- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fractionation and Extraction of Cell Wall Proteins From Candida albicans
(§Deceased) Published: Vol 15, Iss 18, Sep 20, 2025 DOI: 10.21769/BioProtoc.5456 Views: 2840
Reviewed by: Alba BlesaWolf Dieter RötherKyle Stewart Skalenko
Original research article
The authors used this protocol in:

Mar 2018
Advertisement




Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






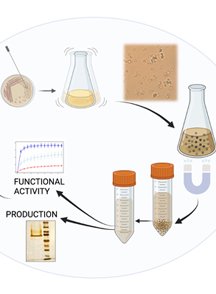

Abstract
Candida albicans is the pathogenic fungus that most frequently causes infections in humans. It is part of the microbiota commonly found in the skin, gastrointestinal tract, and vaginal mucosa. However, certain conditions, including immunosuppression, excessive use of antibiotics, hormonal changes, the use of medical devices in patients, and individual nutritional status, promote the development of opportunistic infections caused by this fungus. One of the main fungal structures interacting with the host is the cell wall, which is principally composed of chitin, glucan, and proteins. The cell wall plays key functions for the cell, such as osmotic protection; it is also responsible for cellular shape and acts as a signaling hub in response to environmental changes. Cell wall proteins participate in diverse cellular functions, such as attachment to surfaces and cell wall structure; some possess catalytic or transport activities. In this protocol, we show the methodology for isolating cell wall proteins covalently linked or not to cell wall components that can be previously labeled with [14C]-L-lysine by the action of the fungal transglutaminase localized in the cell wall. We use an extraction method by mechanical cell disruption and washing with 2 M NaCl, whose ionic strength eliminates contaminating proteins from other organelles, through subsequent serial treatments with SDS, chitinase, and zymolyase.
Key features
• Methodology for obtaining cell walls (CW) by mechanical cell disruption, fractionation, and isolation of CW proteins previously labeled with [14C]-L-lysine by the endogenous CW transglutaminase.
• Extraction of CW noncovalently-linked proteins by sequential treatments with 2% SDS, and those covalently-linked CW proteins by chitinase and zymolyase treatment.
Keywords: Cell fractionationGraphical overview
Background
Globally, Candida albicans is a pathogenic fungus that frequently affects humans, causing mild skin infections to severe invasive infections. This fungus is part of the normal microbiota of the skin, gastrointestinal tract, and vaginal mucosa. However, several factors, including immunosuppression, excessive use of antibiotics, hormonal changes, usage of medical devices, and individual nutritional status, promote the development of opportunistic infections caused by this fungus. At the cellular level, one of the main structures that has contact with the host is the cell wall, which performs various essential functions for the fungus, such as osmotic protection, signaling center, morphogenesis, and reprogramming of cell wall components in response to the availability of carbon, blood, serum, high temperatures, and environmental acidity [1–3]. The response to these factors promotes changes in the content of cell wall components such as glucan, chitin, and proteins (Figure 1).
The mannosylated protein coat, covalently linked to cell wall polysaccharides, mediates adherence to both the host and medical devices. It also prevents the immune system from recognizing the glucan layer, promotes biofilm formation, and favors communication between the host and C. albicans yeast cells [4].
Given the importance of the C. albicans cell wall proteins, several strategies have been implemented for their extraction and analysis. In a study carried out by our group, cell wall proteins previously labeled with [14C]-L-lysine by the action of the fungal transglutaminase were sequentially extracted with 2% SDS, chitinase, and zymolyase, and identified by tandem mass spectrometry. The zymolyase treatment released 37 proteins, and 41 were identified after incubation with chitinase [1].
The proteins extracted with 2% SDS showed an apparent molecular mass of less than 50 kDa. On the contrary, the proteins released by the zymolyase and chitinase treatments showed a molecular mass greater than 180 kDa. Bioinformatics analysis revealed that the proteins extracted with 2% SDS were reportedly involved in catalytic, binding, structural, and transport functions. The proteins extracted with zymolyase have structural and catalytic functions. In the case of proteins extracted with chitinase, proteins have catalytic, binding, structural, and transport roles. Of the entire universe of identified proteins, 24 were extracted by the three conditions. Moreover, only sixteen have been documented and localized in the cell wall, with Eno1, Pgk1, and Als1 being immunodominant proteins [5–7].
This protocol aims to describe the methodology for extracting cell wall proteins by sequential treatments with 2% SDS to extract noncovalently linked proteins, and extracting those covalently linked to chitin and glucan with chitinase and zymolyase, respectively.
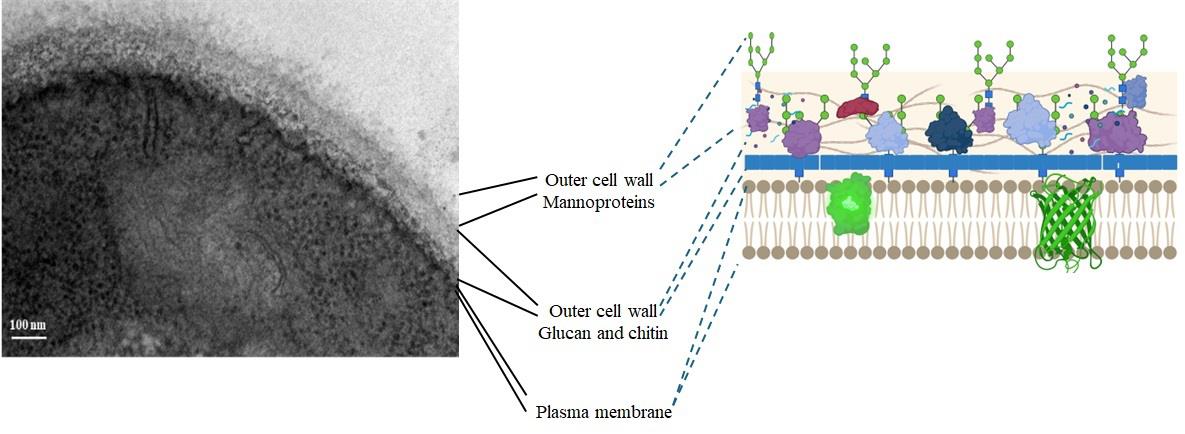
Figure 1. Ultrastructure of the Candida albicans CAI4 yeast cell wall (our results)
Materials and reagents
Biological materials
1. Candida albicans CAI4 strain (Δura3:: λimm434/Δura3:: λimm434) [8]
Reagents
1. Yeast extract (BD, catalog number: 211929)
2. Bacto peptone (MCD LAB, catalog number: 9081)
3. Glucose (Merck, catalog number: 1083371000)
4. Uridine (Sigma-Aldrich, catalog number: U3003-50G)
5. Glass beads, 0.5 mm diameter (BioSpec Products, catalog number: 11079105)
6. CaCl2 (Sigma-Aldrich, catalog number: C4901)
7. Trizma base (Sigma-Aldrich, catalog number: T1503-1KG)
8. HCl (Sigma-Aldrich, catalog number: 258148)
9. Sodium dodecyl sulfate (SDS) (Bio-Rad, catalog number: 161-0302)
10. Zymolyase 20T (MP, catalog number: 0832092)
11. Chitinase from Streptomyces griseus (Sigma-Aldrich, catalog number: C6137)
12. Trichloroacetic acid (TCA) (Sigma-Aldrich, catalog number: T0699-100ML)
13. [14C(U)]-lysine (326 mCi/mL, 3.7 MBq/mL, 50 μCi) Perkin Elmer (NEC-280E), uniformly labeled (all carbon atoms labeled)
14. Phenylmethanesulfonyl fluoride (Sigma-Aldrich, catalog number: P7626)
15. KH2PO4 (Merck, catalog number: 7778-77-0)
16. K2HPO4 (Merck, catalog number: 7758-11-4)
17. NaCl (Sigma-Aldrich, catalog number: S9888)
18. Pepstatin A (Sigma-Aldrich, catalog number: P5318-25MG)
19. Absolute ethanol (J.T. Baker, catalog number: 9000-03)
20. Methanol (J.T. Baker, catalog number: 9070)
21. Developer and replenisher (GBX, Carestream Dental, catalog number: 1900943)
22. Fixer and replenisher (GBX, Carestrean Dental, catalog number: 190 1875)
23. Ready Protein Liquid Scintillation Cocktail (Beckman, catalog number: PN 510814-AA)
24. Coomassie Brilliant Blue R-250 (Bio-Rad, catalog number: 1610400)
25. Isopropyl alcohol (J.T. Baker, catalog number: 49084-02)
26. Acetic acid (J.T. Baker, catalog number: 4908-05)
27. Amplify fluorographic reagent (Amersham, Cytiva, catalog number: 95040-024)
28. N,N-dimethyl casein from bovine milk (Sigma-Aldrich, catalog number: C9801)
29. Distilled deionized water (ddH2O)
Solutions
1. YPD (see Recipes)
2. 50 mM Tris/HCl pH 7.4 (see Recipes)
3. 1 M PMSF (see Recipes)
4. 1 mg/mL pepstatin A (see Recipes)
5. Buffer A (see Recipes)
6. 1 μg/mL pepstatin A (see Recipes)
7. 2 M NaCl (see Recipes)
8. 50 mM phosphate buffer (pH 7.4) (see Recipes)
9. 10 mM CaCl2 (see Recipes)
10. SDS, 2% (w/v) (see Recipes)
11. 5% (v/v) trichloroacetic acid (TCA) (see Recipes)
12. 10% (v/v) TCA (see Recipes)
13. 70% ethanol solution (see Recipes)
14. 75% ethanol solution (see Recipes)
15. Staining solution for protein polyacrylamide gels (see Recipes)
Recipes
1. YPD (1 L)
| Reagent | Final concentration | Quantity or volume |
| Yeast extract | 1% (w/v) | 10 g |
| Bacto peptone | 2% (w/v) | 20 g |
| Glucose | 2% (w/v) | 20 g |
| ddH2O | n/a | 1 L |
Sterilize the uridine stock solution through a sterile 0.22 μm (pore size), 33 mm diameter polyethersulfone filtration device.
Sterilize the YPD medium at 121 °C for 20 min and supplement with 25 μg/mL Uridine, previously sterilized, after cooling the YPD medium to approximately 37–40 °C.
If solid media is required, add 1.5% (w/v) agar to liquid YPD and sterilize by autoclaving at 121 °C for 20 min.
2. 50 mM Tris/HCl pH 7.4 (100 mL)
Mix 0.6057 g of Trizma base in approximately 80 mL of ddH2O. Adjust pH to 7.4 by adding 1 M HCl dropwise. Adjust the volume to 100 mL with ddH2O in a volumetric flask.
3. 1 M PMSF
Dissolve 0.17419 g of phenylmethanesulfonyl fluoride in 1 mL of absolute ethanol and store at -20 °C.
4. 1 mg/mL pepstatin A
Weigh 1 mg of pepstatin A and dissolve in 1 mL of 10% (v/v) acetic acid in methanol.
5. Buffer A (100 mL)
Mix 99.9 mL of 50 mM Tris/HCl pH 7.4 with 0.1 mL of 1 M PMSF. Use immediately.
6. 1 μg/mL pepstatin A
Add 0.1 mL of pepstatin A (1 mg/mL) to 99.9 mL of buffer A.
7. 2 M NaCl
Dissolve 11.688 g of NaCl in 100 mL of ddH2O.
8. 50 mM phosphate buffer (pH 7.4)
Solution A: 13.6 g of KH2PO4 per 1 L (0.1 M) of ddH2O.
Solution B: 17.42 g of K2HPO4 per 1 L (0.1 M) of ddH2O.
Mix 19.0 mL of solution A and 81 mL of solution B for 100 mL of buffer at pH 7.4.
9. 10 mM CaCl2
Dissolve 1.1 mg of CaCl2 per 1 mL of ddH2O.
Add 200 μL to the reaction mixture for labeling proteins with [14C]-L-lysine at 1 mL final volume.
10. SDS, 2% (w/v)
Dissolve 2 g of SDS (sodium dodecyl sulfate) in 100 mL of ddH2O with a low stirring rate to avoid air bubbles. Filter the solution to remove solid contaminants using a 0.22 μm nitrocellulose membrane.
11. 5% (v/v) TCA
Mix 5 mL of 100% TCA solution in 95 mL of ddH2O.
12. 10% (v/v) TCA
Mix 10 mL of 100% TCA solution in 90 mL of ddH2O.
13. 70% ethanol solution
For 100 mL, mix 70 mL of absolute ethanol with 30 mL of ddH2O.
14. 75% ethanol solution
For 100 mL, mix 75 mL of absolute ethanol with 25 mL of ddH2O.
15. Staining solution for protein polyacrylamide gels
For 1 L, dissolve 2 g of Coomassie Brilliant Blue R-250 in a solution of isopropyl alcohol/acetic acid/water (1/1/8, v/v/v).
Laboratory supplies
1. Falcon high-clarity polypropylene 50 mL centrifuge tube (Corning, catalog number: 352070)
2. Microcentrifuge tubes (1.5 mL) (Eppendorf, catalog number: Z606340)
3. Gilson Pipetman pipettes (P1000, P200, P20, P2)
4. Axygen Micropipette tips [1,000 µL (catalog number: T-1000-B), 200 µL (catalog number: T-200-y), 20 µL (catalog number: T-200-y), and 2 µL (catalog number: T-300)]
5. Glass slides (VELAB, catalog number: DBKVEP-20)
6. Glass coverslips (VELAB, catalog number: DBKVE-22)
7. Pyrex Erlenmeyer flasks (100 mL) (Corning, catalog number: CLS4280100)
8. Nitrocellulose filter membrane, pore size 0.45 μm, 47 mm diameter (Millipore, catalog number: N0271)
9. Polyethersulfone filtration device, 0.22 μm (pore size), 33 mm diameter (Millipore, catalog number: LGPM33RS)
10. Open-top thin-wall polypropylene tube, 16 mm × 76 mm (Beckman Coulter, catalog number: 326814)
11. Pyrex Erlenmeyer flasks, 250 mL (Merk, Corning, catalog number: CLS5100250)
12. Whatman glass microfiber grade GF/C filter discs 1.2 μm pore size, 24 mm diameter (Merck, catalog number: WHA1822024)
13. Carestream X-OMAT LS autoradiography film (Sigma-Aldrich, catalog number: F1149)
14. Glass beads 0.5 mm in diameter (BioSpec products, catalog number: 11079105)
15. WHEATON liquid scintillation vial with attached cap (20 mL) (WHEATON, catalog number: DWK986546)
16. 50 mL polypropylene Falcon tubes (Corning, catalog number: CLS352098)
17. Pyrex® VISTA culture tube, rimless, 16 mm diameter × 125 mm in length (Merck, catalog number: CLS7082016X)
18. Cotton swabs (El Crisol, catalog number: LAUKA00904) or Q-tips cotton swabs
19. White Foamboard 5 mm thick, A4 size (Cathedral)
Equipment
1. Water bath incubator (New Brunswick Scientific)
2. Eppendorf 5810R centrifuge
3. Type 90 Ti fixed-angle titanium rotor (Beckman Coulter)
4. NanoDrop 2000 spectrophotometer (Thermo Scientific)
5. Vortex-2 Genie mixer (Sigma-Aldrich)
6. BioSpec BeadBeater homogenizer
7. Epoch 2 ELISA microplate spectrophotometer (BioTek, Agilent Technologies)
8. GelDoc TM EZ imaging system (Bio-Rad)
9. Beckman Coulter LS6500 liquid scintillation counter
10. Ultracentrifuge (Beckman Coulter, model: Optima L-100 XP Class S)
11. Light microscope (Nikon)
12. Analytical balance (Denver Instruments, model: APX-200)
13. Vacuum 1225 Sampling Manifold (Millipore, model: XX2702550)
14. Gel dryer (Bio-Rad, model: 583)
15. Geiger-Müller counter (Dosimeter, model: 3700A)
Software and datasets
1. NanoDrop 2000 software (Thermo ScientificTM, version 1.6)
2. Beckman Coulter LS6500 Scintillation System, using a Motorola 6800 Series microprocessor, a Digital Signal Processor, and a 32,768 channel multichannel analyzer
Procedure
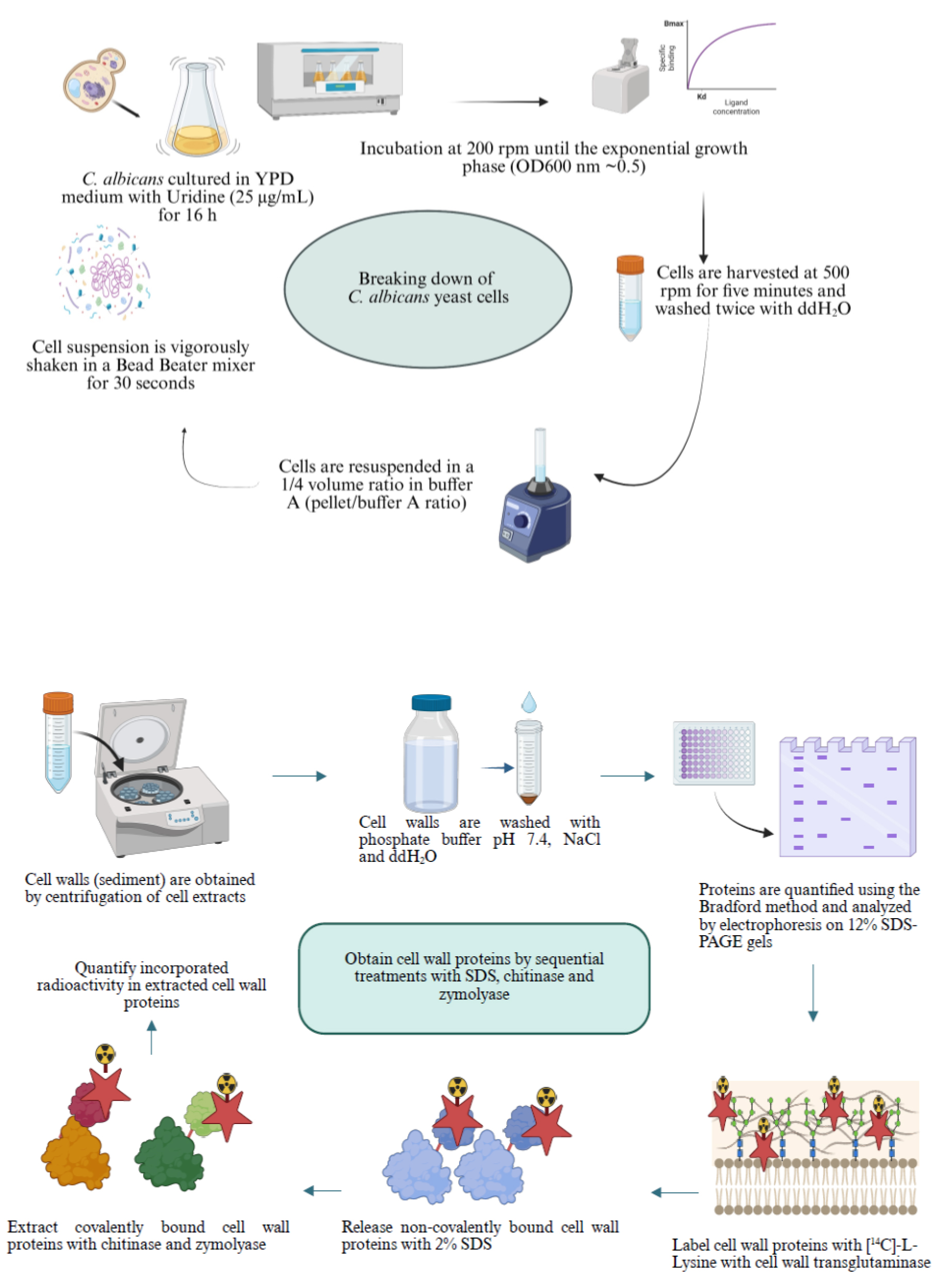
A. Obtaining C. albicans cell walls
1. Inoculate a fresh colony of C. albicans CAI4 yeast cells in 10 mL of liquid YPD medium supplemented with uridine (25 μg/mL) and incubate overnight (approximately 16 h) at 200 rpm in a water bath incubator at 28 °C.
2. Take the amount necessary from the overnight culture to inoculate 100 mL of YPD medium supplemented with 25 μg/mL uridine in an Erlenmeyer flask to obtain an initial optical density at 600 nm (OD600 nm) of 0.1 as determined using a Nanodrop 2000 system or any other spectrophotometer. Incubate the culture in a water bath incubator at 200 rpm until reaching an early exponential growth phase (OD600 nm ~0.5).
3. Harvest the cells by centrifugation at 500 rpm (42× g) for 5 min in an Eppendorf 5810R centrifuge and wash them twice with sterile ddH2O.
4. Determine the wet weight of the cell pellet and resuspend cells in buffer A using a volume ratio (pellet to buffer A) of 1/4. Add 4 g of glass beads (0.5 mm in diameter) per gram of cell pellet.
5. Shake the cell suspension vigorously (approximately 15–20 times) with the glass beads and buffer A for 30 s in a Bead Beater mixer or Vortex-2 Genie and chill for 1 min on ice. Repeat this process until the cells are completely disrupted in a sample observed under a light microscope. Take 2–5 μL of suspension before (as a control) and after the lysis procedure, deposit it on a clean glass slide, cover with a coverslip, and observe through the light microscope using the 40× objective.
6. Centrifuge the cell extracts at 1,200× g for 10 min at 4 °C in an Eppendorf 5810R centrifuge.
7. Wash the pellet corresponding to cell walls (CW) by centrifugation at 1,200× g at 4 °C twice with 50 mM phosphate buffer (pH 7.4), twice with 2 M NaCl, and twice with ddH2O. Resuspend the CW in buffer A using a 1/4 volume ratio (pellet/buffer A). Store the samples at 4 °C.
8. Centrifuge the supernatant at 105,000× g in a Beckman Coulter Optima TM L-100 XP centrifuge using a Type 90 Ti rotor for 1 h at 4 °C without brake. Use the open-top thinwall polypropylene tubes to obtain the soluble fraction (S-35K, which means supernatant obtained at 35,000 rpm, equivalent to 105,000× g, for the rotor used) and the pellet that corresponds to the mixed membrane fraction (MMF).
9. Resuspend the MMF in buffer A with protease inhibitors (in the same volume as that obtained for the S-35K, meaning that this volume must be measured) and keep it at 4 °C, with the help of a pipette tip with its tip sealed and rounded by heat.
10. Quantify protein content in the fractions obtained using the Bradford method [9].
11. Analyze 100 μg or other amounts of proteins in CW, MMF, and S35-K cell fractions by electrophoresis on 12% SDS-PAGE gels and stain them with Coomassie Brilliant Blue [10]. As an example of the proteins found in the different fractions, please see Figure 2.
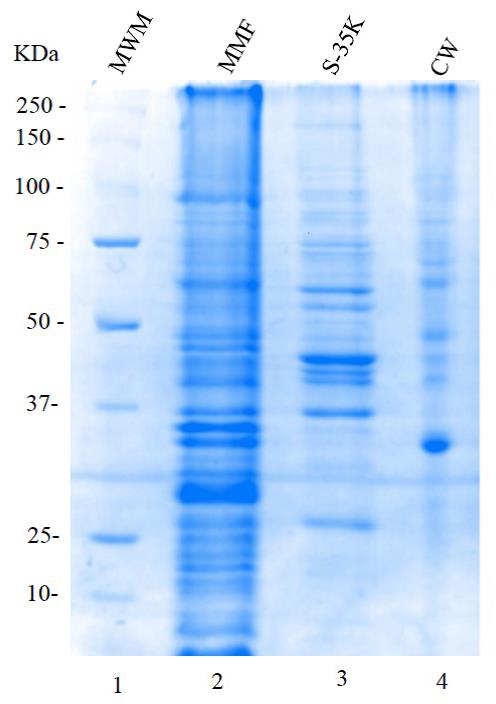
Figure 2. Electrophoresis analysis on a 12% SDS-PAGE gel, stained with Coomassie brilliant blue, corresponding to different cellular fractions of Candida albicans. Lane 1: molecular weight markers (MWM); Lane 2: mixed membrane fraction MMF (100 μg); Lane 3: soluble fraction S-35K (20 μg); Lane 4: cell wall fraction CW (20 μg).
B. Sequential extraction of [14C]-L-lysine-labeled proteins from the cell wall with 2% SDS, zymolyase, and chitinase
1. Label cell wall proteins with [14C]-L-lysine using the endogenous transglutaminase in a 1 mL reaction mixture of buffer A containing 2.5 μCi of [14C(U)]-L-lysine (326 mCi/mmol specific activity) and 2 mM CaCl2, and incubate with gentle shaking for 4 h at 30 °C. Stop the labeling reaction by adding 2 mL of 10% (w/v) TCA, keeping it on ice for 2 h.
2. To measure the incorporated radioactivity, place the sample on ice for 2 h and centrifuge at 3,000× g for 10 min at 4 °C. Then, resuspend the pellet in a glass tube with 1.0 mL of 10% TCA, boil for 10 min, and centrifuge at 3,000× g for 10 min. Resuspend the pellet with 5 mL of 10% TCA by vortexing. Pour the content into a well of a vacuum manifold containing glass fiber filters, connect to vacuum, and wash the precipitate twice with 5 mL of 10% TCA, twice with 5 mL of 5% TCA, and twice with 5 mL of absolute ethanol. Finally, air dry the filters with the help of a pin, avoiding contact of the filter with the surface of a White Foam board 5 mm thick A4 size.
3. To release noncovalently bound proteins, incubate the sample in 2% SDS for 10 min in a boiling water bath. To recover solubilized proteins, centrifuge the sample at 3,000× g for 10 min at room temperature.
4. Recover the supernatant (solubilized material) and keep it on ice. Wash the pellet (insoluble material) by centrifugation at 3,000× g for 10 min at 4 °C twice with ddH2O, twice with distilled water, twice with 70% ethanol, and twice with distilled water.
5. To release glucan-bound proteins, incubate the pellet from step B4 with 1 mg/mL Zymolyase 20T containing 1 mM PMSF for 3 h at 30 °C and recover the solubilized material by centrifugation at 3,000× g for 10 min. Store the supernatant on ice.
6. Subsequently, treat the pellet from the previous step with 0.5 mg/mL chitinase in 10 mM phosphate buffer, pH 7.0, at 30 °C for 3 h. Centrifuge the mixture at 3,000× g for 10 min and keep the supernatant on ice.
7. Precipitate the 2% SDS-extracted samples with 75% ethanol at 4 °C and centrifuge at 3,000× g for 10 min. Wash the pellet three times with 70% ethanol.
8. Quantify the incorporated radioactivity in each fraction extracted using a liquid scintillation counter. Take known amounts (100 μL) from each fraction and put them separately in 2–5 mL of Ready Protein Liquid Scintillation Cocktail in glass vials. Put each vial on a rack and put it in the Counting system using the 14C channel, counting for 2 min each vial. In the case of filter fibers with radioactive material, put each dry filter in one glass vial, add 2–5 mL of Ready Protein Liquid Scintillation Cocktail, and proceed as mentioned before.
9. Electrophorese samples containing 10,000 counts per minute (cpm) per well on a 10% SDS-polyacrylamide gel.
10. Stain the polyacrylamide gel with Coomassie brilliant blue, incubate with 30–50 mL of Amplify solution (Amersham) as recommended by the manufacturer, dry the gel in a gel dryer, and expose it to an X-Omat S film at -70 °C for 1 month, protected from light.
11. Leave the cassette containing the film to reach room temperature. Reveal the film in a dark room with red light, first by completely immersing the film with wooden forceps in a tray containing 200–300 mL of developer dilution [28% (v/v) in water], gently moving the tray until signal appearance. Then, transfer the film with wooden forceps to a tray containing water for 5 min and agitate the tray gently. Then, transfer the film to 200–300 mL of fixer solution [28% (v/v) in water] for 5–10 min. Finally, air-dry the film.
Critical: Protein labeling with [14C]-L-lysine is to exclusively visualize protein sizes after separation by SDS-PAGE and autoradiography. To analyze extracted proteins with the different treatments described in this protocol, the transglutaminase reaction must be omitted.
Data analysis
Once all subcellular fractions were obtained, we determined transglutaminase activity by incubating 100 μL of each fraction with 2 mM CaCl2, 2.5 μL of [14C]-L-lysine (0.25 μCi/μL), and 100 μg of N, N-dimethyl casein as the exogen acceptor in a final volume of 500 μL. The reaction mix was incubated for 4 h at 30 °C. The labeling reaction was stopped by adding 2 mL of 10% (w/v) TCA, keeping it on ice for 2 h. To determine the incorporated radioactivity (Table 1), follow the procedure described in steps B2 and B8.
Table 1. Distribution of transglutaminase activity in subcellular fractions of Candida albicans (unpublished results)
| Enzymatic fraction | Specific activity [cpm·h-1 (protein mg)-1] | Total activity (cpm·h-1) | Relative activity (% of total activity) | Total activity with cystamine in the reaction mix (cpm·h-1) | Inhibition or activation (%) |
| CW | 17,753 | 2,103,018 | 73.81 | 681,168 | -67.61 |
| MMF | 689 | 339,872 | 11.93 | 145,657 | -57.14 |
| Cytosol | 182 | 406,295 | 14.26 | 554,308 | +36.43 |
| Total | 2,849,185 | 100.0 |
CW: cell wall, pellet obtained by centrifugation at 1,200× g, after disruption of yeast cells. MMF: membrane mixture fraction corresponds to the pellet obtained at 100,000× g for 1 h. Cytosol (S-35K): soluble fraction obtained at 100,000× g for 1 h.
As the primary interest was the transglutaminase enzyme present in the cell wall, we determined a kinetic curve of radioactivity incorporation (Figure 3). Extracted labeled proteins with the different treatments are shown in Figure 4.
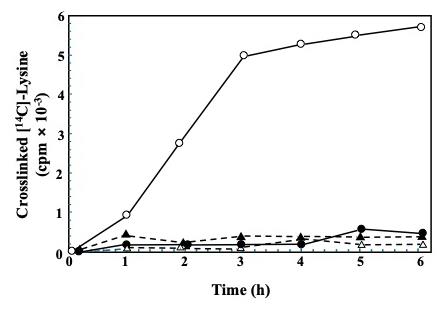
Figure 3. Transglutaminase-mediated incorporation of [14C]-L-lysine into endogenous cell wall proteins in a course-time experiment. Four reaction mixtures series were set, each containing similar aliquots of cell walls and 2.5 μCi of [14C]-L-lysine: 1) without cystamine, the specific inhibitor of transglutaminase (open circles), 2) with 50 mM cystamine (filled circles), 3) with the enzymatic source inactivated by boiling (open triangles), or 4) containing 2 mM EDTA to chelate Ca2+ (filled triangles). All reaction mixtures were incubated for the indicated times. Radioactivity in the TCA-precipitable material was further quantified in a liquid scintillation counter system [7]. Distributed under a Creative Commons Attribution-NonCommercial-NoDerivs (CC BY-NC-ND).
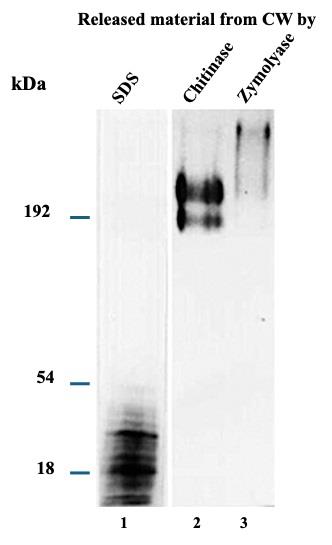
Figure 4. [14C]-L-lysine-labeled proteins released through sequential treatments with SDS, zymolyase, and chitinase were analyzed by electrophoresis in 10% SDS-PAGE gels and autoradiography. 10,000 cpm of labeled fractions were loaded in each lane. Lane 1: SDS-released material; lane 2: zymolyase-released material; lane 3: chitinase-released material [7]. Distributed under a Creative Commons Attribution-NonCommercial-NoDerivs (CC BY-NC-ND).
Validation of protocol
This protocol has been implemented and validated in the following research article:
Reyna-Beltrán et al. [7]. The Candida albicans ENO1 gene encodes a transglutaminase involved in growth, cell division, morphogenesis, and osmotic protection. Journal of Biological Chemistry, 293(12), 4304–4323. doi: 10.1074/jbc.m117.810440.
General notes and troubleshooting
1. For cell disruption, use a volume lower than 50 mL of cells in buffer A in a 50 mL Falcon tube and use the vortex for disruption. If the disruption volume is greater than 50 mL, use the Bead Beater.
2. After cell disruption, a lot of material remains attached to the glass beads. Consider performing an extra wash with buffer A without losing the 1:4 ratio.
3. After cell disruption, protease activity promotes the loss of enzymatic activity of many proteins. Therefore, samples must be stored frozen at -70 °C with the protease inhibitor cocktail in buffer A.
4. Safety considerations when handling radioactive material: Always wear disposable nitrile gloves and a lab coat. Before beginning to work, monitor the exclusive working radioactive station with a Geiger counter throughout the entire working area to determine potential radioactive contamination by 14C, 32P, or 33P, if working with these isotopes, as is the case in our work. When working with tritium, use cotton swabs to contact the working area at various points and count the radioactivity using a scintillation counter. Use a vertical Lucite L-shaped screen to protect yourself by blocking the radioactivity emitted by 14C. This isotope emits low-energy beta radiation, which is not dangerous to the user and barely penetrates the skin. However, the primary concern is the possibility of internal exposure through accidental ingestion or inhalation of the labeled material. Cover the working area with disposable, one-sided plasticized absorbent pads. All Eppendorf tubes containing radioactive material must be manipulated with a Lucite rack. The stock of radioactive material must be thawed at room temperature and kept on ice. Always add the proper amount of radioactive material last to the reaction mix. Put the radioactive pipette tip in a plastic bag in a glass beaker. Incubate the reaction mix at the desired temperature using a multiblock heater. After finishing the labeling reaction, monitor the working area with the Geiger counter. Dispose of the radioactive material by closing the bag and proceeding as recommended by institutional regulations. Immediately freeze the radioactive material in an appropriately labeled Lucite container in the freezer. It is strongly recommended to use a Film Badge Radiation Dosimeter to determine if user exposure is within safe limits. Proceed according to institutional regulations.
Acknowledgments
This work was funded by the Center for Research and Advanced Studies (Cinvestav) to Dr. Juan Pedro Luna-Arias at the Department of Cell Biology. This protocol was used in [7].
Competing interests
The authors declare that they have no financial conflicts or competing interests.
References
- Reyna-Beltrán, E., Isaac Bazán Méndez, C., Iranzo, M., Mormeneo, S. and Pedro Luna-Arias, J. (2019). The Cell Wall of Candida albicans: A Proteomics View. In: Candida Albicans. Sandai, D. (Ed.). IntechOpen. e82348. https://doi.org/10.5772/intechopen.82348
- Ibe, C. and Munro, C. A. (2021). Fungal Cell Wall Proteins and Signaling Pathways Form a Cytoprotective Network to Combat Stresses. J Fungi. 7(9): 739. https://doi.org/10.3390/jof7090739
- Parambath, S., Dao, A., Kim, H. Y., Zawahir, S., Alastruey Izquierdo, A., Tacconelli, E., Govender, N., Oladele, R., Colombo, A., Sorrell, T., et al. (2024). Candida albicans—A systematic review to inform the World Health Organization Fungal Priority Pathogens List. Med Mycol. 62(6): e1093/mmy/myae045. https://doi.org/10.1093/mmy/myae045
- d'Enfert, C., Kaune, A. K., Alaban, L. R., Chakraborty, S., Cole, N., Delavy, M., Kosmala, D., Marsaux, B., Fróis-Martins, R., Morelli, M., et al. (2020). The impact of the Fungus-Host-Microbiota interplay upon Candida albicansi nfections: current knowledge and new perspectives. FEMS Microbiol Rev. 45(3): e1093/femsre/fuaa060. https://doi.org/10.1093/femsre/fuaa060
- Ruiz-Herrera, J., Iranzo, M., Elorza, M. V., Sentandreu, R. and Mormeneo, S. (1995). Involvement of transglutaminase in the formation of covalent cross-links in the cell wall of Candida albicans. Arch Microbiol. 164(3): 186–193. https://doi.org/10.1007/s002030050253
- Iranzo, M., Aguado, C., Pallotti, C., Cañizares, J. V. and Mormeneo, S. (2002). Transglutaminase activity is involved in Saccharomyces cerevisiae wall construction. Microbiology. 148(5): 1329–1334. https://doi.org/10.1099/00221287-148-5-1329
- Reyna-Beltrán, E., Iranzo, M., Calderón-González, K. G., Mondragón-Flores, R., Labra-Barrios, M. L., Mormeneo, S. and Luna-Arias, J. P. (2018). The Candida albicans ENO1 gene encodes a transglutaminase involved in growth, cell division, morphogenesis, and osmotic protection. J Biol Chem. 293(12): 4304–4323. https://doi.org/10.1074/jbc.m117.810440
- Fonzi, W. A. and Irwin, M. Y. (1993). Isogenic strain construction and gene mapping in Candida albicans. Genetics. 134(3): 717–728. https://doi.org/10.1093/genetics/134.3.717
- Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 72: 248–254. https://doi.org/10.1016/0003-2697(76)90527-3
- Gallagher, S. R. (2012). SDS‐Polyacrylamide Gel Electrophoresis (SDS‐PAGE). Curr Protoc Essent Lab Tech. 6(1): eet0703s06. https://doi.org/10.1002/9780470089941.et0703s06
Article Information
Publication history
Received: Feb 14, 2025
Accepted: Jul 17, 2025
Available online: Sep 2, 2025
Published: Sep 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Reyna-Beltrán, E., Iranzo, M., Mormeneo, S., Bazán-Méndez, C. I., Labra-Barrios, M. L., Hernandez-Martínez, E. and Luna-Arias, J. P. (2025). Fractionation and Extraction of Cell Wall Proteins From Candida albicans. Bio-protocol 15(18): e5456. DOI: 10.21769/BioProtoc.5456.
- Reyna-Beltrán, E., Iranzo, M., Calderón-González, K. G., Mondragón-Flores, R., Labra-Barrios, M. L., Mormeneo, S. and Luna-Arias, J. P. (2018). The Candida albicans ENO1 gene encodes a transglutaminase involved in growth, cell division, morphogenesis, and osmotic protection. J Biol Chem. 293(12): 4304–4323. https://doi.org/10.1074/jbc.m117.810440
Category
Microbiology > Microbial cell biology > Organelle isolation
Biochemistry > Protein > Isolation and purification
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.