- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Preparation and Characterization of Lipid Nanoparticles Co-loaded With DNA and Nitro-Oleic Acid
Published: Vol 15, Iss 18, Sep 20, 2025 DOI: 10.21769/BioProtoc.5450 Views: 3882
Reviewed by: Andrea GramaticaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2025
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Lipid nanoparticles (LNPs) are powerful carriers for nucleic acid delivery, but plasmid DNA-loaded LNPs (pDNA-LNPs) have been limited by inflammation and toxicity. We showed that standard pDNA-LNPs activate the cGAS–STING pathway, leading to severe immune responses and mortality in mice. To overcome this, we co-loaded nitro-oleic acid (NOA), an endogenous STING inhibitor, into pDNA-LNPs. NOA-pDNA-LNPs mitigated inflammation, enabled safe in vivo delivery, and supported sustained gene expression for months. Here, we present a detailed protocol for producing and characterizing NOA-pDNA-LNPs to facilitate safer, long-term gene delivery applications.
Key features
• Provides a step-by-step protocol to produce plasmid DNA–LNPs co-loaded with nitro-oleic acid (NOA), optimized for both in vitro and in vivo applications.
• Includes methods for quantitative assessment of DNA and NOA encapsulation efficiencies, particle size, and quality control metrics.
Keywords: Lipid nanoparticlesGraphical overview
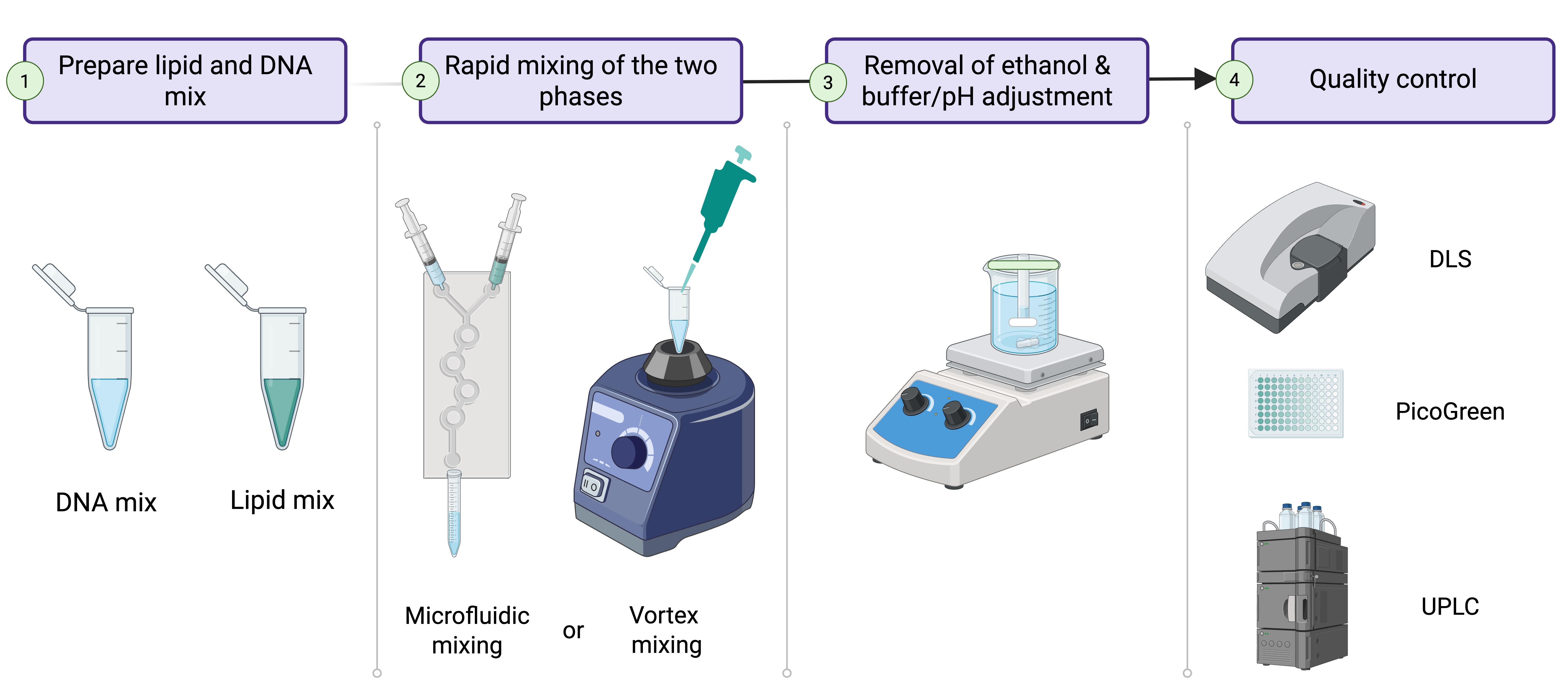
Lipid nanoparticle (LNP) production process. 1. Prepare lipid and DNA mixes in separate 2 mL Eppendorf tubes. 2. Mix the two phases using either a microfluidic chip or a vortex mixer. 3. Dialyze the formed LNPs against 1× PBS, pH 7.4, for 2 h to remove remaining ethanol and adjust pH. 4. Validate LNP quality before use or storage.
Background
The success of COVID-19 mRNA vaccines showcased the power of lipid nanoparticles (LNPs) to deliver nucleic acids and achieve high protein expression [1]. While most LNP applications focus on mRNA or siRNA, these cargos are short-lived and lack promoter control. In contrast, DNA offers longer-term expression (months to years), cell type–specific promoter regulation, and greater stability, making it ideal for expressing therapeutic proteins, gene editors, or shRNAs for long-term effects [2–4].
DNA-loaded LNPs combine durable expression with the advantages of LNPs: high delivery efficiency, low immunogenicity (compared to viral vectors), and flexible cargo capacity. However, their development has lagged due to safety concerns. We found that plasmid DNA-LNPs (pDNA-LNPs) trigger strong inflammation in naive mice, leading to 100% mortality at standard doses [5]. Using knockout mouse models and in vitro assays, we identified the cGAS–STING pathway as the main driver. This innate immune pathway detects cytosolic DNA and activates pro-inflammatory cytokines like type I interferons and IL-6.
To overcome this, we took inspiration from DNA viruses that evade STING-mediated immunity. We loaded pDNA-LNPs with nitro-oleic acid (NOA), an endogenous STING inhibitor. These NOA-pDNA-LNPs eliminated inflammation in vitro and prevented mortality in vivo, without compromising transgene expression.
Together, these results demonstrate that pDNA-LNPs co-loaded with bioactive lipids like NOA can enable safe, long-term gene expression. Here, we present a detailed protocol for producing NOA-pDNA-LNPs to support a broader application of this promising gene delivery platform.
Materials and reagents
Reagents
1. SM-102 (Echelon Biosciences, catalog number: N-1020); store at -20 °C
2. Cholesterol (Avanti Polar Lipids, catalog number: 700100P); store at -20 °C
3. 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) (Avanti Polar Lipids, catalog number: 850365); store at -20 °C
4. 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG 2000) (Avanti Polar Lipids, catalog number: 880151); store at -20 °C
5. Nitro-oleic acid (NOA) (Echelon Biosciences, catalog number: L-0112); store at -20 °C
6. pALD-CV77-Luciferase (pDNA) (Aldevron, catalog number: 5078-5); store at 4 °C short-term and at -20 °C long-term
7. 200 proof ethanol (100% ethanol) (Millipore Sigma, catalog number: 459836-100ML); store at room temperature
8. DEPC water (Invitrogen, catalog number: AM9916); store at 4 °C
9. Phosphate-buffered saline (1× PBS), filter-sterilized (Thermo Fisher Scientific, catalog number: J61196.AP)
10. Acetonitrile (HPLC grade) (Fisher Scientific, catalog number: A998SK-1)
11. Trifluoroacetic acid (TFA) (Millipore Sigma, catalog number: 8082600026)
12. Quant-iT PicoGreen dsDNA assay (Invitrogen, catalog number: P11496); store at 4 °C short-term and at -20 °C long-term
13. Sodium citrate dihydrate (Millipore Sigma, catalog number: W302600)
14. Citric acid (Millipore Sigma, catalog number: 251275)
15. TE buffer (Thermo Fisher Scientific, catalog number: 12090015)
Laboratory supplies
1. 2 mL Eppendorf tubes (Fisher Scientific, catalog number: 05-402-7)
2. 15 mL conical Tubes (Thermo Fisher Scientific, catalog number: 339650)
3. 1–300 μL pipette tips (Fisher Scientific, SureOneTM Micropoint Pipette Tips, catalog number: 02-707-410)
4. 500 mL beaker (Corning, catalog number: 1000J-500)
5. Dialysis tubing 12–14 kD (Repligen, catalog number: 132697)
6. Dialysis tubing 55 mm and 35 mm closures (Repligen, catalog numbers: 132734 and 132760, respectively)
7. Disposable cuvettes (GMBH + Co KG, catalog number: 759075D)
8. Zeba columns (Thermo Fisher Scientific, catalog number: 89877)
9. UPLC vials (Thermo Fisher Scientific, catalog number: 6ESV9-04PP)
Equipment
1. P20 pipette (Gilson, catalog number: F144056M)
2. P200 pipette (Gilson, catalog number: F144058M)
3. Ultrasonic bath (Branson, catalog number: BRANCPX-952-538R)
4. NanoAssemblrTM IgniteTM instrument (Cytiva, catalog number: NIN0001)
5. NanoAssemblrTM IgniteTM cartridges (Cytiva, catalog number: NIN0061)
6. Vortex-Genie 2 (Fisher Scientific, catalog number: 50-728-002)
7. Zetasizer Pro ZS (Malvern Panalytical)
8. MiniMag Magnetic Stirrer (NETA/Sigma-Aldrich, catalog number: Z742293)
9. Acquity I-Class UPLC system (Waters)
10. Cortecs C18 column (1.6 μm particle size, 2.1 × 50 mm)
Procedure
A. Preparation of lipid mix
1. Thaw lipids to room temperature prior to opening the vial.
2. Dissolve the lipids using 100% ethanol to the indicated stock concentrations (Table 1).
a. Follow the pipetting protocol at https://www.addgene.org/protocols/pipetting/.
b. Some lipids may require brief heating to 50–60 °C and sonication for complete dissolution.
Caution: Ensure cholesterol crystals are completely dissolved.
c. Store at -20 °C after aliquoting the stock solutions.
d. This protocol is designed for a 40:1 w/w ratio of total lipids to DNA (excluding NOA in the total lipid weight).
i. 25 mM lipid mix for microfluidics.
ii. 6.25 mM lipid mix for vortex mixing.
3. Prepare a 2 mL Eppendorf tube and pipette the indicated volume of each lipid/ethanol (Table 1).
Notes:
1. Vortex mixing is typically used for producing LNPs for in vitro experiments, and microfluidics is used for in vivo experiments.
2. The “L” in NOA:L indicates the total lipid content excluding NOA.
Table 1. Components in the lipid mix tube
| Component | Name | Stock concentration (mg/mL) | Molar (%) in LNP formulation | Volume (μL) for 0.4 mL of LNPs (microfluidics) | Volume (μL) for 0.16 mL of LNPs (vortex) |
|---|---|---|---|---|---|
| Ionizable lipid | SM-102 | 50 | 50 | 35.5 | 4.4 |
| Cholesterol | Cholesterol | 11.6 | 38.5 | 64.2 | 8.0 |
| Helper lipid | DSPC | 10 | 10 | 19.8 | 2.5 |
| PEG lipid | DMG-PEG 2000 | 10 | 1.5 | 18.8 | 2.4 |
| Ethanol | - | - | - | 37.5 | 79.69 |
| Anti-inflammatory lipid | NOA | 13.5 | 0.2 NOA:L (mol:mol) | 24.2 | 3.03 |
B. Preparation of DNA mix
1. Dissolve DNA to 1 mg/mL using nuclease-free water (TE buffer works as well).
2. Prepare a 2 mL Eppendorf tube and pipette the indicated volume of DNA and buffer (Table 2).
Note: To prepare a 50 mM citrate buffer at pH 4, dissolve 496 mg of sodium citrate dihydrate and 636 mg of citric acid in 100 mL of water. Adjust pH using HCl or NaOH if needed.
Table 2. Components in the DNA mix tube
| Component | Name | Composition | Volume (μL) for 0.4 mL of LNPs (microfluidics, in vivo) | Volume (μL) for 0.16 mL of LNPs (vortex, in vitro) |
|---|---|---|---|---|
| DNA | pALD-CV77-Luciferase | 1 mg/mL in water | 50.7 | 3.9 |
| Buffer | Citrate buffer | 50 mM, pH 4 | 348.3 | 116.1 |
C. Production of LNPs using microfluidics
1. Insert the microfluidic cartridge into the NanoAssemblrTM IgniteTM (Figure 1).

Figure 1. Microfluidic chip for NanoAssemblrTM IgniteTM
2. Load one 1 mL BD syringe with the lipid mix and another with the DNA mix (Figure 2).
Tip: To remove bubbles, invert the syringe and gently tap/flick until the bubble flows to the top. Then, push the liquid until it reaches the top of the tip.
Critical: Ensure the volumes are > 0.3 mL in the DNA syringe and > 0.1 mL in the lipid syringe.
3. Add waste (left) and LNP collection (right) 15 mL conical tubes to the right side of the cartridge/syringes (Figure 2).

Figure 2. Use 1 mL BD syringes to load lipid and DNA mixes into the microfluidic chip
4. Enter various formulation parameters (Figure 3):
a. Syringe type: BD 1 mL.
b. Flow rate ratio: 3:1 (DNA to lipid).
c. Total LNP volume: 0.4 mL.
d. Total flow rate: 12 mL/min.
e. Waste: 0.02 mL for start and end waste.
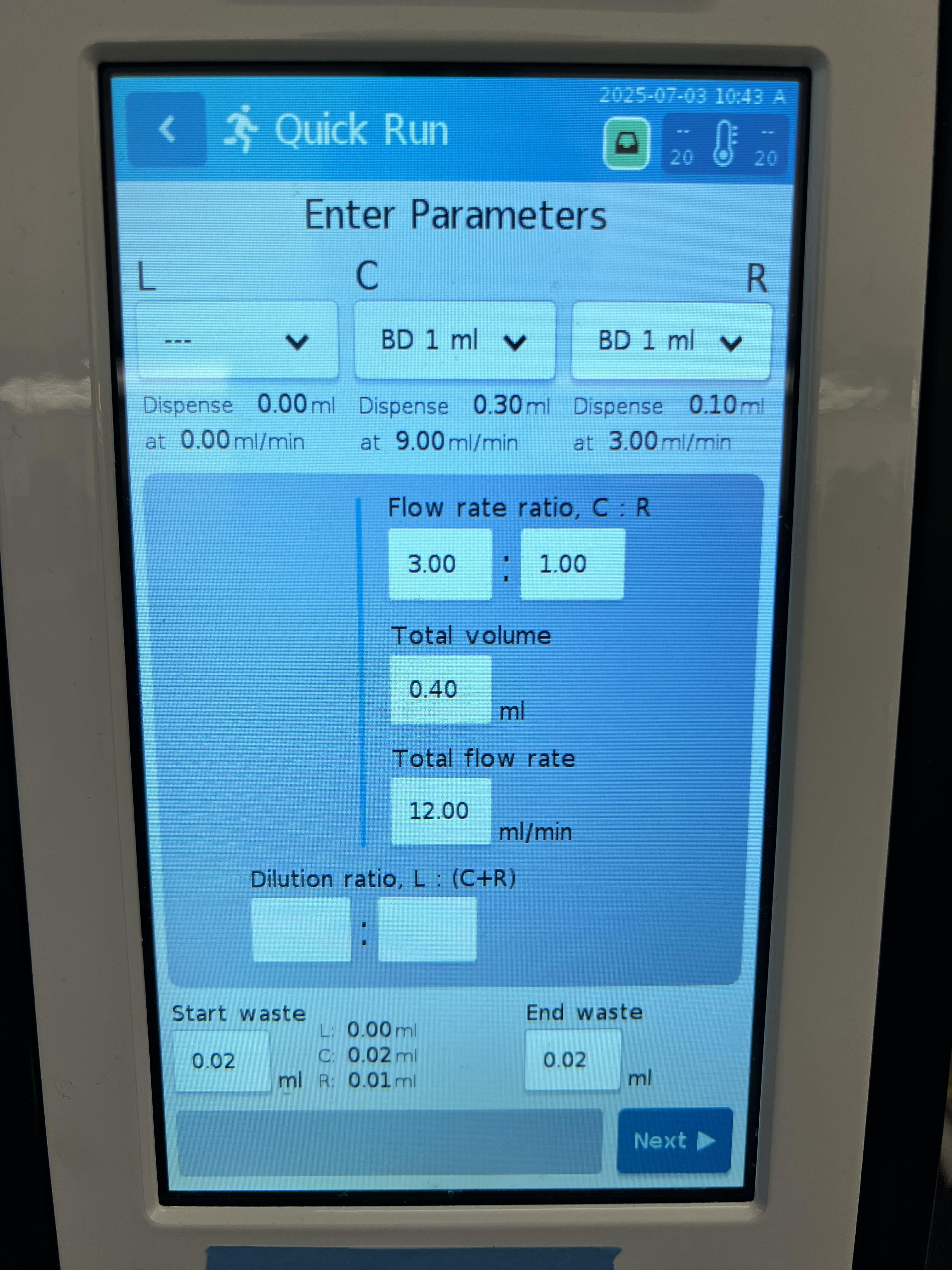
Figure 3. Lipid nanoparticle (LNP) formulation parameters
Note: A detailed protocol and machine function/troubleshooting can be found in the NanoAssemblrTM IgniteTM manual.
5. Continue to Section E after the machine finishes its run.
D. Production of LNPs using vortex mixing
1. Pipette the various lipids into a 2 mL Eppendorf tube (Table 1).
Tip: Add ethanol first as it has the highest volume.
2. Pipette the DNA and citrate buffer into another 2 mL Eppendorf tube (Table 2).
3. While vortexing the DNA mix tube at maximum speed (setting 10 on the Vortex-Genie 2), quickly pipette 40 μL of the lipid mix all at once (Figure 4). The pipette tip should be positioned just above the surface of the DNA mix, not submerged.
4. Continue to vortex for 10 s after adding the lipid mix, then proceed to Section E.

Figure 4. Vortex mixing for lipid nanoparticle (LNP) production
E. Dialysis of LNPs (Figure 5)
1. Cut and pre-wet the dialysis tubing (10 kDa MW cutoff) for 1 min.
2. Clip the bottom end of the tubing using the 35 mm weighted clip and pipette in LNPs.
Tip: Squeeze the tubing from the sides first for easier LNP loading.
3. Clip the top end of the tubing using a 55 mm unweighted clip to secure the LNPs inside the tubing.
4. Submerge in 1× PBS, pH 7.4, and stir for 2 h at room temperature.
Caution: Ensure the LNP solution is completely submerged in the dialysis bath.
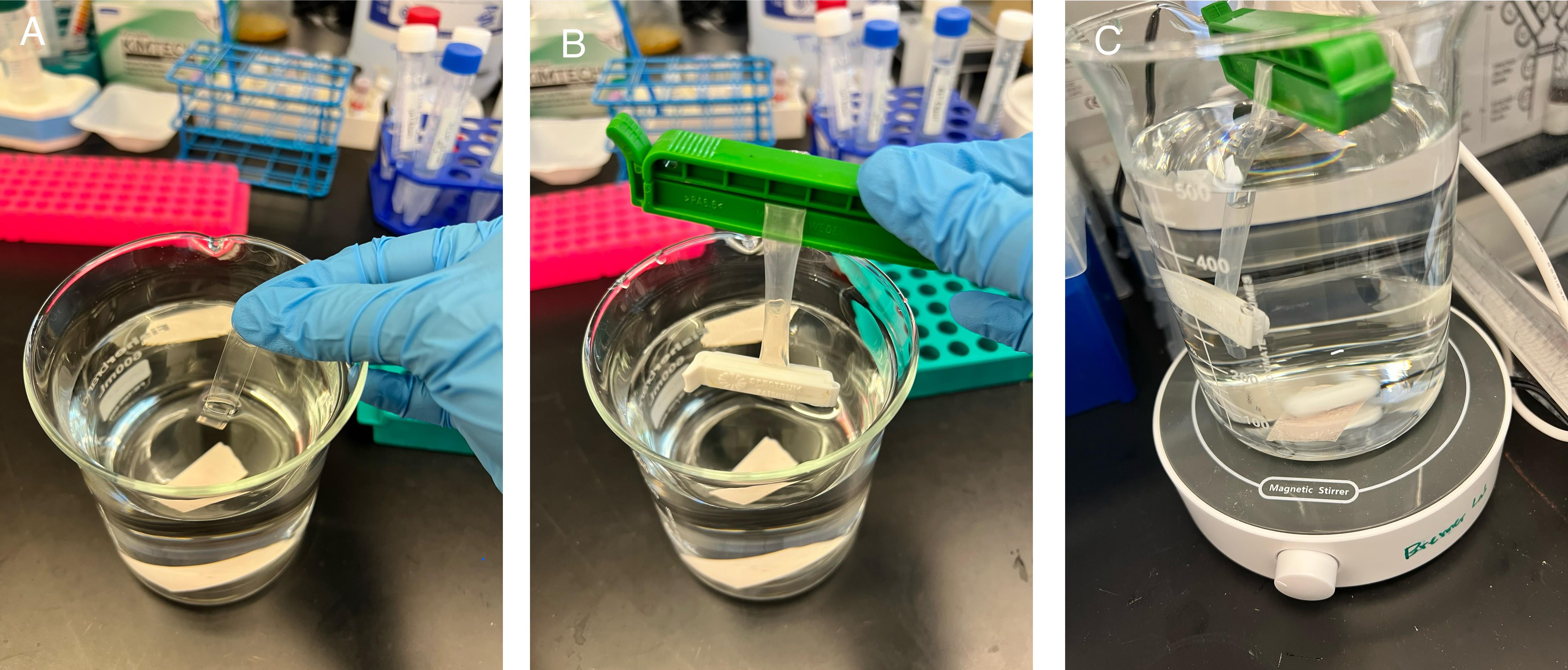
Figure 5. Dialyzing lipid nanoparticles (LNPs) against 1× PBS. (A) Pre-wetted dialysis tubing. (B) LNPs loaded in the tubing with both ends clipped. (C) Dialysis of LNPs with stirring.
5. After dialysis, remove the top clip and pipette out LNPs into a fresh 2 mL Eppendorf tube.
Pause point: Store the LNPs at 4 °C or proceed to Section F.
Caution: It is best practice to use the LNPs within a couple of days.
F. Characterization of LNPs
F1. Measure particle size using dynamic light scattering (DLS)
1. Dilute the LNPs in 1× PBS solution directly in the disposable DLS cuvettes.
a. For vortex-made LNPs: 20 μL of LNPs into 480 μL of 1× PBS.
b. For microfluidic-made LNPs: 5 μL of LNPs into 495 μL of 1× PBS.
2. Insert in the Zetasizer Pro ZS and conduct size and PDI measurement of the LNPs.
Note: A detailed protocol can be found in the Zetasizer Pro ZS (Malvern) manual.
F2. Measure DNA encapsulation
1. Follow the exact protocol from the Quant-iTTM PicoGreenTM dsDNA Assay kits and dsDNA reagents from ThermoFisher Scientific.
2. Typical LNP dilutions:
a. For vortex LNPs, dilute 40 μL of LNPs in 210 μL of TE buffer.
b. For microfluidic LNPs, dilute 10 μL of LNPs in 240 μL of TE buffer.
F3. Measure NOA encapsulation
1. Purify out unencapsulated NOA using Zeba size exclusion columns.
Note: A detailed protocol can be found in the Zeba column manual (Thermo Fisher).
a. Remove Zeba column storage solution by centrifuging at 1,500× g for 1 min.
b. Wash the column using 300 μL of 1× PBS, three times, centrifuging at 1,500× g for 1 min between each wash.
c. Load 70 μL of LNPs and collect in a fresh 2 mL Eppendorf tube.
2. Dilute LNPs (before Zeba column purification and after purification) 10× into 2 mL polypropylene screw-top microvials containing 50% water and 50% acetonitrile: 10 μL of LNP into 45 μL of water + 45 μL of acetonitrile. Briefly vortex to mix.
3. Run the two samples on an Acquity I-Class UPLC system with Cortecs C18 column (1.6 μm particle size, 2.1 × 50 mm).
a. Run with mobile phase A [water with 0.1 vol% trifluoroacetic acid (TFA)] and mobile phase B (acetonitrile with 0.1 vol% TFA).
Caution: TFA is a strong, corrosive, and volatile acid
b. Set the flow rate to 0.5 mL/min with a gradient of 10% B from 0 min to 0.4 min, linearly increasing to 90% B from 0.4 min to 0.8 min, holding at 90% B from 0.8 min to 1.3 min, and then linearly decreasing to 10% B from 1.3 min to 1.4 min.
c. Set the sample injection volume to 8 μL.
4. Using a standard curve for NOA, calculate the total concentration of NOA in LNP.
5. Based on the pre-Zeba and post-Zeba NOA concentrations, determine the NOA encapsulation efficiency.
Data analysis
1. DLS analysis showed that the average size for NOA-pDNA-LNPs (Figure 6) is 80.7 ± 2.0 nm, based on triplicate measurements.
Note: LNPs made via the vortex method typically are larger than those made with microfluidics. Inefficient mixing may lead to aggregation of LNPs (Figure 7). The DNA encapsulation efficiency and transfection efficiency may be slightly lower as well.
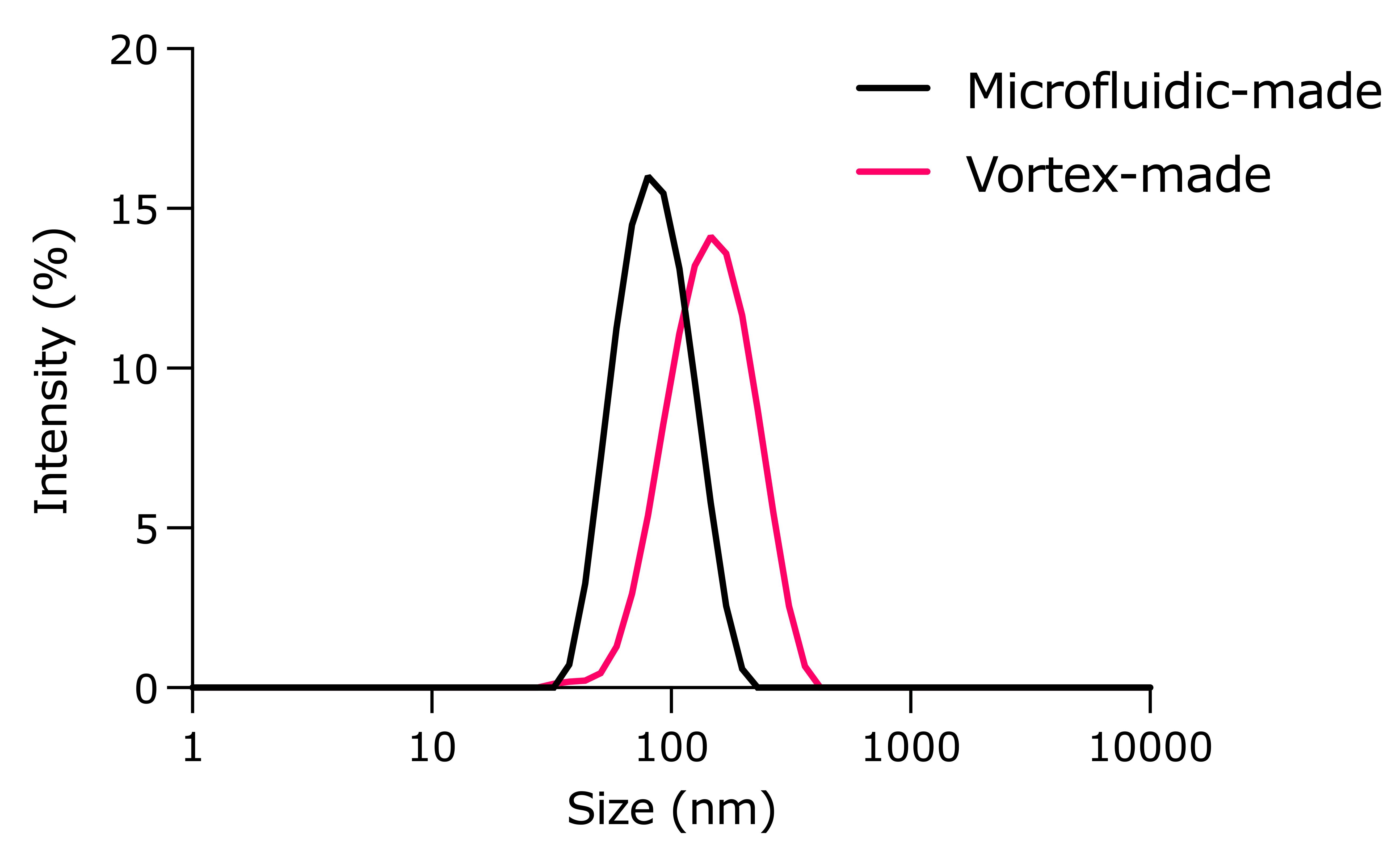
Figure 6. Size measurement of nitro-oleic acid (NOA)-plasmid DNA (pDNA)-loaded lipid nanoparticles (LNPs) using dynamic light scattering (DLS)
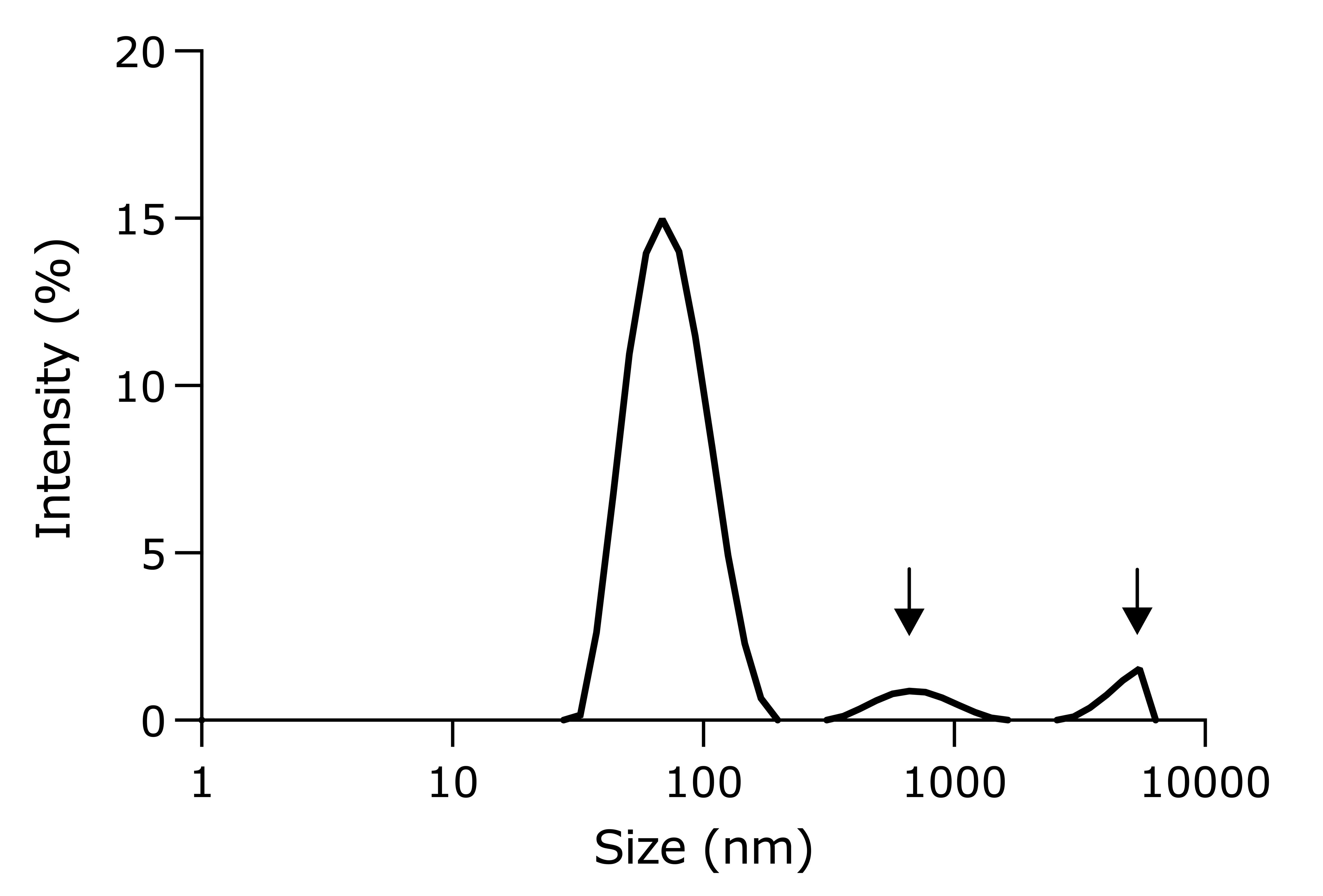
Figure 7. An example of a rejected lipid nanoparticle (LNP) sample. Arrows indicate the presence of aggregates.
2. The PicoGreen analysis showed that the average pDNA encapsulation for NOA-pDNA-LNPs is 87.7% ± 1.67%, based on triplicate measurements.
3. The ultra-performance liquid chromatography (UPLC) analysis showed that the average NOA encapsulation for NOA-pDNA-LNPs is 82.8% ± 1.72%, based on triplicate measurements.
Note: Elution of NOA (1.5 min, wavelength: 262 nm) is depicted in Figure 8 when using the gradient method, as outlined in Section F3. This elution time should be similar for both vortex and microfluidic LNPs.
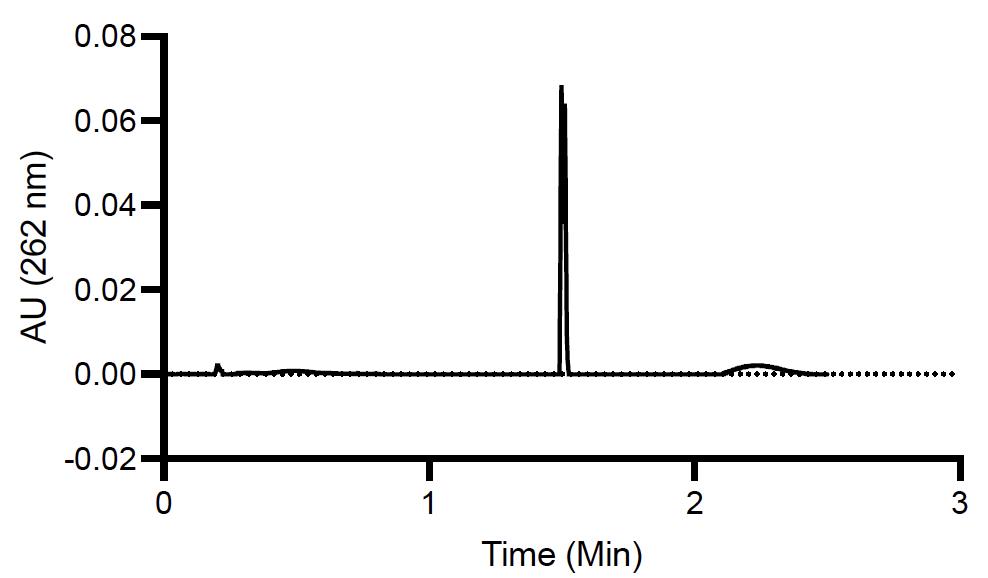
Figure 8. Ultra-performance liquid chromatography (UPLC) chromatogram of nitro-oleic acid (NOA) content within NOA-pDNA-lipid nanoparticles (LNPs)
Validation of protocol
This protocol (or parts of it) has been used and validated in the following research article:
• Patel et al. [5]. Safer non-viral DNA delivery using lipid nanoparticles loaded with endogenous anti-inflammatory lipids. Nature Biotechnology (Figure 3b).
General notes and troubleshooting
General notes
1. Batch-to-batch variability in LNP properties (size, encapsulation efficiency) may occur due to slight differences in mixing technique, lipid lot quality, or ambient temperature. It is recommended to validate each new lipid lot and calibrate the mixing process when switching operators.
2. This protocol was optimized for the delivery of pDNA using the pALD-CV77-Luciferase plasmid (Aldevron). While it may be applicable to other plasmids or DNA types (e.g., minicircle, dbDNA, or ssDNA), optimization of lipid ratios and buffer conditions may be required.
3. The stability of LNPs is limited. It is best to use freshly prepared LNPs within 2–3 days of production. Extended storage at 4 °C may reduce transfection efficiency due to LNP aggregation or hydrolysis.
4. pDNA-LNPs containing NOA show significantly reduced inflammation compared to standard LNPs. However, further validation is recommended before clinical or immunologically sensitive applications.
5. Downstream quantification assays (e.g., PicoGreen and UPLC for NOA) are sensitive to contamination. Use nuclease-free and LC/MS-grade reagents, clean pipettes, and avoid repeated freeze-thaw cycles of standards or reagents.
6. NanoAssemblrTM IgniteTM provides reproducible LNPs, but settings may require tuning when switching to different nucleic acids, buffers, or lipid compositions. Refer to the manufacturer's manual and consider pilot runs for each new formulation.
7. Both vortex- and microfluidic-produced LNPs can be used for in vitro and in vivo studies, though microfluidic LNPs are preferred for better reproducibility.
8. Typical acceptance criteria are as follows: size < 200 nm; PDI < 0.25; and DNA encapsulation efficiency > 80%. Failure in one or more of these criteria warrants rejection of the LNP batch.
9. This protocol is amenable to different pDNA sizes (tested for 3–7 kb) and different NOA percentages (tested for 0.1–1 NOA/L).
Troubleshooting
Problem 1: Low LNP yield after microfluidic mixing.
Possible cause: Incomplete loading of syringes or improper flow rate settings.
Solution: Ensure DNA and lipid syringe volumes exceed 0.3 and 0.1 mL, respectively, and verify if flow rate settings on the NanoAssemblrTM Ignite are correctly entered (TFR: 12 mL/min, FRR: 3:1).
Problem 2: Cloudy or phase-separated LNP solution.
Possible cause: Incomplete lipid dissolution before mixing.
Solution: Briefly heat and sonicate lipid stock (especially cholesterol) to ensure full solubilization before preparing the ethanol lipid mix.
Problem 3: Large or polydisperse particle sizes in DLS analysis.
Possible cause: Improper mixing technique or ethanol not fully removed post-dialysis.
Solution: Verify if a correct and vigorous vortexing or microfluidic mixing was used. Ensure a full 2-h dialysis in 1× PBS and that the LNPs are fully submerged and stirred.
Problem 4: Low DNA encapsulation efficiency.
Possible cause: Incorrect lipid-to-DNA ratio or degraded DNA.
Solution: Confirm 40:1 w/w lipid:DNA ratio was used, and that the DNA stock is intact (check via gel electrophoresis if needed). Use freshly prepared or properly stored DNA at 1 mg/mL.
Problem 5: Difficulty loading LNPs into dialysis tubing.
Possible cause: Tubing was not pre-wet, or tubing collapsed during loading.
Solution: Pre-wet tubing with water before use, and pinch sides gently to open the channel for easier pipetting.
Problem 6: No peak or inconsistent NOA signal on UPLC.
Possible cause: Incorrect gradient or injection parameters; TFA evaporation or contamination.
Solution: Double-check mobile phase composition and gradient timing. Prepare fresh mobile phases with 0.1% TFA.
Problem 7: Bubbles in syringes during microfluidic mixing.
Possible cause: Air trapped during syringe filling.
Solution: Invert syringes and gently flick to move bubbles to the top. Push the plunger slowly to expel bubbles before insertion into the NanoAssemblrTM.
Acknowledgments
Research reported in this publication was supported by the American Heart Association under grant 24PRE1195406 (to M.N.P.), grants R01-HL-153510, R01-HL160694, and R01-HL164594 (to J.S.B.), and grant R01AI153064 (to N.P.). This protocol was used in [5].
Competing interests
M.N.P., S.T., and J.S.B. have filed a patent for the DNA-LNP technology described herein. The other authors declare no competing interests.
References
- Hou, X., Zaks, T., Langer, R. and Dong, Y. (2021). Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 6(12): 1078–1094. https://doi.org/10.1038/s41578-021-00358-0
- Hughes, T. S., Langer, S. J., Johnson, K. W., Chavez, R. A., Watkins, L. R., Milligan, E. D. and Leinwand, L. A. (2009). Intrathecal Injection of Naked Plasmid DNA Provides Long-term Expression of Secreted Proteins. Mol Ther. 17(1): 88–94. https://doi.org/10.1038/mt.2008.230
- Zhu, Y., Shen, R., Vuong, I., Reynolds, R. A., Shears, M. J., Yao, Z. C., Hu, Y., Cho, W. J., Kong, J., Reddy, S. K., et al. (2022). Multi-step screening of DNA/lipid nanoparticles and co-delivery with siRNA to enhance and prolong gene expression. Nat Commun. 13(1): e1038/s41467–022–31993–y. https://doi.org/10.1038/s41467-022-31993-y
- Chen, Z. Y., He, C. Y., Ehrhardt, A. and Kay, M. A. (2003). Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther. 8(3): 495–500. https://doi.org/10.1016/s1525-0016(03)00168-0
- Patel, M. N., Tiwari, S., Wang, Y., O’Neill, S., Wu, J., Omo-Lamai, S., Espy, C., Chase, L. S., Majumder, A., Hoffman, E., et al. (2025). Safer non-viral DNA delivery using lipid nanoparticles loaded with endogenous anti-inflammatory lipids. Nat Biotechnol.: e1038/s41587–025–02556–5. https://doi.org/10.1038/s41587-025-02556-5
Article Information
Publication history
Received: Jul 15, 2025
Accepted: Aug 20, 2025
Available online: Sep 1, 2025
Published: Sep 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Patel, M. N., Tiwari, S. and Brenner, J. S. (2025). Preparation and Characterization of Lipid Nanoparticles Co-loaded With DNA and Nitro-Oleic Acid. Bio-protocol 15(18): e5450. DOI: 10.21769/BioProtoc.5450.
Category
Biological Engineering > Biomedical engineering > Drug Delivery
Molecular Biology > Nanoparticle > Plan-derived nanoparticles
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.