During incubation, screw the cap onto the outlet nozzle of the QIAfilter cartridge. Place the cartridge into a fresh 50 mL conical tube using the provided tip holder.
f. Add 6 mL of pre-chilled buffer P3 to the lysate and mix well by inverting 4–6 times.
g. Remove the cap, insert the plunger, and filter the solution through the syringe filter into a fresh 50 mL conical tube.
h. Add 1 mL of buffer ER (from the EndoFree Mega kit) to the filtered solution, mix, and incubate on ice for 30 min.
i. Ten minutes before the end of the incubation period, use a blue tip holder to place a HiSpeed tip into a fresh 50 mL conical tube. Equilibrate the tip by adding 4 mL of buffer QBT and allowing it to flow through via gravity.
j. After the 30 min incubation, apply the incubated solution from step E16h to the QBT-equilibrated HiSpeed Tip and allow it to flow through via gravity.
k. Wash the HiSpeed tip with 10 mL of buffer QC, allowing the buffer QC to flow through via gravity.
l. Wash the HiSpeed tip a second time with another 10 mL of buffer QC.
m. Place the HiSpeed tip over a fresh 50 mL conical tube and elute by applying 5 mL of buffer QN (from the EndoFree Mega kit).
n. Add 3.5 mL of isopropanol to the eluted solution. Mix by inverting and incubate at room temperature for 5 min.
o. During incubation, remove the plunger from a 20 mL syringe and attach the QIAprecipitator module onto the outlet nozzle.
p. Place the QIAprecipitator over a fresh 50 mL conical tube. Transfer the eluate mixture into the syringe and insert the plunger. Filter the mixture using constant pressure.
Critical: The QIAprecipitators are fragile and crack easily. Hold the QIAprecipitator firmly in place while pushing the eluate through so that the QIAprecipitator does not detach from the syringe, but avoid pushing the QIAprecipitator back against the syringe, as strong pressure will result in cracks and subsequent loss of sample.
q. Remove the QIAprecipitator from the syringe and pull out the plunger. Reattach the QIAprecipitator and add 2 mL of 70% ethanol to the syringe. Insert the plunger and push the 70% ethanol through.
r. Remove the QIAprecipitator from the syringe and pull out the plunger. Attach the QIAprecipitator again and insert the plunger. Dry the membrane by pressing air through the QIAprecipitator. Repeat this step several times.
s. Dry the outlet nozzle of the QIAprecipitator with a Kimwipe.
t. Remove the plunger from a new 5 mL syringe, attach the QIAprecipitator to the syringe, and hold the outlet over a 1.5 mL microcentrifuge tube. Add 1 mL of buffer TE to the syringe. Insert the plunger and elute the DNA into the collection tube using constant pressure.
u. Remove the QIAprecipitator from the 5 mL syringe and pull out the plunger. Reattach the QIAprecipitator to the syringe.
v. Transfer the eluate from step E16t to the 5 mL syringe and elute for a second time into the same 1.5 mL tube.
17. The final elution should be performed using TE buffer to maximize recovery of plasmid DNA. However, the TE buffer is not suitable for microinjections of Drosophila embryos, and the plasmid needs to be concentrated as well. Perform the following ethanol precipitation to concentrate the donor plasmid and exchange buffers.
a. Estimate the volume of DNA solution and add 1/10 volume of 3 M sodium acetate pH 5.2. Mix well.
b. Add 3 volumes of 100% molecular-grade ethanol.
c. Incubate at -80 °C for 30 min.
d. Centrifuge at 16,000 rcf for 15 min at 4 °C. Remove supernatant.
Note: Split the solution into multiple 1.5 mL microcentrifuge tubes if necessary. Process separate tubes in parallel until the final pellet resuspension.
e. Add 800 μL of cold 70% ethanol to wash each pellet. Vortex briefly and centrifuge at 16,000 rcf for 5 min at 4 °C. Remove the supernatant.
f. Repeat the wash in step E17e.
g. Remove the supernatant and allow pellet to air-dry for 10 min. Ensure that all residual ethanol has been removed.
h. Resuspend each pellet in 15–20 μL of nuclease-free water. Pipette to mix. If DNA from the same donor plasmid prep was split into multiple tubes before step E17d, combine all DNA solutions into a single tube. Purified donor plasmid DNA can be stored at -20 °C.
18. Measure the final concentration of the purified donor plasmid using either a spectrophotometer (e.g., NanoDrop) or a fluorometer (e.g., Qubit). The final concentration should be ~240 nM or higher for the microinjection of Drosophila embryos.
19. Sequence the purified donor plasmid using a commercial Oxford Nanopore Sequencing (ONT) service before injections.
F. Integration of the ribozyme cassette into a target lncRNA gene via Cas9-mediated homology-directed repair
Microinjection of Drosophila embryos to integrate the ribozyme cassette into a target gene proceeds similarly to microinjection of embryos to validate sgRNA efficiency (Section C), with several modifications detailed below.
1. Microinjections for ribozyme integration should be performed in the same Drosophila stock used for testing sgRNA cleavage efficiency. Prepare embryo collection cages as described in Section C.
2. Perform at least two pre-clears of stored embryos (at least 1 h each) on the day of injection, as previously described.
3. While collecting embryos for injection, prepare the RNP mix, also including donor plasmid DNA:
RNP mix:
1.19 μL of Alt-R S.p. Cas9 nuclease V3 (10 μg/μL)
0.38 μL of 2 M KCl
2.36 μg of sgRNA
0.60 pmol of donor plasmid DNA
to 5 μL of nuclease-free water
Incubation conditions: Room temperature for 10 min.
Note: 5 μL of RNP mix is typically sufficient for injecting ~300 embryos. Double the volumes if injecting more embryos.
4. Centrifuge the RNP mix at 16,000 rcf for 10 min. Transfer 4 μL of supernatant into a fresh tube to be loaded into injection needles. Keep at room temperature until needle loading.
5. Load individual needles with the RNP mix.
6. Place embryos for injection on 18 mm × 18 mm cover glasses as described previously, but line up as many embryos as possible across the entire cover glass (typically 80–90 embryos).
7. Cover embryos with a layer of halocarbon oil mix and inject ~100 pL of RNP mix into each pre-cellularized embryo, as before. Collect fresh embryos every 45–60 min and repeat injections. Injecting a total of 300–350 embryos is typically sufficient to produce at least one or more germline HDR events.
8. Remove as much halocarbon oil as possible from the embryos with Kimwipes and a razor blade.
Critical: Take care when using a razor blade to prevent cuts.
9. Gently insert the edge of the cover glass with injected embryos into the food in a standard Drosophila food vial so that the injected embryos are situated just above the food. Keep the vial in a humid chamber at 25 °C overnight. The vials can be removed from the humid chamber after 24 h and placed in a standard 25 °C incubator. The cover glass can remain in the vial.
10. Once the G0 adults eclose, cross them individually to virgin females or males from the same stock used for injections. Crossing to the same line will ensure a consistent genetic background.
11. Screen the G1 adult offspring from each G0 cross for two eye phenotypes: DsRed fluorescence and small rough eyes. G1 adults may have one, both, or neither of these phenotypes. Keep all G1 offspring that have normal-sized DsRed fluorescent eyes (DsRed+ eya+), since these are likely to result from successful integration of the ribozyme cassette via HDR. G1 offspring with small rough DsRed fluorescent eyes (DsRed+ eya-) are likely the result of nonhomologous integration of the entire donor plasmid into the genome. These should be discarded. See Troubleshooting if no DsRed+ G1 individuals are detected.
12. Cross each DsRed+ eya+ G1 adult to an appropriate balancer chromosome stock and establish a balanced line.
13. Once lines are established, verify successful integration of the ribozyme cassette into the targeted gene via PCR of the genome, followed by Sanger sequencing of the PCR products. Because imprecise edits and spurious recombination events are possible, make sure to sequence the entire region, including sequences spanning both homology arms and their flanks. Use primers that anneal outside of the homology arms in combination with internal primers. Use internal primers for sequencing the remainder of the homology arms, the 3xP3-DsRed marker, and the ribozyme/insertion site.
Critical: PCR amplification across the ribozyme cassette is more difficult than standard PCR due to its secondary structure. To facilitate this reaction, make the following modifications to the standard PCR reaction: (1) add DMSO to a final concentration of 3% in the PCR reaction mix, (2) increase the initial denaturation time from 2 to 5 min, and (3) increase the denaturation time within each cycle from 15 s to 30 s. See Troubleshooting if the ribozyme cassette was not successfully integrated into DsRed+ individuals.
G. Scarless excision of the DsRed integration marker using piggyBac transposase
Once successful integration of the ribozyme cassette via HDR has been verified by sequencing, the 3xP3-DsRed transformation marker can be precisely excised from the genome, leaving no sequence “scars” other than insertion of the ribozyme cassette.
1. Cross virgin females from verified N79+ DsRed+ lines to males from the stock containing transgenes α-tubulin-piggyBac transposase and 3xP3-eCFP.
2. Select males in the F1 generation that have adult eyes, both DsRed positive and eCFP positive. The piggyBac transposase is only weakly efficient, so DsRed fluorescence will be visible albeit mosaic in these F1 adults. Cross the selected F1 males to 10–20 virgin females carrying an appropriate balancer chromosome.
3. If the DsRed marker is on an autosome, select F2 flies that have the appropriate balancer chromosome and whose adult eyes are both DsRed-minus and eCFP-minus. Cross single F2 flies to the appropriate balancer chromosome line and establish a balanced stock.
Note: For autosomal insertions, collecting both F2 males or virgin females is acceptable, though for practical purposes, it is easier to screen only males.
4. If the DsRed marker is on the X chromosome, select virgin female F2 flies whose adult eyes are both DsRed-minus and eCFP-minus. Cross single F2 virgin females to males from the appropriate balancer line and establish a balanced stock.
5. Verify that the ribozyme cassette is still present and that excision of the 3xP3-DsRed marker was scarless using genomic PCR and Sanger sequencing across the insertion region, as described previously.
Note: piggyBac excision of the cassette is almost always scarless, though in rare cases, indels may occur.
H. Validation of ribozyme-mediated lncRNA knockdown via RT-qPCR
Reverse transcription quantitative PCR (RT-qPCR) can be used to quantitatively assess the efficiency of knockdown in various regions of the lncRNA transcript.
1. Identify regions of the lncRNA transcript that will be assessed for knockdown efficiency. Generally, 100 bp or more is needed for reliable qPCR amplicons.
Note: Current evidence indicates that RNA knockdown is highly efficient for 3' cleavage fragments but more variable for 5' cleavage fragments. Therefore, we recommend targeting at least one region in the 3' cleavage fragment.
2. Design multiple primer pairs, both from the region(s) of interest within the lncRNA transcript and from several suitable reference genes (e.g., Act5C, eIF2, RpL32), using Primer3Plus (https://www.primer3plus.com) with the following settings:
Product size range: 80–130 bp
Optimal primer size: 20 nt
Optimal Tm: 60 °C
GC content: 30%–80%
GC clamp: 0
Concentration of monovalent cations: 65
Concentration of divalent cations: 2.6
Concentration of dNTPs: 0.35
Annealing oligo concentrations: 2.5 for an abundant RNA, 0.25 for an average RNA, 0.025 for a rare RNA
Note: lncRNAs tend to be expressed at lower levels than mRNAs, especially in bulk RNA samples from whole-body animals.
3. Isolate total RNA from whole-body or isolated tissues of interest from both wild-type and ribozyme-knockdown individuals using TRIzol reagent following the manufacturer's protocol for RNA extraction. Resuspend pellets in nuclease-free water.
4. Measure the concentration of total RNA using a NanoDrop spectrophotometer. Purified RNA should have a 260/280 ratio of ~2.0 and a 260/230 ratio between 2.0 and 2.3.
Pause point: Purified RNA can be safely stored at -80 °C until ready to proceed.
5. Remove any residual genomic DNA from the RNA sample with DNase. Set up the following reaction:
Reaction mix (per μg of RNA):
1 μL of RQ1 DNase 10× reaction buffer
1 μg of total RNA
1 μL of RQ1 DNase (1 unit/μL)
to 10 μL of nuclease-free water
Incubation conditions: 37 °C for 30 min.
Notes:
1. Depending on the nature of the samples, this reaction can be scaled up. DNase treatment of 5–10 μg is recommended for total RNA from whole-body flies or large tissues like gonads.
2. Precipitates often form in the RQ1 DNase 10× reaction buffer. Vortex vigorously and incubate at elevated temperatures (e.g., 70 °C) to resuspend precipitates. Cool on ice before adding to the reaction mix.
6. Immediately purify the DNase-treated RNA using the Monarch Spin RNA Cleanup kit (50 μg), eluting in 20 μL of nuclease-free water.
7. Measure the concentration of the DNase-treated RNA using a NanoDrop spectrophotometer.
Pause point: Purified DNase-treated RNA can be safely stored at -80 °C until ready to proceed.
8. Prepare cDNA from DNase-treated RNA using the following reverse transcription (RT) reaction. Also, perform a negative control reaction in parallel by replacing the reverse transcriptase with water (NoRT).
a. For each RT-qPCR reaction (both RT and NoRT), anneal primers to the RNA, making master mixes if necessary:
Annealing mix:
0.5–5 μg of DNase-treated total RNA
1 μL of 10 mM dNTPs
0.33 μL of 50 μM random 9mer oligos
0.67 μL of 50 μM anchored oligo(dT) primers
to 13 μL of nuclease-free water
Incubation conditions: (1) 65 °C for 5 min; (2) cool to 4 °C.
b. Spin down tubes briefly and place on ice.
c. Make enzyme mixes, adding SuperScript III Reverse Transcriptase for the RT reactions and nuclease-free water for the NoRT reactions. Add 7 μL of either enzyme mix to each annealing mix from above:
Enzyme mix:
4 μL of 5× first-strand buffer
1 μL of 0.1 M DTT
1 μL of RNaseOUT (40 units/μL)
1 μL of SuperScript III Reverse Transcriptase (200 units/μL) OR nuclease-free water
Incubation conditions: (1) 25 °C for 5 min; (2) 50 °C for 60 min; (3) 70 °C for 15 min.
Pause point: cDNA can be safely stored at 4 °C (short-term) or -20 °C (long-term) until ready to proceed.
9. Before performing qPCR, perform test PCR reactions using standard Taq DNA polymerase and analyze by running on a 1% (w/v) agarose gel to verify that candidate qPCR primer pairs produce single, strong amplicons at the concentration of cDNA that will be used for subsequent qPCR reactions. Use similar thermocycling conditions as the qPCR reaction (see below). If candidate qPCR primer pairs fail to give single, strong bands, either increase the concentration of cDNA or design and test new primer pairs.
Note: We often dilute the initial 20 μL of cDNA generated in step H8 1:4 or 1:5 in nuclease-free water for qPCR reactions, though the optimal concentration will depend on the abundance of the target lncRNA in the total RNA.
10. Design a qPCR plate, considering the following:
a. qPCR reactions must be performed on both RT and NoRT control samples.
b. To determine the amplification efficiencies of individual primer pairs, a dilution series must be performed for each RNA of interest (both target lncRNAs and reference RNAs) using cDNA from wild-type samples. At least four dilutions should be used. Serial dilutions of 1:5 (i.e., 1, 1:5, 1:25, 1:125) work well.
c. Each qPCR reaction should be performed in triplicate to estimate technical variance.
d. Multiple biological replicates for each condition (e.g., wild type and ribozyme-knockdown) should also be performed.
e. Ideally, all qPCR reactions should be run on a single plate. If that is not possible, consider reducing the number of technical replicates for NoRT reactions from three to two. If all reactions still cannot be run on a single plate, then use multiple plates, splitting reactions by target RNA (e.g., all lncRNA target reactions on plate 1 and all reference RNA reactions on plate 2).
11. Pipette 3 μL of RT or NoRT sample into each well of an appropriate 96-well reaction plate for qPCR.
12. Set up the following master mixes for each primer pair:
Per reaction:
7.5 μL of 2× TB Green Premix Ex Taq II
0.38 μL of forward primer (10 μM)
0.38 μL of reverse primer (10 μM)
to 12 μL of nuclease-free water
13. Add 12 μL of reaction master mix to the 3 μL of RT or NoRT sample in each well. Pipette each reaction up and down at least 10 times to mix thoroughly.
14. Seal the plate with optical adhesive film, and then briefly spin the plate down.
15. Perform the qPCR reactions on a real-time PCR system using the following thermocycling conditions:
Initial denaturation
(1) 95 °C for 30 s.
40 cycles of amplification
(2) 95 °C for 10 s.
(3) 55 °C for 15 s.
(4) 72 °C for 20 s followed by fluorescence detection
(5) To step 2, 39 times
Melt curve analysis
(6) 95 °C for 1 min.
(7) 55 °C for 5 s.
(8) Ramp to 95 °C at 0.5 °C/s with fluorescence detection
16. The software should provide Cq (quantification cycle) values for each reaction, indicating the cycle number where the fluorescence signal rose above the background. Assess the reliability of the data using these Cq values.
a. Successful amplification of cDNA typically gives Cq values between 15 and 30. If Cq values for wildtype samples are higher than 30, then increase the concentration of cDNA in the reactions.
b. Technical variance among Cq values of triplicates should be low, ideally less than 0.2.
c. Cq values for RT samples should be at least 5 and ideally 10 cycles smaller than Cq values for corresponding NoRT controls.
d. For the dilution series, plot the Cq value (y-axis) vs. the log10 (cDNA dilution factor) (x-axis), which should result in a linear relationship with the R2 of a linear model approaching 1.0.
17. See Data analysis for analysis of RT-qPCR data.
I. Validation of ribozyme-mediated RNA knockdown via single-molecule RNA FISH
Ribozyme-mediated lncRNA knockdown can also be spatially assessed using single-molecule RNA fluorescent in situ hybridization (smFISH) of tissues that normally express the lncRNA (e.g., imaginal discs, ovaries, testes, brains, embryos) [30,31]. The fluorescent probes are oligos arranged to anneal in a tandem array across the RNA, with each oligo conjugated to a single Atto fluor at its 3’ end.
1. Manually design smFISH oligo probe sets of 24–48 oligonucleotides for each target lncRNA using IDT's OligoAnalyzer Tool (https://www.idtdna.com/calc/analyzer). The oligonucleotide sequence must be the reverse complement of the transcribed lncRNA sequence. Use a target type of RNA and Na+ concentration of 660 mM, and design primers with melting temperatures of 68–70 °C. Adjacent oligos should be separated from each other by at least 1 nt and preferably 2 nt. Order the set as standard DNA oligos and resuspend each oligo in nuclease-free water to a final concentration of 100 μM.
Note: The length of each oligo will vary depending on its sequence composition. A larger number of oligos is more likely to result in robust smFISH signals, though we have been able to detect lncRNA smFISH signals using a probe set as small as 19 oligos.
2. Conjugate Atto 633 to amino-11-ddUTP with the following reaction:
Reaction mix:
20 μL of 10 mM amino-11-ddUTP
20 μL of 33.4 mM Atto 633 NHS ester in DMSO
5 μL of 1 M NaHCO3 pH 8.3
5 μL of nuclease-free water
Incubation conditions: Room temperature for 3 h, shielded from light.
Note: Atto 633 is typically the superior fluorophore for smFISH, though Atto 565 also works well should smFISH probes with different fluorophores be desired for dual-smFISH.
3. Add 0.5 μL of 1 M Tris-HCl pH 7.4 to the above reaction to quench. Store the ddUTP-Atto 633 at -20 °C, shielded from light.
4. For each probe set, make an equimolar pool of oligonucleotides from the 100 μM oligo stocks generated in step I1.
5. Conjugate ddUTP-Atto 633 to the 3' ends of oligonucleotides with the following reaction:
Reaction mix:
20 μL of pooled oligonucleotides
1.5 μL of ddUTP-Atto 633
6 μL of 5× TdT buffer
0.6 μL of terminal deoxynucleotidyl transferase (TdT) (20 units/μL)
1.8 μL of nuclease-free water
Incubation conditions: 37 °C overnight (~14–16 h)
6. Add 20 μL of nuclease-free water to the reaction, mixing well.
7. Purify the labeled oligos from free oligos using microspin G-25 columns according to the manufacturer's protocol.
8. Further purify the labeled oligos using HPLC. Add 100 mM TEAA buffer to column-purified labeled oligos to obtain sufficient volume for loading into the HPLC system. Run samples through a C18 column using a gradient from 95% TEAA buffer/5% acetonitrile to 50% TEAA buffer/50% acetonitrile over 30 min. Place the eluate containing the purified oligos in a SpeedVac and evaporate the organic solvent until a gel-like pellet has formed. Take care not to over-dry.
9. Resuspend the pellet to a final concentration of 25 μM in nuclease-free water. The probes are now ready for smFISH.
Note: HPLC purification of labeled probes is not strictly necessary, though higher concentrations of column-purified probes should be used in the subsequent hybridization step, as the background may be higher.
Pause point: Labeled probes can be stored at -20 °C until ready to proceed.
10. Isolate tissues of interest from both wild-type and ribozyme-knockdown animals and fix them in 1 mL of PFA fixative for 20 min at room temperature with gentle rocking.
Note: Specific protocols for tissue preparation and fixation will vary from tissue to tissue and may need to be optimized. The approach detailed here is a generic approach that we have validated for Drosophila ovaries [23].
11. Remove the PFA fixative and wash tissue with 1 mL of PBT for 5 min at room temperature with gentle rocking.
12. Repeat step I11 two more times, for a total of three washes.
13. Remove wash solution and incubate the tissue in 1 mL of methanol for 30 min at room temperature with gentle rocking.
14. While incubating, dilute HPLC-purified probes 1:50 or column-purified probes 1:20 in 50 μL of RNA smFISH hybridization buffer. Preheat probe solution at 62 °C, shielded from light. Also, prepare appropriate and preheated negative control solutions in RNA smFISH hybridization buffer (e.g., no probes or nonspecific probes) if desired.
Note: Optimal probe concentrations may need to be empirically determined.
15. After incubation, remove methanol and add 1 mL of RNA smFISH wash buffer to tissues. Incubate for 5 min at room temperature with gentle rocking.
16. Remove as much wash buffer as possible and add 50 μL of the prewarmed probe or control solution to each sample. Hybridize the probes to the target RNAs for 1 h in a hybridization oven at 62 °C with gentle rocking, shielded from light.
Critical: Labeled samples should be shielded from light from this point forward.
17. While incubating, preheat RNA smFISH wash buffer to 62 °C.
18. Remove probe solution and add 1 mL of preheated RNA smFISH wash buffer to tissues. Incubate for 5 min at 62 °C with gentle rocking.
Note: Diluted probe solutions can be reused up to four times. Store them at -20 °C, shielded from light.
19. Remove wash buffer and add 1 mL of DAPI solution (2.5 μg/mL DAPI in PBT) to tissues. Incubate for 5 min at room temperature with gentle rocking.
20. Remove DAPI solution and add 1 mL of PBT to tissues. Incubate for 5 min at room temperature with gentle rocking.
21. Remove PBT and mount labeled tissues on a microscope slide in Vectashield Plus with a 22 mm × 22 mm No. 1.5H Zeiss cover glass. Seal with clear nail polish.
Pause point: Mounted microscope slides can be stored at 4 °C in the dark for several days until imaging.
22. Image labeled tissues using a confocal microscope tuned to detect DAPI and Atto633 fluorescence in the tissue samples.
Data analysis
Analyze RT-qPCR data according to MIQE (minimum information for publication of quantitative real-time experiments) guidelines as detailed in Taylor et al. (see Figure 5a) [32]. According to the MIQE guidelines, Cq is the preferred term for the quantification cycle, which was traditionally termed cycle threshold (Ct). See Table 1 for symbols and their full meanings.
Table 1. Symbols for RT-qPCR analysis and their full meanings
| Symbol | Full meaning |
|---|---|
| Cq | Quantification cycle |
| E | Amplification efficiency |
| m | Slope of the fitted linear curve |
| ΔCq | Difference in Cq values between each biological replicate and the mean of the control samples for a given gene |
| RQ | Relative quantity of each gene before normalization to the reference gene(s) |
1. Using the dilution series data, determine amplification efficiency for each primer pair by plotting the mean Cq values (y-axis) vs. the log10 (cDNA dilution factor) (x-axis). Use least-squares linear regression to calculate the slope (m) of the fitted curve. Calculate the amplification efficiency E using the following equation:
E = -1 + 10(-1/m)
2. Calculate the mean Cq value among the technical replicates of a sample. Calculate these values for all genes assayed, all biological replicates, and all conditions.
3. For each gene assayed, calculate the mean Cq value among biological replicates for the control condition (i.e., wild type for the lncRNA gene).
4. For each gene assayed, calculate the ΔCq for each biological replicate in both the control and the ribozyme conditions. This is calculated by subtracting the mean Cq among technical replicates (from a single biological replicate) from the mean Cq among biological replicates for the control condition from step 3.
5. Calculate the relative quantity (RQ) using the following equation:
RQ = (1 + E)ΔCq
6. If a single reference gene was used, the normalization factor for each sample is the RQ value for that reference gene. If multiple reference genes were used, calculate the normalization factor for each sample by taking the geometric mean of RQ values for all reference genes.
7. Calculate the normalized expression value of the lncRNA gene for each biological replicate by dividing the RQ of the target gene by the normalization factor.
8. Perform appropriate statistical tests (e.g., ANOVA or t-tests in R) on log2-transformed normalized expression values [33].
Validation of protocol
This protocol (or parts of it) has been used and validated in the following research articles:
Nyberg et al. [27]. A pipeline for precise and efficient genome editing by sgRNA-Cas9 RNPs in Drosophila. Fly (Figures 2–4 and Table 1).
Nyberg et al. [23]. Robust and heritable knockdown of gene expression using a self-cleaving ribozyme in Drosophila. Genetics (Figures 1–8 and Supplemental Figure 1).
General notes and troubleshooting
General notes
1. RNA tends to be less stable than DNA due to the ubiquity of RNases. To minimize RNase-mediated degradation of RNA, (1) spray down bench tops at least daily using 0.5% SDS solution, (2) keep RNA on ice unless the protocol explicitly indicates otherwise, (3) use RNase-free pipette tips, tubes, and solutions (including DEPC water in replacement of double-distilled water), (4) store RNA at -80 °C when not in use, (5) always wear gloves when handling consumables, solutions, and samples.
2. Nucleic acid clean-up kits other than New England Biolab's Monarch Spin kit should also work well; however, ensure that the elution buffer does not contain EDTA, as that may inhibit subsequent enzymatic reactions. Nuclease-free water is always a good choice for an elution buffer.
3. High 230 absorbance values on the NanoDrop spectrophotometer are likely due to contamination from organic solvents like ethanol. Take care to fully dry pellets before resuspending in nuclease-free water. If using a nucleic acid column clean-up kit, we recommend an extra spin after the wash steps to remove any residual wash buffer. In addition, columns can be air-dried with caps open for ~2 min before adding the nuclease-free water for elution.
4. The authors have successfully used the software listed in this protocol for the stated purposes. Should any of these online tools not be available in the future, other tools could be used as alternatives. For example, Benchling has tools for both Gibson assembly and oligonucleotide design, SnapGene (https://www.snapgene.com) can be used for visualization and computational manipulation of DNA, as well as Gibson assembly design, and Eurofins Genomics has tools for both oligonucleotide design (Oligo Analysis Tool, https://eurofinsgenomics.com/en/resources/tools/oligo-analysis-tool/) and qPCR primer pair design (qPCR Primer and Probe Design Tool, https://eurofinsgenomics.eu/en/ecom/tools/qpcr-assay-design/).
Troubleshooting
Problem 1: No DsRed+ G1 individuals were observed after microinjections with sgRNA/Cas9 RNPs and the donor repair plasmid.
Possible cause (A): Lethality was high after injections, reducing the probability of obtaining a successful HDR event.
Solution (A): An injection round of 300–350 G0 embryos should produce 50–100 surviving and fertile G0 adults. If the mortality is significantly higher, inject an additional round of embryos, taking care to handle the embryos gently. Transfer them to cover glasses as quickly as possible and without intense light exposure at the microscope to minimize moisture loss.
Possible cause (B): The sgRNA was not sufficiently efficient.
Solution (B): Standard CRISPR/Cas9 protocols in Drosophila do not typically call for empirical validation of the cleavage efficiency of sgRNAs. While most sgRNAs will work sufficiently well, we have found that a significant minority of tested sgRNAs did not result in any cleavage, which would doom a CRISPR/Cas9 HDR effort from the start. Thus, we strongly recommend empirical validation of sgRNA cleavage efficiency before usage. We have described the T7E1 assay previously, which provides such empirical testing.
Problem 2: DsRed+ G1 individuals were observed after microinjection, but the ribozyme cassette was not integrated into the target genome region.
Possible cause: Homologous recombination occurred between the scarless 3xP3-DsRed cassette and the ribozyme cassette.
Solution: While homology arms of ~1,000 bp in length have been shown to be optimal for HDR, the recombination event itself typically happens in close proximity to the DSB. Thus, the DSB, the scarless 3xP3-DsRed cassette, and the ribozyme cassette should all be ideally within 30 bp of each other.
Acknowledgments
The authors would like to thank the Biological Imaging Facility (RRID:SCR_017767) and the Keck Biophysics Facility (RRID:SCR_012203) at Northwestern University and Dr. Rachael Bakker for assistance in optimizing RNA smFISH protocols. The specific contributions of each author to this work were as follows: conceptualization, K.G.N. and R.W.C.; writing—original draft, K.G.N.; writing—review & editing, R.W.C.; funding acquisition, K.G.N. and R.W.C.; supervision, R.W.C. This work was funded by the National Institutes of Health (R35GM118144 to R.W.C. and F32GM12239 to K.G.N.). The work was also funded by the National Science Foundation (2235451) and Simons Foundation (MP-TMPS-00005320) to R.W.C. The CRISPR/Cas9 methodology in this work was originally described in Nyberg et al., 2020 [27], and the ribozyme knockdown methodology was originally described in Nyberg et al. [23]. Figure 5 was generated using icons provided free for any use from the NIAID NIH BioArt Source (https://bioart.niaid.nih.gov/).
Competing interests
The authors declare no conflicts of interest.
References
- Mattick, J. S., Amaral, P. P., Carninci, P., Carpenter, S., Chang, H. Y., Chen, L. L., Chen, R., Dean, C., Dinger, M. E., Fitzgerald, K. A., et al. (2023). Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol. 24(6): 430–447. https://doi.org/10.1038/s41580-022-00566-8
- Nemeth, K., Bayraktar, R., Ferracin, M. and Calin, G. A. (2023). Non-coding RNAs in disease: from mechanisms to therapeutics. Nat Rev Genet. 25(3): 211–232. https://doi.org/10.1038/s41576-023-00662-1
- Brockdorff, N., Ashworth, A., Kay, G. F., McCabe, V. M., Norris, D. P., Cooper, P. J., Swift, S. and Rastan, S. (1992). The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell. 71(3): 515–526. https://doi.org/10.1016/0092-8674(92)90519-i
- Penny, G. D., Kay, G. F., Sheardown, S. A., Rastan, S. and Brockdorff, N. (1996). Requirement for Xist in X chromosome inactivation. Nature. 379(6561): 131–137. https://doi.org/10.1038/379131a0
- Amrein, H. and Axel, R. (1997). Genes Expressed in Neurons of Adult Male Drosophila. Cell. 88(4): 459–469. https://doi.org/10.1016/s0092-8674(00)81886-3
- Franke, A. and Baker, B. S. (1999). The rox1 and rox2 RNAs Are Essential Components of the Compensasome, which Mediates Dosage Compensation in Drosophila. Mol Cell. 4(1): 117–122. https://doi.org/10.1016/s1097-2765(00)80193-8
- Meller, V. H., Wu, K. H., Roman, G., Kuroda, M. I. and Davis, R. L. (1997). roX1 RNA Paints the X Chromosome of Male Drosophila and Is Regulated by the Dosage Compensation System. Cell. 88(4): 445–457. https://doi.org/10.1016/s0092-8674(00)81885-1
- Gutschner, T., Hämmerle, M., Eißmann, M., Hsu, J., Kim, Y., Hung, G., Revenko, A., Arun, G., Stentrup, M., Groß, M., et al. (2013). The Noncoding RNA MALAT1 Is a Critical Regulator of the Metastasis Phenotype of Lung Cancer Cells. Cancer Res. 73(3): 1180–1189. https://doi.org/10.1158/0008-5472.can-12-2850
- Ji, P., Diederichs, S., Wang, W., Böing, S., Metzger, R., Schneider, P. M., Tidow, N., Brandt, B., Buerger, H., Bulk, E., et al. (2003). MALAT-1, a novel noncoding RNA, and thymosin β4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 22(39): 8031–8041. https://doi.org/10.1038/sj.onc.1206928
- Malakoti, F., Targhazeh, N., Karimzadeh, H., Mohammadi, E., Asadi, M., Asemi, Z. and Alemi, F. (2022). Multiple function of lncRNA MALAT1 in cancer occurrence and progression. Chem Biol Drug Des. 101(5): 1113–1137. https://doi.org/10.1111/cbdd.14006
- Lennox, K. A. and Behlke, M. A. (2016). Mini-review on current strategies to knockdown long non-coding RNAs. Int J Rare Dis Treat Dis. 1(3): 66–70. https://doi.org/10.29245/2572-9411/2016/3.1066.
- Zhao, Y., Teng, H., Yao, F., Yap, S., Sun, Y. and Ma, L. (2020). Challenges and Strategies in Ascribing Functions to Long Noncoding RNAs. Cancers (Basel). 12(6): 1458. https://doi.org/10.3390/cancers12061458
- Perkins, L. A., Holderbaum, L., Tao, R., Hu, Y., Sopko, R., McCall, K., Yang-Zhou, D., Flockhart, I., Binari, R., Shim, H. S., et al. (2015). The Transgenic RNAi Project at Harvard Medical School: Resources and Validation. Genetics. 201(3): 843–852. https://doi.org/10.1534/genetics.115.180208
- Kulkarni, M. M., Booker, M., Silver, S. J., Friedman, A., Hong, P., Perrimon, N. and Mathey-Prevot, B. (2006). Evidence of off-target effects associated with long dsRNAs in Drosophila melanogaster cell-based assays. Nat Methods. 3(10): 833–838. https://doi.org/10.1038/nmeth935
- Ma, Y., Creanga, A., Lum, L. and Beachy, P. A. (2006). Prevalence of off-target effects in Drosophila RNA interference screens. Nature. 443(7109): 359–363. https://doi.org/10.1038/nature05179
- Tuck, A. C. and Bühler, M. (2020). Long Non-coding RNA Depletion Using Self-Cleaving Ribozymes. Methods Mol Biol. 2167: 287–301. https://doi.org/10.1007/978-1-0716-0716-9_16
- Tuck, A. C., Natarajan, K. N., Rice, G. M., Borawski, J., Mohn, F., Rankova, A., Flemr, M., Wenger, A., Nutiu, R., Teichmann, S., et al. (2018). Distinctive features of lincRNA gene expression suggest widespread RNA-independent functions. Life Sci Alliance. 1(4): e201800124. https://doi.org/10.26508/lsa.201800124
- Yen, L., Svendsen, J., Lee, J. S., Gray, J. T., Magnier, M., Baba, T., D'Amato, R. J. and Mulligan, R. C. (2004). Exogenous control of mammalian gene expression through modulation of RNA self-cleavage. Nature. 431(7007): 471–476. https://doi.org/10.1038/nature02844
- Zhong, G., Wang, H., He, W., Li, Y., Mou, H., Tickner, Z. J., Tran, M. H., Ou, T., Yin, Y., Diao, H., et al. (2019). A reversible RNA on-switch that controls gene expression of AAV-delivered therapeutics in vivo. Nat Biotechnol. 38(2): 169–175. https://doi.org/10.1038/s41587-019-0357-y
- Juan, T., Molina, T., Xie, L., Papadopoulou, S., Cardoso, B., Jha, S. G. and Stainier, D. Y. (2025). A recombinase-activated ribozyme to knock down endogenous gene expression in zebrafish. PLos Genet. 21(2): e1011594. https://doi.org/10.1371/journal.pgen.1011594
- Fang, J., Wang, J., Wang, Y., Liu, X., Chen, B. and Zou, W. (2023). Ribo-On and Ribo-Off tools using a self-cleaving ribozyme allow manipulation of endogenous gene expression in C. elegans. Commun Biol. 6(1): 816. https://doi.org/10.1038/s42003-023-05184-4
- Jacobsen, T., Yi, G., Al Asafen, H., Jermusyk, A. A., Beisel, C. L. and Reeves, G. T. (2020). Tunable self-cleaving ribozymes for modulating gene expression in eukaryotic systems. PLoS One. 15(4): e0232046. https://doi.org/10.1371/journal.pone.0232046
- Nyberg, K. G., Navales, F. G., Keles, E., Nguyen, J. Q., Hertz, L. M. and Carthew, R. W. (2024). Robust and heritable knockdown of gene expression using a self-cleaving ribozyme in Drosophila. Genetics. 227(4): e1093/genetics/iyae067. https://doi.org/10.1093/genetics/iyae067
- Win, M. N. and Smolke, C. D. (2007). A modular and extensible RNA-based gene-regulatory platform for engineering cellular function. Proc Natl Acad Sci USA. 104(36): 14283–14288. https://doi.org/10.1073/pnas.0703961104
- Bruckner, J. J., Zhan, H., Gratz, S. J., Rao, M., Ukken, F., Zilberg, G. and O’Connor-Giles, K. M. (2016). Fife organizes synaptic vesicles and calcium channels for high-probability neurotransmitter release. J Cell Biol. 216(1): 231–246. https://doi.org/10.1083/jcb.201601098
- Nyberg, K. G. and Carthew, R. W. (2022). CRISPR-/Cas9-Mediated Precise and Efficient Genome Editing in Drosophila. Methods Mol Biol. 2540: 135–156. https://doi.org/10.1007/978-1-0716-2541-5_6
- Nyberg, K. G., Nguyen, J. Q., Kwon, Y. J., Blythe, S., Beitel, G. J. and Carthew, R. (2020). A pipeline for precise and efficient genome editing by sgRNA-Cas9 RNPs in Drosophila. Fly (Austin). 14: 34–48. https://doi.org/10.1080/19336934.2020.1832416
- Bier, E., Harrison, M. M., O’Connor-Giles, K. M. and Wildonger, J. (2018). Advances in Engineering the Fly Genome with the CRISPR-Cas System. Genetics. 208(1): 1–18. https://doi.org/10.1534/genetics.117.1113
- Lorenz, R., Bernhart, S. H., Honer Zu Siederdissen, C., Tafer, H., Flamm, C., Stadler, P. F. and Hofacker, I. L. (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. https://doi.org/10.1186/1748-7188-6-26.
- Bakker, R., Mani, M. and Carthew, R. W. (2020). The Wg and Dpp morphogens regulate gene expression by modulating the frequency of transcriptional bursts. eLife. 9: e56076. https://doi.org/10.7554/elife.56076
- Little, S. C. and Gregor, T. (2018). Single mRNA Molecule Detection in Drosophila. Methods Mol Biol. 1649: 127–142. https://doi.org/10.1007/978-1-4939-7213-5_8
- Taylor, S. C., Nadeau, K., Abbasi, M., Lachance, C., Nguyen, M. and Fenrich, J. (2019). The Ultimate qPCR Experiment: Producing Publication Quality, Reproducible Data the First Time. Trends Biotechnol. 37(7): 761–774. https://doi.org/10.1016/j.tibtech.2018.12.002
- R Core Team (2025). R: A language and environment for statistical computing.
Article Information
Publication history
Received: Jul 22, 2025
Accepted: Sep 9, 2025
Available online: Sep 22, 2025
Published: Oct 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Nyberg, K. G. and Carthew, R. W. (2025). Ribozyme-Mediated Knockdown of lncRNA Gene Expression in Drosophila. Bio-protocol 15(20): e5477. DOI: 10.21769/BioProtoc.5477.
Category
Molecular Biology > RNA > RNA interference
Developmental Biology > Genome editing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.