- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Detection of Plant RNA–Protein Interactions Using GFP-tag for Immunoprecipitation

Published: Vol 15, Iss 19, Oct 5, 2025 DOI: 10.21769/BioProtoc.5466 Views: 1670
Reviewed by: Noelia ForesiDevendra Pratap SinghHarsimranjit Sekhon
Original research article
The authors used this protocol in:

Jun 2020

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The study of RNA metabolism involves understanding how RNA molecules interact with specific RNA-binding proteins (RBPs). In plants, these interactions have traditionally been investigated using a variety of in vivo and in vitro approaches, such as electrophoretic mobility shift assays or the analysis of knockout mutants. More recently, immunoprecipitation-based techniques have been developed. Most of the available protocols rely on crosslinking procedures, magnetic beads, and RNA-seq as the final endpoint analysis. Here, we present a protocol developed to identify specific RNA targets that directly interact with known plant RBPs using GFP-Trap® agarose (ChromoTek) for immunoprecipitation without the need for crosslinking or RNA-seq. Briefly, a GFP-tagged RNA-binding protein is expressed in plant tissue, protein extracts are incubated with the GFP-Trap® agarose matrix, and the resulting complexes are isolated. Co-purified RNAs, specifically mRNAs, are then analyzed by RT-PCR to detect bound transcripts. This protocol was first implemented for the study of RNA–protein interaction in Arabidopsis thaliana. This approach presents high potential for analysis in other plant species as well as several advantages, such as its high specificity and low cost. Even though GFP-Trap® magnetic agarose (ChromoTek) has been used in plant systems to detect RNA–protein interactions, the protocol presented here consists of an alternative that is straightforward to implement when both candidate RNAs and RNA-binding proteins are known, and it can be broadly applied to study RNA–protein interactions in other plant systems.
Key features
• Can be used to confirm predicted RNA–protein interactions.
• Suitable for validating RNA–protein interactions when candidate transcripts and RBPs are already known.
• Compatible with downstream analysis by RT-PCR; can be adapted to RNA-seq if high-throughput data is needed.
• Does not require crosslinking or specialized equipment beyond standard molecular biology tools for direct and strong RNA–protein interactions.
Keywords: Plant RNA binding proteinsGraphical overview
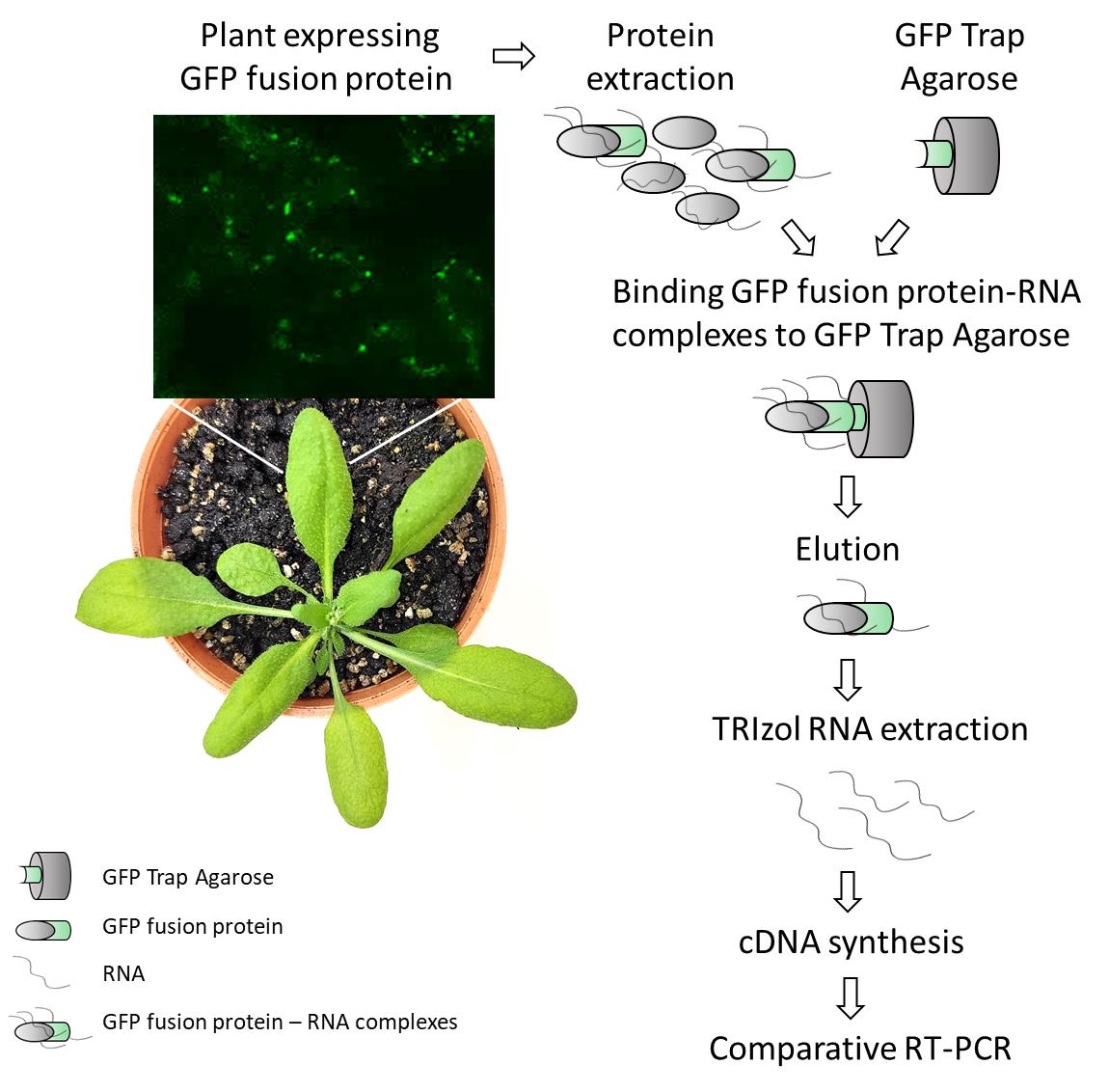
Detection of plant RNA–protein interactions using GFP-tag for immunoprecipitation from Arabidopsis thaliana leaf
Background
The field of protein–RNA interactions gained significant attention due to the complexity of RNA metabolism and the essential roles these interactions carry out to control RNA stability, localization, and translation. Different strategies have been employed to decipher such interactions, including a broad range of in vivo, in vitro, and in silico approaches [1,2]. In plants, initial attempts to demonstrate these interactions were carried out using methods such as nucleic acid-binding assays or mutant and knockout screening (cited in Burjoski et al. [3]). More recently, co-immunoprecipitation-based techniques have started to be developed. Even though these approaches have been developed primarily for mammalian systems, their application in plants is becoming increasingly common and useful (summarized in Burjoski et al. [3]; Mateos and Staiger [4]).
RNA immunoprecipitation (RIP) followed by RNA-seq (RIP-seq) is one of the most widely used techniques to study RNA–protein interactions in plants. A recent example is a report published in Bio-protocol, which demonstrated the identification of small RNA targets of GFP-tagged proteins by Small RNA-seq using GFP-Trap® magnetic agarose (ChromoTek) [5]. Here, we present a protocol for use when candidate transcripts and RNA-binding proteins (RBPs) are already known. Using the original protocol of GFP-Trap® agarose (developed for mammalian applications), we efficiently performed co-immunoprecipitation of RNA–protein complexes from Arabidopsis thaliana leaf. After elution, RNAs are extracted and reverse-transcribed for RT-PCR detection of specific mRNA targets. In addition to its high specificity and simplicity, the method described in this protocol offers other advantages, such as low cost, versatility, and quick steps, allowing a complete procedure within a few days, resulting in the identification of candidate RNA targets of plant-specific GFP-tagged RBPs. Although this protocol is primarily designed for the detection of known candidate RNAs, the in planta RBP co-purified RNA can also be subjected to RNA-seq for transcriptome-wide analysis. This makes it a valuable tool for targeted studies that can later be expanded to broader investigations, not only for leaf tissue but also for other plant organs.
Materials and reagents
Biological materials
1. Arabidopsis leaves from GFP-tagged protein of interest–expressing lines (protein of interest-GFP) and GFP-expressing lines (control line, see Note 2).
Notes:
1. In this protocol, leaves from 20-day-old Arabidopsis plants overexpressing GFP-tagged proteins were used. However, the protocol can be adapted for the analysis of RNA-protein interaction in seedlings and roots, as well as other plant species.
2. A control of a GFP protein is needed in order to discard nonspecific targets that can be bounded to GFP itself after co-IP. A useful control could be the overexpression of the GFP alone (for example, 35S::GFP, ideally with the same vector you selected for the overexpression of your protein of interest). An alternative would be overexpressing a GFP-tagged protein not belonging to the protein family of your protein of interest. In this protocol, ATG8a-GFP overexpressing lines were used as a negative control.
3. To be considered, the saturation of the GFP-Trap® agarose according to manufacturer recommendations corresponds to 12 µg of recombinant GFP protein per 10 µL of GFP-Trap® agarose.
Reagents
1. GFP-Trap® agarose (ChromoTek, catalog number: gta-20)
2. Tri (hydroxymethyl) aminomethane (Tris) (Genbiotech, catalog number: RU2510)
3. Ethylenediamine tetraacetic acid disodium salt (EDTA) (Biopack, catalog number: 1604.07)
4. Sodium chloride (NaCl) (J.T. Baker®, catalog number: 3624-19)
5. Sodium dodecyl sulfate (SDS) (Anedra, catalog number: 7211)
6. Triton X-100 (Biopack, catalog number: 2002.08)
7. Deoxycholate (Fluka, catalog number: 30970)
8. Magnesium chloride (MgCl2) (Promega, catalog number: A351H)
9. Phenylmethylsulphonyl fluoride (PMSF) (ICN Biochemicls Inc., catalog number: 195381)
10. Natrium azide (Na-Azide) (Merk, catalog number: S28222 917)
11. Glycine (Merck, catalog number: 4201)
12. Diethyl pyrocarbonate (DEPC) (Sigma, catalog number: 016K3726)
13. (Iso) propyl alcohol (anhydrous) (Mallinckrodt, catalog number: 3032-08)
14. Ethanol (Baker)
15. Chloroform (Biopack, catalog number: 1651.08)
16. Boric acid (Cicarelli, catalog number: 771214)
17. Agarose (Gene Biotech LE-Agarose 1200, catalog number: RU1010)
18. Ethidium bromide (Biobasic, catalog number: DB0195)
19. Hydrochloric acid (HCl) 36.5%–38% (Cicarelli, catalog number: 918110)
20. Liquid N2
21. M-MLV kit retrotranscriptase (Inbio, catalog number: K1601)
22. dNTPs set (Promega, catalog number: U1330)
23. Random primers (Macrogen Inc.)
24. Polymerase Taq-Pegasus kit (PB-L, catalog number: EA0102)
25. Specific primers designed to analyze putative targets (see Table 1)
Table 1. Primer list
| Primer name | Sequence | Citation | |
| atp8 | Fw | ACACTGACGACATGGTTCTACATTTAGCACTGGTGTATCCTATATGT | Takenaka et al. [6] |
| Rv | TACGGTAGCAGAGACTTGGTCTTGCTTCCTTGGCCATGTACA | Takenaka et al. [6] | |
| nad2int3ex4 | Fw | GGCGAATTTCAAACTTGTGG | Koprivova et al. [7] |
| Rv | TACGGTAGCAGAGACTTGGTCTTCCTTCTATGGTTCCACATGAGA | Takenaka et al. [6] | |
| nad2ex4 | Fw | ACACTGACGACATGGTTCTACACTTTCTATTGCGCCTAAAATCTCT | Takenaka et al. [6] |
| Rv | TACGGTAGCAGAGACTTGGTCTTCCTTCTATGGTTCCACATGAGA | Takenaka et al. [6] | |
26. 100 base pairs molecular marker (PB-L, catalog number: MA02)
27. TRIzol (Ambion by life technologies, catalog number: 15596018)
Solutions
1. Tris-HCl, pH 7.5, 1 M (see Recipes)
2. EDTA, pH 8, 0.5 M (see Recipes)
3. SDS 10% (see Recipes)
4. RIP A buffer (ChromoTek) (see Recipes)
5. Washing buffer (ChromoTek) (see Recipes)
6. Elution buffer (ChromoTek) (see Recipes)
7. Neutralization buffer (ChromoTek) (see Recipes)
8. DEPC water (see Recipes)
9. TBE buffer 5× (see Recipes)
Recipes
1. Tris-HCl, pH 7.5, 1 M
Weigh 12.11 g of Tris, add 75 mL of sterile water, adjust to pH 7.5 with HCl, and fill with sterile water to a final volume of 100 mL.
2. EDTA, pH 8, 0.5 M
Weigh 18.6 g of EDTA, add 75 mL of sterile water, adjust to pH 8 using 10 M NaOH, and stir until EDTA is completely dissolved. Solubility improves while alkalinity increases. Complete with sterile water to a 100 mL final volume.
3. SDS 10%
Dissolve 10 g of SDS in 100 mL of sterile water.
4. RIP A buffer (ChromoTek, adapted recipe)
10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.1% SDS, 1% TritonTM X-100, 1% deoxycholate, 0.09% Na-Azide, 2.5 mM MgCl2, 1 mM PMSF (adjust the pH to 7.5 at 4 °C).
5. Washing buffer (ChromoTek, adapted recipe)
10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA (adjust the pH to 7.5 at 4 °C), 0.018% Na-Azide, 1 mM PMSF.
6. Elution buffer
200 mM glycine pH 2.5 (adjust the pH at 4 °C).
7. Neutralization buffer
1 M Tris pH 10.4 (adjust the pH at 4 °C).
8. DEPC water
1 mL DEPC/L water; stir overnight and autoclave.
9. TBE buffer 5×
Dissolve 54 g of Tris and 27.5 g of boric acid in 500 mL of sterile water. Add 20 mL of EDTA pH 8, 0.5 M (Recipe 2) and complete the final volume up to 1 L of stock solution. For electrophoresis and gels, make a 10× dilution to obtain a 0.5× TBE buffer.
Notes:
1. Sterile water is obtained by autoclaving 40–70 µS/cm water. DEPC water is used for RNA extraction, cDNA synthesis, and RT-PCR steps.
2. We followed the manufacturer’s recommendations to adjust the pH of solutions at 4 °C. The procedure presented in this protocol is mainly performed at cold temperatures or on ice to prevent RNA or protein degradation, so the pH of solutions must be adjusted at 4 °C as the working temperature for the protocol.
Laboratory supplies
1. Mortar
2. Spatula
3. Centrifuge tubes 1.5 mL (Henso, conical bottom, sterile)
4. PCR tubes 0.2 mL (Henso, DNAse and RNAse free)
Equipment
1. Table centrifuge (Eppendorf, model: centrifuge 5418)
2. PCR machine (Applied BiosystemTM, Veriti 96 wells, 4375786)
3. Agarose gel electrophoresis equipment (Bio-Rad, model: PowerPacTM Basic Power Supply, Horizontal Electrophoresis Systems)
Procedure
Before starting the protocol, ensure you have a plant line that overexpresses your gene of interest tagged with GFP, as well as an appropriate control line expressing GFP alone (ideally) or a tagged gene belonging to another protein family than the gene of interest. In the case of Marchetti et al. [8], we used a transgenic line expressing AtATG8a fused to GFP as a control [9], which was available in our lab at that time.
Part I: GFP fusion protein purification
A. Plant protein extraction
1. Weigh 200 mg of leaf tissue from plants expressing the line of interest and the GFP control line. When collecting all the samples and starting the procedure, keep the tissue frozen in liquid N2.
2. Mortars must be cold to prevent unfreezing of the tissue while making powder (next step). Cold mortar is prepared by carefully pouring liquid N2 on it until placing the plant tissue to grind.
3. Place plant tissue on cold mortar (each sample in the corresponding mortar). Carefully add N2 to make the leaves float on N2 and be completely frozen. With the mortar, grind the tissue until the plant tissue is transformed into powder.
4. With a spatula, transfer the powder to a 1.5 mL centrifuge tube and place the samples on ice.
5. Add 300 µL of cold RIP A buffer and mix by manually hitting the bottom of the centrifuge tube until you see that powder and buffer are mixed. Mixing can also be performed by pipetting; however, this may lead to a loss of plant material retained in the tip.
6. Incubate for 30 min on ice, mixing by pipetting every 10 min (pipetting is recommended in this step to ensure proper mixing).
7. Centrifuge samples at 20,000× g for 10 min at 4 °C.
8. Keep supernatant by pipetting and add 200 µL of washing buffer to dilute the sample as per the manufacturer’s recommendations for co-IP steps. Samples can be stored at -80 °C.
B. Co-IP GFP-protein from the GFP-Trap® agarose
While working with protein and RNA samples, as well as molecular interactions, maintain samples on ice and use cold buffers to prevent RNA or protein degradation. For co-IP, we followed the manufacturer’s recommendations as described below:
B1. Equilibrate GFP-Trap® agarose
1. Vortex (gently, 1,200 rpm) GFP-Trap® agarose.
2. For each sample (200 mg plant tissue), use 25 µL of GFP-Trap® agarose. The volume of GFP-Trap® agarose can be adjusted for higher quantities of plant tissue if higher volumes of cDNA than those described in this protocol are needed.
3. Mix the GFP-Trap® agarose with 500 µL of cold washing buffer to equilibrate the GFP-Trap® agarose from its storage solution.
4. Centrifuge at 2,500× g for 2 min at 4 °C to pellet the GFP-Trap® agarose.
5. Repeat steps B1.3–4 one time. Remove the supernatant by pipetting and keep the pellet (equilibrated GFP-Trap® agarose).
B2. Binding GFP-protein to the GFP-Trap® agarose
1. Add the sample obtained in step A8 (whole volume, containing GFP-protein of interest) to the tube of step B1.5 (containing equilibrated GFP-Trap® agarose). Mix gently by pipetting.
2. Mix gently for 1 h, preferably placing samples in a laboratory rocker, at 4 °C.
3. Centrifuge at 2,500× g for 2 min at 4 °C.
4. Remove the supernatant and keep the pellet of GFP-Trap® agarose containing GFP-proteins. Keep on ice.
B3. Washing
1. Add 500 µL of cold washing buffer to the tube prepared in step B2.4 containing GFP-Trap® agarose and GFP-proteins. You can mix manually by gently hitting the bottom of the centrifuge tube. No incubation is needed.
2. Centrifuge at 2,500× g for 2 min at 4 °C.
3. Repeat steps B3.1–2 at least 3 times. In case there is green coloration still visible at the bottom of the tube, the number of washings can be increased up to 6 times (that was the maximum amount of washing tested by us).
4. GFP-Trap® agarose is visible at the bottom of the centrifuge tube, which makes it easier to distinguish it from the washing buffer. Remove all the supernatant by pipetting and keep the pelleted GFP-Trap® agarose containing GFP-proteins on ice.
B4. Elution
Immediately after the washing steps, proceed with elution as described below:
1. Add 50 µL of cold elution buffer.
2. Mix by pipetting for no longer than 30 s.
3. Centrifuge at 2,500× g for 2 min at 4 °C.
4. Transfer the supernatant to a new centrifuge tube by pipetting carefully. Avoid pipetting the visible GFP-Trap® agarose. Keep on ice.
5. Add 5 µL of neutralization buffer to the supernatant you recovered in the new centrifuge tube.
6. With the tube containing the remainder GFP-Trap® agarose after elution, apply steps B4.1–4. This helps to increase elution efficiency.
7. Pipette the second elution into the tube containing the first elution. Add 5 µL more of neutralization buffer.
8. Add 500 µL of TRIzol to the elution volume to prevent RNA from degradation (500 µL of TRIzol was used for the total elution volume of 100 µL, approximately)
Note: In the original protocol provided by the manufacturer, the elution step involves resuspension of the GFP-Trap® Agarose directly in 2× SDS sample buffer, as the GFP-tagged protein remains bound and can be loaded onto an SDS-PAGE gel directly because the main focus is the purification of GFP-tagged protein. Since RNA-protein interactions are the aim of the protocol presented here, different steps in the elution procedure were applied to recover RNA.
Part II: RNA extraction
RNA extraction steps were performed following the manufacturer’s recommendations (https://assets.thermofisher.com/TFS-Assets/LSG/manuals/trizol_reagent.pdf) and are described below.
1. Incubate the tube obtained in step B4.8 for 5 min at room temperature (15–30 °C, simply take tubes from ice).
2. Add 200 µL of chloroform and vortex for 15 s.
3. Incubate for 3 min at room temperature.
4. Centrifuge at 11,200× g for 15 min at 4 °C.
5. Transfer aqueous phase to a new tube by pipetting.
6. Add 0.7 volumes of (Iso) propyl alcohol.
7. Incubate for 10 min at room temperature.
8. Centrifuge at 11,200× g for 10 min at 4 °C.
9. Discard the supernatant by inverting the tube. Keep the pellet.
10. Add 1 mL of ethanol 75%. Shake the tube manually.
11. Centrifuge at 7,500× g for 5 min at 4 °C.
12. Discard the supernatant by inverting the tube. Keep the pellet containing RNA.
13. Dry the pellet (optional: keep the tube inverted or incubate at 37 °C to accelerate drying until no liquid remains in the tube).
14. Resuspend pellet in 15 μL of DEPC water (optional: heating the sample at 55 °C improves resuspension).
Note: RNA was resuspended in 15 µL of DEPC water as the final volume. The amount of RNA used for the following step corresponds to the whole volume of the 15 µL used to resuspend RNA in the RNA extraction step. As a comparative parameter for the protocol presented here, the same amount of plant tissue between samples was used, as well as the same amount of GFP-Trap® agarose, which represents the amount of protein for co-IP.
Part III: cDNA synthesis and RT-PCR
cDNA synthesis was performed using the M-MLV kit, following the manufacturer’s protocol (https://drive.google.com/file/d/1fH513GRiC522UjIxh26BfWycPdOgYKYK/view) to obtain 20 µL of cDNA from RNAs that interact with the GFP-fusion protein isolated by co-immunoprecipitation using the GFP-Trap® agarose. As described above, before starting this protocol, you need to know candidate RNA targets that interact with your protein of interest, as well as a negative control to include in your analysis. In our case, we identified the putative RNA candidates by a comparative transcriptome analysis between the wild type and plants lacking the protein of interest [8]. For example, we analyzed the relative expression of the mRNA affected in the mutant line that lacks the protein of interest. In addition, we conducted an in vitro analysis to detect RNA interactions of the protein of interest expressed in a heterologous system when incubated with total plant RNA, followed by co-IP with MBP tag and RT-PCR analysis to identify specific RNA targets. In comparison with the three experiments, we could approach putative RNA targets of the protein of interest. Once you select the specific and nonspecific primers, as negative controls that support the specificity of the RNA–protein interaction as well as the correct isolation of your co-IP complexes, RT-PCR is carried out. We used Taq Pegasus polymerase reaction as follows: 0.25 µL of primer forward (10 µM), 0.25 µL of primer reverse (10 µM), 1 µL of dNTPs (2 mM), 2 µL of Buffer Tag Pegasus 5×, 0.15 µL of Taq Pegasus polymerase, and 1 µL of cDNA to a final volume of 10 µL. For each RT-PCR reaction (primers listed in Table 1), 1 µL of pure cDNA was used as template for a 40-cycle PCR reaction. The whole final volume of the RT-PCR reaction was stained with ethidium bromide 2 mg/mL and loaded and run onto a 1% agarose gel, using 0.5× TBE buffer (see Recipes), both for gel and running. Finally, this was run at 90 V for 30 min. Results were comparatively analyzed considering negative and positive PCR controls (Figure 1).
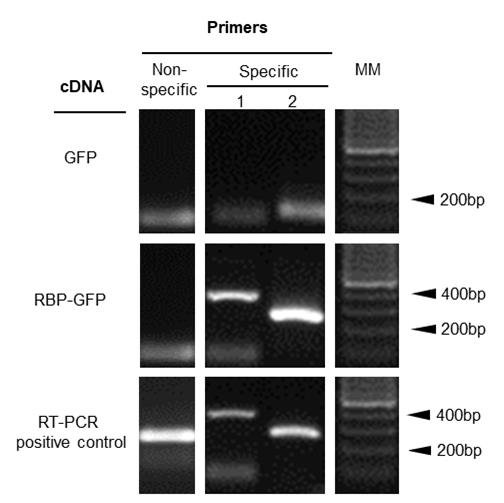
Figure 1. Identification of plant mRNA targets bound to the GFP tag protein. RT-PCR amplification of candidate mRNAs interacting with the plant RBP-GFP fusion protein. Total RNA was extracted from RNA–protein complexes that were co-immunoprecipitated using GFP-Trap® agarose. cDNA was synthesized from the purified RNA using TRIzol reagent and used as a template for RT-PCR to detect specific mRNA targets. Specific primer 1 corresponds to nad2, a mitochondrial transcript putative target of the protein of interest [8]. Specific primer 2 corresponds to another region of the mRNA nad2 transcript. Non-specific primer corresponds to atp8, another mitochondrial transcript not bound to PPR. Primer sequences are listed in Table 1. Note that the smooth band that is observed under 100 pb corresponds to primer dimmers. MM: 100 base pairs (bp) molecular marker.
Data analysis
This protocol can be considered as a slanted analysis of RNA–protein interaction because knowing putative RNA targets that interact with your protein of interest is needed to design the selection of the primers you are interested in. To obtain a positive result, you need to follow three simple steps: check that your primers work well using cDNA from leaves (RT-PCR positive control), that the reaction is negative when amplifying from cDNA corresponding to RNA co-IP coming from your control lines, and that they are positive only in samples that correspond to cDNA from the GFP-tagged protein of interest.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
• Marchetti et al. [8]. Mitochondrial Pentatricopeptide Repeat Protein, EMB2794, Plays a Pivotal Role in NADH Dehydrogenase Subunit nad2 mRNA Maturation in Arabidopsis thaliana. Plant Cell Physiol. 61(6): 1080–1094. https://doi.org/10.1093/pcp/pcaa028
General notes and troubleshooting
General notes
This protocol was originally developed to identify RNA–protein interactions in plant tissue by RT-PCR analysis of RNA-GFP fusion protein complexes purified using GFP-Trap® agarose. Since its development, the manufacturer has released updated buffer recipes tailored specifically for plant tissue to optimize fusion protein purification. However, the protocol presented here consists of experimental evidence of the manufacturer’s earlier recommendations, modified to efficiently purify RNA from RNA-GFP fusion protein complexes as well as to facilitate the identification of associated mRNAs previously known as putative targets. Although there are differences between the immunoprecipitation (IP) buffer used in the published Bio-Protocol employing GFP-Trap® magnetic agarose by Fan et al. [5] and the RIP-A buffer presented here, both protocols have proven effectiveness in extracting RNA and yielding high-quality samples suitable for RNA analysis. These findings support the flexibility of GFP-Trap®-based approaches across different buffer conditions and experimental contexts. This method holds broad potential for simplifying buffer compositions that were initially designed for mammalian cell extractions and for expanding the downstream research applications of the purified RNAs.
Regarding buffer compositions, although the original buffer included sodium azide, which is commonly used for sample preservation and antiseptic purposes, it can be omitted when preparing fresh buffers and when working with plant tissue. The same applies to deoxycholate, which is generally used to enhance cell lysis. This adaptation reflects a shift toward plant-specific lysis conditions, as seen in recent GFP-Trap® buffer recommendations. However, to our knowledge, those newer protocols have not yet been validated for RNA-based applications, which would be worthwhile to test in the future. The protocol presented here successfully identifies strong RNA–protein interactions in Arabidopsis thaliana using GFP-tagged proteins. Due to the extraction buffer composition and the usage of DEPC water, together with the in vivo extraction of RNA–protein interactions, no extra components or requirements were applied to prevent RNA degradation. Current careful manipulation consists of avoiding possible RNase contamination in the samples and maintaining cold temperatures during the procedure. In our hands, this worked well to detect strong interactions. However, we do not discard the possibility that this protocol could be modified from this original version and improved in the future. For the analysis, a positive RT-PCR result indicates that the corresponding RNA directly interacts with the protein of interest. Conversely, a negative RT-PCR result does not necessarily rule out indirect or low-affinity RNA–protein interactions, which may require more sensitive or crosslinking-based approaches for detection, for an in-depth analysis. In summary, building on the previous evidence, our protocol could be considered as a powerful tool for analyzing post-transcriptional regulatory networks in plants.
Acknowledgments
Acknowledgments to CONICET and ANPCyT for funding, as well as to the Institute IIB-CONICET-UNMDP, together with Mar del Plata National University. This protocol was used in Marchetti et al. [4].
Competing interests
There are no conflicts of interest or competing interest.
References
- Ramanathan, M., Porter, D. F. and Khavari, P. A. (2019). Methods to study RNA–protein interactions. Nat Methods. 16(3): 225–234. https://doi.org/10.1038/s41592-019-0330-1
- Shen, W. J., Cui, W., Chen, D., Zhang, J. and Xu, J. (2018). RPiRLS: Quantitative Predictions of RNA Interacting with Any Protein of Known Sequence. Molecules. 23(3): 540. https://doi.org/10.3390/molecules23030540
- Burjoski, V. and Reddy, A. S. N. (2021). The Landscape of RNA-Protein Interactions in Plants: Approaches and Current Status. Int J Mol Sci. 22(6): 2845. https://doi.org/10.3390/ijms22062845
- Mateos, J. L. and Staiger, D. (2022). Toward a systems view on RNA-binding proteins and associated RNAs in plants: Guilt by association. Plant Cell. 35(6): 1708–1726. https://doi.org/10.1093/plcell/koac345
- Fan, L., Gao, B. and Chen, X. (2022). Profiling of Single-cell-type-specific MicroRNAs in Arabidopsis Roots by Immunoprecipitation of Root Cell-layer-specific GFP-AGO1. Bio Protoc. 12(24): e4575. https://doi.org/10.21769/bioprotoc.4575
- Takenaka, M., Zehrmann, A., Brennicke, A. and Graichen, K. (2013). Improved Computational Target Site Prediction for Pentatricopeptide Repeat RNA Editing Factors. PLoS One. 8(6): e65343. https://doi.org/10.1371/journal.pone.0065343
- Koprivova, A., des Francs-Small, C. C., Calder, G., Mugford, S. T., Tanz, S., Lee, B. R., Zechmann, B., Small, I. and Kopriva, S. (2010). Identification of a Pentatricopeptide Repeat Protein Implicated in Splicing of Intron 1 of Mitochondrial nad7 Transcripts. J Biol Chem. 285(42): 32192–32199. https://doi.org/10.1074/jbc.m110.147603
- Marchetti, F., Cainzos, M., Shevtsov, S., Córdoba, J. P., Sultan, L. D., Brennicke, A., Takenaka, M., Pagnussat, G., Ostersetzer-Biran, O., Zabaleta, E., et al. (2020). Mitochondrial Pentatricopeptide Repeat Protein, EMB2794, Plays a Pivotal Role in NADH Dehydrogenase Subunit nad2 mRNA Maturation in Arabidopsis thaliana. Plant Cell Physiol. 61(6): 1080–1094. https://doi.org/10.1093/pcp/pcaa028
- Robert, G., Yagyu, M., Koizumi, T., Naya, L., Masclaux‐Daubresse, C. and Yoshimoto, K. (2020). Ammonium stress increases microautophagic activity while impairing macroautophagic flux in Arabidopsis roots. Plant J. 105(4): 1083–1097. https://doi.org/10.1111/tpj.15091
Article Information
Publication history
Received: Jul 11, 2025
Accepted: Sep 2, 2025
Available online: Sep 16, 2025
Published: Oct 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Marchetti, F., Distéfano, A., Pagnussat, G. C. and Zabaleta, E. J. (2025). Detection of Plant RNA–Protein Interactions Using GFP-tag for Immunoprecipitation. Bio-protocol 15(19): e5466. DOI: 10.21769/BioProtoc.5466.
Category
Plant Science > Plant molecular biology
Biochemistry > RNA > RNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.