- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Editing the Serratia proteamaculans Genome Using the Allelic Exchange Method
Published: Vol 15, Iss 18, Sep 20, 2025 DOI: 10.21769/BioProtoc.5448 Views: 1392
Reviewed by: Alba BlesaAnu ThomasAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2022

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
No specific ecological niche has been identified for Serratia proteamaculans. Different strains of the bacterium have been described as opportunistic pathogens of plants, animals, and humans, as plant symbionts, and as free-living bacteria. This makes S. proteamaculans and its particular strains promising models for research, particularly aimed at studying the role of various genes in interspecific interactions. Genome editing is one of the most significant approaches used to study gene function. However, as each bacterial species has its own characteristics, editing methods often need to be adapted. In this study, we adapted a conventional approach based on homologous recombination—the allelic exchange method—to edit the genome of S. proteamaculans, with the aim of examining the biological role of protealysin. Plasmids for recombination were created using the suicidal vector pRE118, and then an auxotrophic Escherichia coli ST18 strain was used to deliver these plasmids to S. proteamaculans through conjugation. This method is valid and can potentially be used to create knockouts, knockins, and point mutations in the S. proteamaculans genome, without the need to insert a selective marker into the genome.
Key features
• The genome editing method for Serratia proteamaculans does not require the insertion of selective markers into the genome.
• The selection strategy allows obtaining 30%–40% of clones with the target mutation at the final stage.
• The method can be adapted to introduce knockouts, knockins, and point mutations into the genomes of other bacteria.
Keywords: Serratia proteamaculansGraphical overview
Background
Genome editing is a widely used technique when working with bacteria. It is used, in particular, to impart specific properties to microorganisms, search for genes responsible for a particular phenotype, and determine the functions of the studied genes. However, current genome editing methods have been developed for a limited number of bacterial species, and these require adaptation when used with new species. This is due to differences in, for example, cultivation conditions or genomic features.
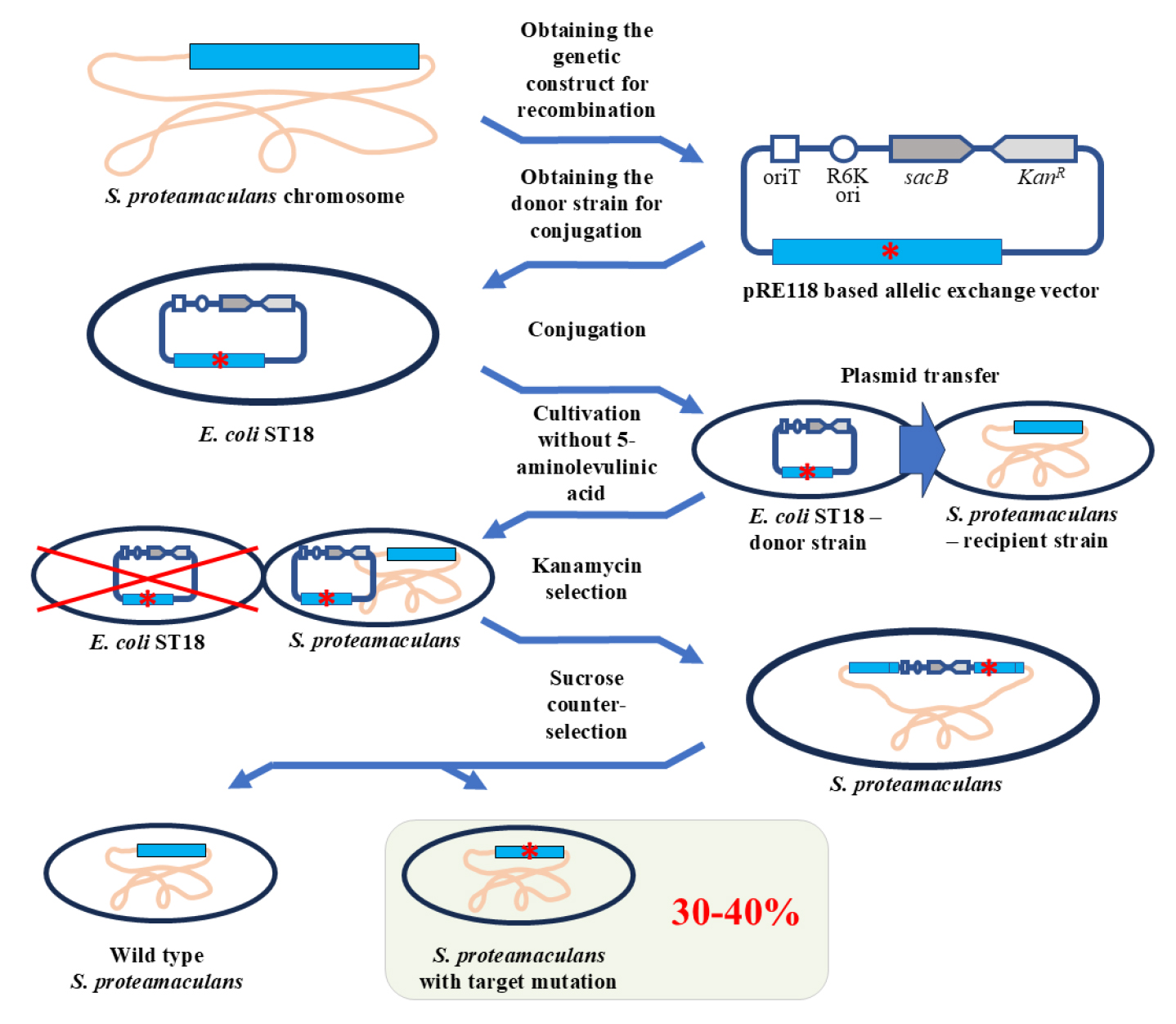
In this protocol, we propose a method for genome editing of Serratia proteamaculans. We developed the protocol to study the biological role of protealysin—a prototype of a new group of proteolytic enzymes, which are widespread among bacteria and are likely involved in pathogenesis [1]. Genome modification methods for S. proteamaculans, the natural producer of protealysin, have not been previously described. Therefore, to edit the S. proteamaculans genome, we adapted a conventional approach based on homologous recombination—the allelic exchange method. The principle of operation of the allelic exchange method is to replace a chromosomal copy with a mutated allele introduced from a plasmid delivered to bacteria. We combined and used the previously described plasmid vector pRE118 [2] and the conjugative E. coli strain ST18 [3]. The vector contains the R6K origin of replication, which only functions in the presence of the pir gene. Since the gene is present in the E. coli ST18 genome, the origin effectively operates in this bacterium, but not in S. proteamaculans. Additionally, pRE118 contains the kanamycin resistance gene for the selection of resistant S. proteamaculans cells in which the plasmid has integrated into the genome through recombination (Figure 1, first crossover). This vector also contains the suicide levansucrase sacB gene, which helps to select cells in which the pRE118 auxiliary elements have been removed from the genome by recombination (Figure 1, second crossover). E. coli ST18 provides plasmid transfer to various recipient bacteria due to the genes from the RP4 plasmid embedded in its chromosome and the transfer origin (oriT) in the pRE118 vector. The advantage of the ST18 strain is that it is auxotrophic for 5-aminolevulinic acid. Thus, the donor strain can be easily removed after conjugation by using a medium without this nutrient, with no need to introduce additional selective markers into the recipient’s genome.
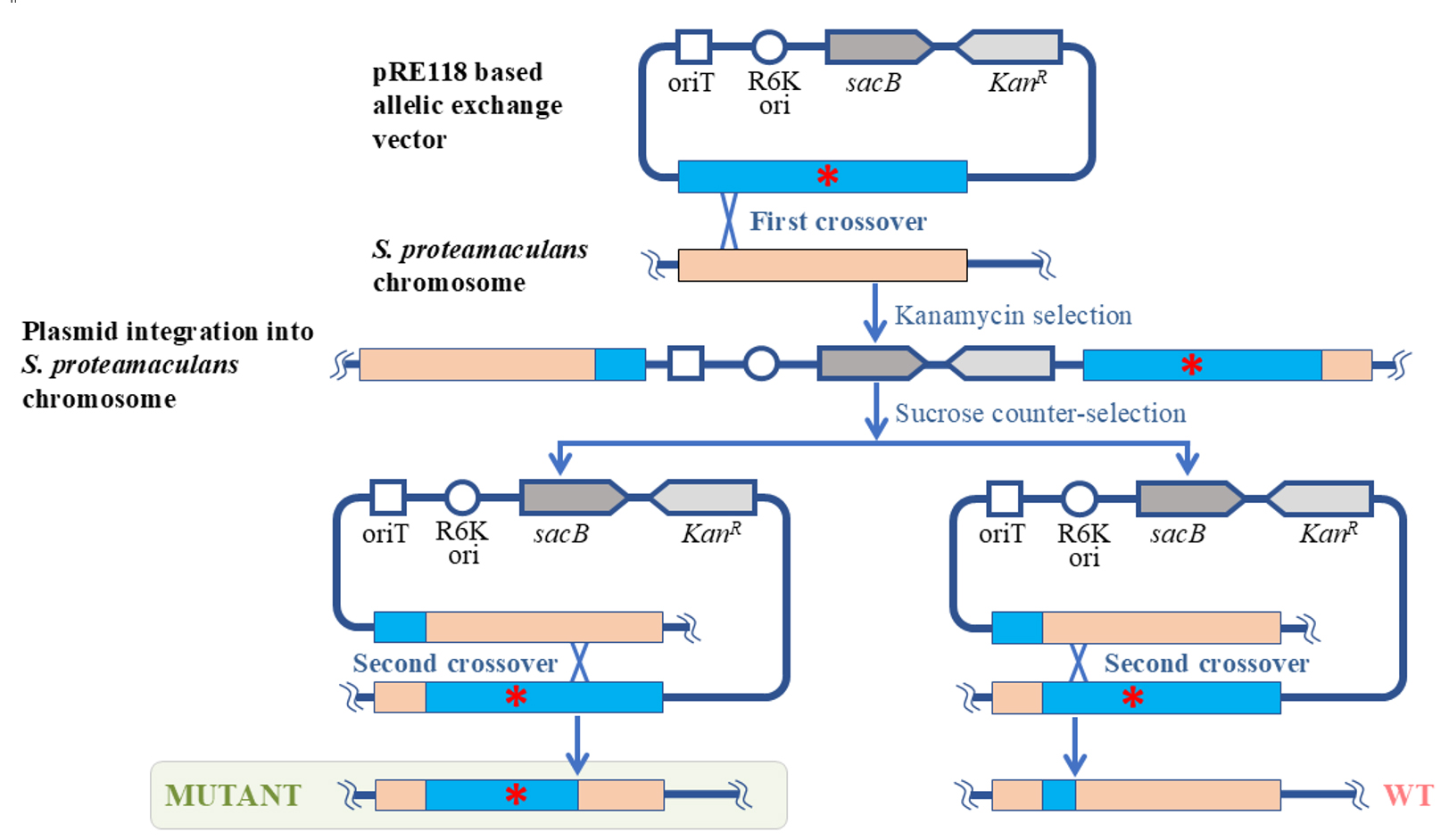
Figure 1. Schematic representation of S. proteamaculans genome editing using the allelic exchange method. The red asterisk denotes any deletion, insertion, or point mutation. oriT: transfer origin; R6K ori: R6K replication origin; sacB: levansucrase gene; KanR: kanamycin resistance gene; WT: wildtype S. proteamaculans.
To delete the part of the S. proteamaculans genome, the homologous recombination cassette (approximately 1,000 bp) was cloned into pRE118. The cassette contained the fragment of genomic DNA with the target deletion. The resulting construct was then introduced using calcium transformation into E. coli ST18 cells, which were further used as donors for plasmid transfer to S. proteamaculans. The first stage of the selection process was carried out on a medium containing kanamycin but no 5-aminolevulinic acid. The second stage took place on a sucrose medium. Sucrose-resistant and kanamycin-sensitive clones were identified and further tested for the target mutations using PCR with the primers specific to the corresponding regions of the bacterial chromosome. Target mutations were detected in 30%–40% of the clones, while the remainder returned to the wild-type allele. The presence of the target mutations was additionally confirmed by sequencing.
We used the described protocol to acquire S. proteamaculans variants with the protealysin gene or protealysin operon deletions [1]. However, this protocol can also be applied to create insertions and point mutations, as was previously described for other bacterial species using a similar approach [4]. Taken together, the proposed protocol allows for successful modification of the S. proteamaculans genome, and it is likely to be applicable to other species from the Serratia genus, as well as other gamma-proteobacteria.
Materials and reagents
Biological materials
1. Strain Serratia proteamaculans 94 [5]
2. Strain Escherichia coli ST18 (DSM 22074) [3]
3. Strain Escherichia coli CC118 (λpir) [6]
4. Plasmid pRE118 (gift from Dieter Schifferli; Addgene plasmid no. 43830) [2]
Reagents
1. Cleanup Standard kit for DNA purification from agarose gels and reaction mixtures (Evrogen, catalog number: BC022S)
2. Plasmid Miniprep kit for plasmid DNA isolation (Evrogen, catalog number: BC221S)
3. Restriction endonucleases XbaI and Psp124BI (an isoschizomer of SacI) and reaction buffer G (SibEnzyme, catalog numbers: SE-E141, SE-E107, and B002)
4. T4 DNA ligase with the reaction buffer (SibEnzyme, catalog number: SE-E329)
5. Q5 High-Fidelity DNA polymerase (New England Biolabs, catalog number: M0491S)
6. Mixture of deoxynucleotide triphosphates (dNTP), 100 mM water solution of each dNTP (SibEnzyme, catalog number: N028)
7. Bacto peptone (Gibco, catalog number: 211820)
8. Yeast extract (Sigma-Aldrich, catalog number: Y1625)
9. Agar powder (BD Difco, catalog number: 281230)
10. Kanamycin sulfate (Gibco, catalog number: 11815024)
11. UltraPure sucrose (Invitrogen, catalog number: 15503022)
12. Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S9888)
13. Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: C4901)
14. Phosphate-buffered saline (PBS) tablets (Gibco, catalog number: 18912014)
15. 5-aminolevulinic acid hydrochloride (Sigma-Aldrich, catalog number: A7793)
16. Molecular weight marker DNA ladder (from 0.25 to 10 kb) (SibEnzyme, catalog number: SE-M11)
17. Certified molecular biology agarose (Bio-Rad, catalog number: 1613102)
18. Tris(hydroxymethyl)aminomethane (Merck, catalog number: 1.08387)
19. Boric acid (Sigma-Aldrich, catalog number: 15663)
20. Ethylenediaminetetraacetic acid (Sigma-Aldrich, catalog number: ED)
Solutions
1. Kanamycin stock solution (50 mg/mL) (see Recipes)
2. 5-aminolevulinic acid stock solution (50 mg/mL) (see Recipes)
3. Sucrose stock solution (50% w/w) (see Recipes)
4. 0.1 M CaCl2 solution (see Recipes)
5. PBS buffer (see Recipes)
6. TBE electrophoresis buffer (see Recipes)
7. Agarose 1% (see Recipes)
8. LB medium (see Recipes)
9. Solid LB medium for agar plates (see Recipes)
Recipes
1. Kanamycin stock solution (50 mg/mL)
Dissolve 500 mg of kanamycin sulfate in 10 mL of deionized H2O and store 1 mL aliquots at -20 °C until use.
2. 5-aminolevulinic acid stock solution (50 mg/mL)
Dissolve 50 mg of 5-aminolevulinic acid hydrochloride in 1 mL of deionized H2O, sterilize the solution by filtration through a 0.22 μm membrane, and store at -20 °C until use.
3. Sucrose stock solution (50% w/w)
Dissolve 100 g of sucrose in 100 mL of deionized H2O, sterilize the solution by filtration through a 0.22 μm membrane, and store at 4 °C until use.
4. 0.1 M CaCl2 solution
Weigh 1.11 g of CaCl2 and add deionized H2O to 100 mL, sterilize the solution by filtration through a 0.22 μm membrane, and store at 4 °C until use.
5. PBS buffer
Dissolve one PBS tablet in 100 mL of deionized H2O, sterilize the solution by filtration through a 0.22 μm membrane, and store at 4 °C until use.
6. TBE electrophoresis buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris(hydroxymethyl)aminomethane | 89 mM | 10.8 g |
| Boric acid | 89 mM | 5.5 g |
| Ethylenediaminetetraacetic acid | 2 mM | 0.58 g |
| H2O | n/a | to 1 L |
Dissolve TBE electrophoresis buffer components in deionized H2O and store at room temperature until use.
7. Agarose 1%
Weigh 1 g of agarose and add TBE electrophoresis buffer (see Recipe 6) to 100 mL. Heat the resulting mixture in a boiling water bath until the agarose has completely dissolved and a homogeneous liquid has been obtained. To create the gel, pour into a mold the solution that has been cooled to 45–50 °C.
8. LB medium
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Peptone | 1% | 10 g |
| Yeast extract | 0.5% | 5 g |
| NaCl | 0.5% | 5 g |
| Kanamycin (optional) | 50 µg/mL | 1 mL of stock solution |
| 5-aminolevulinic acid (optional) | 50 µg/mL | 1 mL of stock solution |
| H2O | n/a | to 1 L |
Dissolve liquid LB medium components in distilled H2O and autoclave the resulting mixture at 121 °C for 45 min. Note that 5-aminolevulinic acid and kanamycin should be added after autoclaving when the medium has been cooled to 40–45 °C.
9. Solid LB medium for agar plates
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Peptone | 1% | 10 g |
| Yeast extract | 0.5% | 5 g |
| NaCl* | 0.5% or 0% | 5 g |
| Agar | 1.5% | 15 g |
| Kanamycin (optional) | 50 µg/mL | 1 mL of stock solution |
| 5-aminolevulinic acid (optional) | 50 µg/mL | 1 mL of stock solution |
| Sucrose (optional)* | 10% | 200 mL of stock solution |
| H2O | n/a | to 1 L |
* If the medium is to be supplemented with sucrose, do not add NaCl.
Add distilled H2O to solid LB medium components and autoclave the resulting mixture at 121 °C for 45 min. Note that sucrose, 5-aminolevulinic acid, and kanamycin should be added after autoclaving when the medium has been cooled to 40–45 °C. Use 20 mL of the medium per 90 × 15 mm Petri plate.
Laboratory supplies
1. Petri dishes, 90 mm × 15 mm (SPL life sciences, catalog number: 10093)
2. Microcentrifuge tubes, 1.5 mL (Biologix, catalog number: 80-1500)
3. PCR tubes, 0.2 mL (Accumax, catalog number: APC-001)
4. Pipette tips (Tarsons, catalog numbers: 521000, 521010, 521020)
5. Cell spreader (SPL life sciences, catalog number: 90050)
6. Centrifuge sterile tube, 15 mL (Accumax, catalog number: ACT-15-R-S)
7. Omnifix Luer Lock Solo syringes, 5 mL (B. Brown)
8. PES sterile syringe filter (0.22 μm) (Alwsci, catalog number: C0000502)
9. Cellulose acetate filters (0.45 μm) (Sartorius, catalog number: 11106-25-N)
10. Syringe filter holders (25 mm) (Sartorius, catalog number: 16517-E)
Note: Sterilize 1.5 mL microcentrifuge tubes and pipette tips by autoclaving at 121 °C for 45 min to work with cell cultures. Assemble the filter holder and membrane filter, wrap the assembled filters in aluminum foil, and sterilize in an autoclave at 121 °C for 45 min. Do not overtighten the filter holder before sterilization to avoid damage to the filter during heating. Tighten it just before transferring the cells to the filter.
Equipment
1. Pipettes (Thermo Fisher Scientific, catalog numbers: 4641010N, 4641040N, 4641070N, 4641100N)
2. Water Purification Systems Proseers (Innova, catalog number: NOVAU)
3. T100 PCR thermal cycler (Bio-Rad, catalog number: 1861096)
4. PowerPac basic power supply (Bio-Rad, catalog number:16450504)
5. Horizontal electrophoresis camera (Helicon, catalog number: SE-2)
6. ChemiDoc EZ imaging system (Bio-Rad, catalog number: 12003153)
7. UV transilluminator, wavelength 302 nm (Analytik Jena US, catalog number: 849-20017-4)
8. Thermostat ThermoMixer C (Eppendorf, catalog number: 5382000015)
9. Water bath (Lab Companion, model name: BW3-05G)
10. High-speed freezing centrifuge (Sigma, model name: 2-16KL)
11. Centrifuge MiniSpin (Eppendorf, catalog number: 022620100)
12. UV-visible absorption spectrometer (Agilent Technologies, model number: 8453) equipped with a fiber-optic ultra-micro measuring cell TrayCell (Hellma, catalog number: 105.800-UVS)
13. Autoclave (Tuttnauer, model type: 2540 ML)
Software and datasets
1. Primer-BLAST [7]
Procedure
A. Obtaining the genetic constructs for recombination by common molecular cloning methods
1. According to the recommendations mentioned in General note 1, select the primers for amplifying the fragments that flank the target deletion region (Figure 2). The outer primers must contain restriction sites along which the recombining DNA fragment will be transferred to the vector. For the current protocol, these are XbaI for the Up_F primer and Psp124BI (SacI) for the Down_R primer. The inner primers (Up_R and Down_F for the current protocol) must frame the deletion region and must be partially or fully complementary to each other. Use Primer-BLAST for primer designing (see General note 2).
Note: Any other suitable restriction sites for cloning in the pRE118 vector can be selected. However, it is important to make sure that the chosen sites are not present inside the fragments to be cloned.
2. Culture S. proteamaculans cells in LB medium overnight (16–18) at 30 °C under intensive stirring conditions (200–220 rpm).
3. Centrifuge 1 mL of the culture at 3,000× g for 10 min. Resuspend the cell pellet in 100 μL of PBS buffer.
4. Heat the suspension at 95 °C for 10 min and centrifuge at 12,000× g for 10 min. Use the supernatant as a PCR template.
5. Mix the reaction components as indicated in Table 1. Prepare two separate PCR mixtures for upstream and downstream fragments using the primer pairs Up_F/Up_R and Down_F/Down_R (Figure 2), respectively, and using S. proteamaculans lysate as a template. Carry out the reaction in accordance with Table 2.
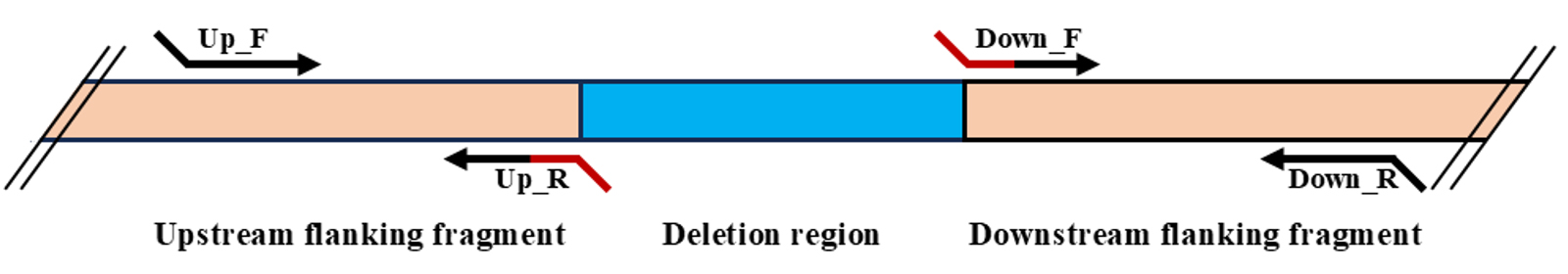
Figure 2. Scheme of primer selection for cloning of fragments for recombination from S. proteamaculans in the case of a deletion mutation. The fragment to be deleted is indicated in blue, and the recombining fragments are indicated in light orange. The arrows indicate the primers, and the overlapping region is shown in red.
Table 1. Components for the PCR mixture
| Component | Volume per reaction | Final concentration |
|---|---|---|
| Polymerase | 0.2 μL | 0.04 U/μL |
| dNTP mix | 4 μL | 0.2 μM (for each dNTP) |
| Polymerase buffer (×10) | 2.5 μL | 1× |
| Forward primer | 1 μL | 0.4 μM |
| Reverse primer | 1 μL | 0.4 μM |
| Template | 1 μL | n/a |
| ddH2O | to 25 μL | n/a |
Table 2. PCR cycling parameters
| Step | Temperature (°C) | Duration | No. of cycles |
| Initial denaturation | 94 | 3 min | 1 |
| Denaturation | 94 | 30 s | 25 |
| Annealing | Ta* | 30 s | |
| Extension | 72 | 1 min per kb | |
| Final extension | 72 | 4 min | 1 |
* The appropriate annealing temperature (Ta) should be estimated for each primer pair.
5. Purify both fragments from the PCR mixtures using 1% agarose gel electrophoresis in TBE electrophoresis buffer followed by gel extraction with a gel purification kit.
6. Measure the absorbance at 260 nm of both PCR products and estimate the DNA concentration. Mix them at a 1:1 molar ratio. Use 16 μL of the resulting mixture as a template for the PCR. Other components should be added according to Table 1. Use the outer primers Up_F and Down_R. Carry out PCR according to Table 2. This will generate the DNA fragment with the target deletion.
7. Purify the PCR product as described in step A4.
8. Digest approximately 3 μg of the pRE118 vector and the purified PCR product with restriction enzymes. Use 40 U of each enzyme per reaction.
9. Purify the digested pRE118 vector and PCR product as described in step A4.
10. Measure the absorbance of the purified digested vector and PCR product at 260 nm and estimate the DNA concentration.
11. Mix the purified digested pRE118 vector and PCR product in a 1:1 molar ratio. Add 200 U of T4 DNA ligase to 8 μL of the resulting mixture and incubate at 16 °C for 2 h or at 4 °C overnight.
12. Transform the E. coli CC118 (λpir) cells with 5 μL of the ligation mixture using the calcium method according to the standard protocol [8].
13. Grow an overnight culture of transformed bacteria in 2 mL of LB with kanamycin.
14. Purify the plasmid from the overnight culture using the plasmid DNA isolation kit according to the instructions. Perform the sequence analysis of the generated plasmid.
B. Transformation by conjugation
1. Introduce the generated pRE118 derivate in E. coli ST18 cells using the calcium transformation method [8].
2. Prepare the donor strain cells. Inoculate E. coli ST18, bearing the pRE118 derivate, in 5 mL of LB medium supplemented with kanamycin and 5-aminolevulinic acid and cultivate overnight at 37 °C under intensive stirring conditions (200–220 rpm).
3. Prepare the recipient strain cells. Inoculate S. proteamaculans in 5 mL of LB medium and cultivate overnight at 30 °C under intensive stirring conditions (200–220 rpm).
4. Dilute 100 times the overnight cultures of both strains (add 50 µL of the overnight culture to 5 mL of fresh medium) and continue the cultivation under the corresponding conditions up to OD600 0.6–0.8.
5. Centrifuge 3 mL of E. coli ST18 culture and 1 mL of S. proteamaculans culture at 8,500× g for 1 min at 4 °C and remove the supernatant.
6. Resuspend each cell pellet in 1 mL of PBS buffer solution.
7. Combine the cell suspensions of the donor strain E. coli ST18 and the recipient strain S. proteamaculans at a 1:1 volume ratio. Connect a sterile syringe to a sterile filter holder that contains a 0.45 μm cellulose acetate membrane filter. Transfer 4 mL of the mixture onto the filter using the syringe. Place the filter on an LB agar plate (without 5-aminolevulinic acid).
Note. A detailed description of how to build and use a mating filter apparatus can be found elsewhere [4].
8. Incubate the plate at 30 °C for 24 h.
С. Selection
1. Wash the grown cells from the cellulose acetate filter with 500 µL of LB medium.
2. Make a series of dilutions (10, 100, 1,000, and 10,000 times) and sow 100 µL of each obtained suspension onto LB agar plates containing kanamycin. Incubate the plates overnight at 30 °C. For further work, select a plate with single colonies.
3. Select a colony, transfer it to liquid LB medium containing kanamycin, and culture overnight at 30 °C with intensive stirring.
4. Sow 100 μL of the resulting culture suspension onto an LB agar plate containing 10% sucrose. Incubate overnight at 30 °C.
5. Select several clones (approximately 15–20) and transfer them simultaneously to an LB agar plate with kanamycin and to an LB agar plate with sucrose.
6. Choose all the clones that grow on sucrose but not on kanamycin.
7. Design the PCR primers to confirm the presence of target deletion on the S. proteamaculans chromosome. For primer design, use Primer-BLAST (see General note 2).
Note. These primers are usually designed outside the upstream and downstream flanking deletion fragments.
8. Mix the PCR components in accordance with Table 1, using the cell lysate of the clones chosen in step C6 as PCR template and the primers designed in step C7. Perform PCR in accordance with the program described in Table 2.
9. Analyze the amplified fragments by electrophoresis on a 1% agarose gel and select three clones with the correct fragment length.
10. Purify the fragments of selected clones using 1% agarose gel electrophoresis in TBE electrophoresis buffer, followed by gel extraction with a gel purification kit.
11. Sequence the purified fragments and ensure that they contain the desired mutation.
Validation of protocol
This protocol has been used and validated in the following research article:
Tsaplina et al. [1] Protealysin targets the bacterial housekeeping proteins FtsZ and RecA. International Journal of Molecular Sciences (Figure 2B).
General notes and troubleshooting
General notes
1. The current protocol can be used for obtaining deletion and, potentially, insertion and point mutations. In all cases, a plasmid for recombination should contain the region to be modified that is flanked by non-modified fragments. The length of the DNA fragment required for successful homologous recombination depends on the size of the region to be modified. For deletion/insertion mutations, if the modified region is up to 1,000 base pairs (bp), the upstream and downstream fragments of this region should be approximately 400–500 bp each. If the region increases, the length of the flanking fragments should be expanded by 50–100 bp for each additional 1,000 bp of the region. For point mutations, the fragment used for recombination should be approximately 1,000 bp long, and the target mutation should be located approximately in the middle of this fragment [4].
2. Primer design using Primer-BLAST tool
A detailed description of the Primer-BLAST tool application can be found in [7]. Additionally, the tool manual can be found at www.ncbi.nlm.nih.gov/tools/primer-blast/search_tips.html.
Primer design features to obtain the genetic constructs for recombination:
a. The inner primers, which flank the region that should be deleted through recombination (here referred to as Up_R and Down_F), should be 26–30 nucleotides in length. Note that these primers should overlap, and the size of the overlapping region should be at least half the length of the primers: more than 13–15 nucleotides.
b. The design of the outer primers (here referred to as Up_F and Down_R) should be carried out in two stages. At the first stage, using the preselected sequences of the inner primers and Primer-BLAST tool, select the sequences of the outer primers among variants suggested by the tool. At the second stage, add to the 5′-end of these sequences the selected restriction sites and additional 3–4 nucleotides (the latter to ensure correct digestion by restriction enzymes). The updated sequences of the outer primers should be checked again using Primer-BLAST tool (in pair with the corresponding inner primers) for specificity to the intended matrix, as well as absence of hairpins and dimers. Make the adjustments if necessary and check the primer pairs again.
c. Additionally, since the outer primers will be used together during the generation of the DNA fragment with the target deletion, this pair should also be checked for specificity to the intended matrix and absence of dimers using the Primer-BLAST tool.
Primers used for the deletion of the protealysin operon or the emfourin gene can be found in Table 1 in the previous work [1].
Troubleshooting
Problem 1: No PCR product is obtained.
Possible cause: Estimated annealing temperature is not optimal.
Solution: Choose the proper annealing temperature empirically using a temperature gradient.
Problem 2: The reaction mixture volume for the pRE118 restriction is too high for further purification of the digested plasmid.
Possible cause: pRE118 is a low-copy plasmid; therefore, the plasmid concentration after purification from bacteria may be low.
Solution: Concentrate the plasmid using ethanol precipitation according to the standard protocol described in the Plasmid Miniprep manual.
Problem 3: Unable to pick a single colony while growing cells on the sucrose medium—no colonies are formed, or the bacterial culture is too dense.
Possible cause: Too low or too high efficiency of the second round of the recombination.
Solution: If no colonies are formed, sow more than 100 mL of the overnight cell suspension onto LB agar; if the culture is too dense, use serial dilutions of the overnight cell suspension to generate a culture with single colonies, as was used for kanamycin selection.
Acknowledgments
Specific contributions of each author: Conceptualization, D.I.V.; Investigation, Ch.K.N., K.M.A., Writing—Original Draft, Ch.K.N.; Writing—Review & Editing, K.A.A., D.I.V., K.M.A.; Funding acquisition, D.I.V.; Supervision, D.I.V.
This work was supported by the Russian Science Foundation (project no. 24-24-00122). This protocol was adapted from the previous work [1].
Competing interests
The authors declare no conflicts of interest.
References
- Tsaplina, O., Khaitlina, S., Chukhontseva, K., Karaseva, M., Demidyuk, I., Bakhlanova, I., Baitin, D., Artamonova, T., Vedyaykin, A., Khodorkovskii, M., et al. (2022). Protealysin Targets the Bacterial Housekeeping Proteins FtsZ and RecA. Int J Mol Sci. 23(18): 10787. https://doi.org/10.3390/ijms231810787
- Edwards, R. A., Keller, L. H. and Schifferli, D. M. (1998). Improved allelic exchange vectors and their use to analyze 987P fimbria gene expression. Gene. 207(2): 149–157. https://doi.org/10.1016/s0378-1119(97)00619-7
- Thoma, S. and Schobert, M. (2009). An improved Escherichia coli donor strain for diparental mating. FEMS Microbiol Lett. 294(2): 127–132. https://doi.org/10.1111/j.1574-6968.2009.01556.x
- Hmelo, L. R., Borlee, B. R., Almblad, H., Love, M. E., Randall, T. E., Tseng, B. S., Lin, C., Irie, Y., Storek, K. M., Yang, J. J., et al. (2015). Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc. 10(11): 1820–1841. https://doi.org/10.1038/nprot.2015.115
- Kostenko, Ju. G., Spitsyna, D. N., et al. (2001). Strain Serratia proteamaculans 94 as producer of collagenase. RU 2175350 C1, October 27, 2001.
- Herrero, M., de Lorenzo, V. and Timmis, K. N. (1990). Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J Bacteriol. 172(11): 6557–6567. https://doi.org/10.1128/jb.172.11.6557-6567.1990
- Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S. and Madden, T. L. (2012). Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. 13(1): 134. https://doi.org/10.1186/1471-2105-13-134
- Mamiatis, T., Fritsch, E. F., Sambrook, J. and Engel, J. (1985). Molecular cloning–A laboratory manual. New York: Cold Spring Harbor Laboratory. Acta Biotechnol. 5(1): 104–104. https://doi.org/10.1002/abio.370050118
Article Information
Publication history
Received: Jun 23, 2025
Accepted: Aug 12, 2025
Available online: Aug 28, 2025
Published: Sep 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Chukhontseva, K., Karaseva, M. A., Komissarov, A. and Demidyuk, I. (2025). Editing the Serratia proteamaculans Genome Using the Allelic Exchange Method. Bio-protocol 15(18): e5448. DOI: 10.21769/BioProtoc.5448.
Category
Microbiology > Heterologous expression system
Molecular Biology > DNA > Mutagenesis
Microbiology > Microbial genetics > Recombination
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.