- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Genetic Engineering of Humanized Telomere Mice
Published: Vol 15, Iss 18, Sep 20, 2025 DOI: 10.21769/BioProtoc.5445 Views: 2587
Reviewed by: Alka MehraAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2025

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Telomere shortening is a hallmark of human aging, and telomerase regulation plays a critical role in cellular proliferation and replicative senescence. In human cells, telomere length imposes a limit on proliferative potential, a phenomenon known as the Hayflick limit. However, species-specific differences in telomere dynamics and telomerase regulation between humans and mice present challenges to using mice as accurate models for human telomere-related research. To address this limitation, we engineered a humanized telomerase gene (hmTert) in mice by replacing the non-coding sequences within the mouse Tert locus (mTert) with corresponding regulatory sequences from the human TERT gene. Breeding of these genetically modified mice resulted in progressive telomere shortening over successive generations, ultimately reaching human-like lengths (below 10 kb). This protocol outlines the development of this humanized telomere mouse model, referred to as HuT mice, offering a robust platform for studying human telomere biology and aging-related diseases.
Key features
• This protocol describes methods to increase the success rates of knocking in large genomic fragments (~47 kb) by integrating CRISPR-Cas9 with homologous recombination.
• It enables precise engineering of a humanized telomerase gene (hmTert), faithfully recapitulating human TERT regulation and telomere length dynamics in mice.
Keywords: Genetic engineeringGraphical overview
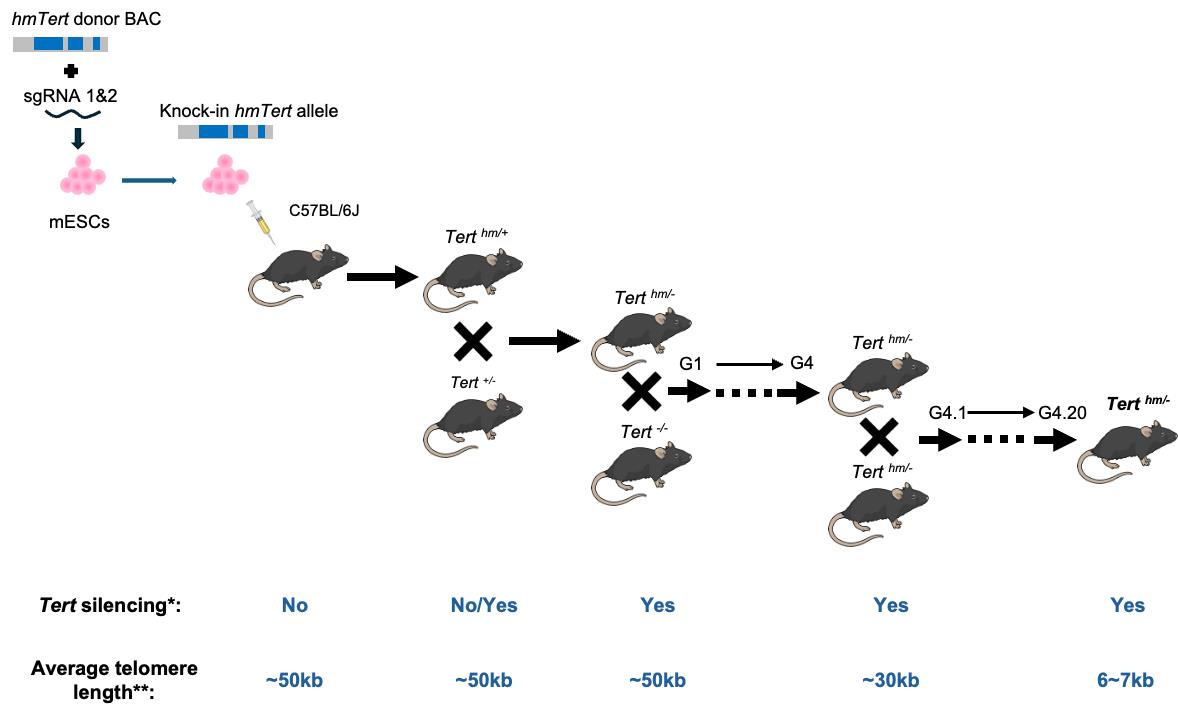
*No indicates that Tert expression is found in many mouse adult tissues; Yes indicates that Tert mRNA is not detected in all adult tissues except for testis, ovary, and thymus.
**Telomere length is measured in splenocytes by Flow-FISH.
Background
Telomeres, repetitive sequences at chromosome ends, are essential for maintaining genomic stability. In most human somatic cells, telomeres progressively shorten with each cell division, a process counteracted by the telomerase enzyme complex, which consists of the TERT protein and TERC RNA subunits. Most human adult tissues contain low or no telomerase, and telomere shortening eventually triggers cell cycle arrest, leading to replicative senescence or the Hayflick limit. Telomerase elongates telomeres, delaying senescence. However, telomere homeostasis and telomerase regulation differ markedly between species. For instance, while telomere lengths in human tissues typically range from 10 to 15 kb, laboratory mouse strains, such as C57BL/6, often exhibit telomeres over ~50 kb. Moreover, telomerase remains active in most adult mouse tissues, unlike in humans, where it is largely repressed [1]. As a result, mouse cells do not exhibit replicative senescence or the Hayflick limit.
Telomerase knockout (KO) mice, including those deficient in Tert or Terc, exhibit limited viability by the sixth generation. Despite lacking telomerase, these mice maintain average telomere lengths exceeding 25 kb—substantially longer than those in humans [2]. Importantly, early-generation telomerase-null mice do not display overt phenotypic abnormalities [3], suggesting that mice have considerable telomere reserves capable of sustaining multiple generations before critical shortening occurs. These species-specific differences significantly limit the usefulness of conventional mice as models for studying human telomere biology and aging-related diseases.
Our laboratory focuses on the regulation of the TERT gene, which encodes the rate-limiting catalytic subunit of telomerase. We have identified key regulatory regions within the human TERT gene—including the 5′ intergenic region, intron 2, and intron 6—that modulate TERT expression [4]. Replacing those genetic contexts on the mouse locus with human counterparts disrupted Tert repression in differentiated mouse ESCs [5]. To investigate the role of these elements in vivo, we engineered a humanized telomerase gene (hmTert) in mouse embryonic stem cells (mESCs) by replacing the corresponding non-coding regions in the mouse Tert locus with a 47 kb hybrid sequence from a bacterial artificial chromosome (BAC) containing the human regulatory elements [6–8]. Initial attempts to introduce this large fragment via homologous recombination were hindered by low efficiency and incomplete integration. To overcome this, we employed CRISPR-Cas9 technology to enhance homologous recombination efficiency, increasing the success rate from 0.05% to 11%, in mESCs [8]. The hmTert gene in mESCs accurately recapitulated human TERT regulation during in vitro differentiation [5]. We subsequently established a mouse strain carrying germline hmTert alleles. Investigation of these mice confirmed human-like Tert regulatory patterns. With successive intercrossing of heterozygous mice, telomere lengths gradually shortened across generations, eventually reaching a human-like length of less than 10 kb. Despite their much shorter telomeres, Terth/h and Terth/- mice maintained normal body weight and tissue homeostasis. These mice, which exhibit physiologically relevant telomerase regulation, serve as a powerful model for studying human telomere biology, aging, and associated diseases.
This protocol outlines the key steps for generating and validating the humanized telomere mouse model, known as HuT mice.
Materials and reagents
Biological materials
1. Mouse embryonic stem cells G4 [9]
2. Drug-resistant (DR4) feeder cells: mouse embryonic fibroblast cells derived from E13.5–14.5 embryos and treated as previously described [7]
3. pSpCas9 (BB)‐2A‐GFP (PX458, Addgene #48138), a gift from Feng Zhang [10]
Reagents
1. 7-AAD (Thermo Fisher, catalog number: 00-6993-50)
2. Agarose (Invitrogen, catalog number: 16500-500)
3. LB broth (Affymetrix, catalog number :75852)
4. Agar (VWR, catalog number: J637)
5. DMEM medium (HyClone, catalog number: SH30243)
6. Penicillin/Streptomycin solution 100× (Gemini, catalog number: 400-109-100)
7. NEAA 100× (HyClone, catalog number: SH30238.01)
8. Fetal bovine serum (FBS) (Atlanta, catalog number: S12450)
9. RPMI medium (HyClone, catalog number: SH30027.LS)
10. Lipofectamine 2000 (Thermo Fisher, catalog number: 11668027)
11. Ladderman DNA Labeling kit (Takara, catalog number: 6046)
12. Isopropanol (MP Biomedical, catalog number: 0219400690)
13. Ethanol absolute, pure (200 Proof) (Avantor, catalog number: 71001-754)
14. Chloroform (MP Biomedical, catalog number: 0219400225)
15. TRI reagent (Ambion, catalog number: 155960118)
16. PowerUpTM SYBRTM Green Master Mix for qPCR (Thermo Fisher, catalog number: A25778)
17. Deoxynucleotide (dNTP) solution mix (NEB, catalog number: N0447)
18. QIAEX II Gel Extraction kit (Qiagen, catalog number: 20021)
19. SuperScriptTM III reverse transcriptase (Thermo Fisher, catalog number: 18080044)
20. TaKaRa LA Taq® DNA polymerase (Takara, catalog number: RR002B)
21. Reprobing charged Nylon (GVS, catalog number: 1226556)
22. Restriction digestion enzyme: all restriction enzymes were purchased from New England Biolabs (NEB)
23. NEB® 5-alpha Competent E. coli (NEB, catalog number: C2987H)
24. Taq DNA polymerase with ThermoPol® buffer (NEB, catalog number: M0267L)
25. T4 polynucleotide kinase (NEB, catalog number: M0201S)
26. T4 DNA ligase (NEB, catalog number: M0202L)
27. TelC-FAM probe (PNA Bio, catalog number: F1001): a PNA probe for leading stand of telomere sequences (CCCTAA)3 conjugated with FAM
28. GenEluteTM Plasmid Miniprep kit (Sigma, catalog number: PLN70)
29. Wizard® Genomic DNA Purification kit (Promega, catalog number: A1125)
30. 32P-dCTP (PerkinElmer, catalog number: BLU513H250UC)
31. Ampicillin sodium salt (Fisher Scientific, catalog number: BP1760-5)
32. Puromycin dihydrochloride (Sigma, catalog number: P8833)
33. Ganciclovir (GCV) (Sigma, catalog number: SML2346)
34. Roche blocking reagent (Roche, catalog number: 11096176001)
35. Trypsin solution (HyClone, catalog number: SH30042)
36. GlutaMaxTM supplement (Gibco, catalog number: 35050061)
37. LIF recombinant mouse protein (Gibco, catalog number: A35935)
38. β-Mercaptoethanol (Sigma, catalog number: M6250)
Solutions
1. Ampicillin solution (see Recipes)
2. 50 mg/mL puromycin (see Recipes)
3. 10 mM GCV (see Recipes)
4. 10% blocking buffer (see Recipes)
5. LB medium (see Recipes)
6. 0.025% trypsin solution (see Recipes)
7. mESCs culture medium (see Recipes)
8. 0.5× TBE (see Recipes)
9. Denature buffer (see Recipes)
10. Neutralization buffer (see Recipes)
11. Church and Gilbert’s hybridization buffer (Church buffer) (see Recipes)
12. 20× SSC (see Recipes)
13. DNA wash solution (see Recipes)
14. Hank's balanced salt solution (HBSS) (see Recipes)
15. PBS (see Recipes)
16. Red blood cell (RBC) lysis buffer (see Recipes)
17. Hybridization buffer (see Recipes)
18. Wash solution I (see Recipes)
19. Wash solution II (see Recipes)
20. Maleic acid buffer (see Recipes)
Recipes
1. Ampicillin solution
A stock solution (100 mg/mL) is prepared by dissolving 100 g of ampicillin sodium salt into 10 mL of Milli-Q water and filtering it through a 0.22 μm filter. The stock solution is added to the LB medium at a final concentration of 100 μg/mL.
2. 50 mg/mL puromycin
A stock solution (50 mg/mL) is prepared by dissolving 0.5 g of puromycin dihydrochloride into 10 mL of Milli-Q water and filtering it through a 0.22 μm filter. The stock solution is added to the mESCs culture medium at a final concentration of 1.5 μg/mL.
3. 10 mM GCV
This stock solution is added to the mESCs culture medium at a final concentration of 50 μM.
4. 10% blocking buffer
Dissolve Roche blocking reagent in maleic acid buffer to a final concentration of 10% (w/v) by shaking and heating on a heating block.
5. LB medium
Dissolve 20 g of LB broth in 1 L of Milli-Q water and autoclave. Prior to autoclaving, add 15 g of agar to 1 L of LB medium to prepare the bacterial culture plate.
6. 0.025% Trypsin solution
Dilute 10 mL of 0.25% Trypsin in 40 mL of autoclaved PBS.
7. mESCs culture medium
| Reagent | Final concentration |
|---|---|
| FBS | 15% |
| GlutaMaxTM supplement (100×) | 1× |
| NEAA (100×) | 1× |
| LIF recombinant mouse protein | 10 ng/mL |
| β-mercaptoethanol | 55 µM |
| Penicillin/Streptomycin solution (100×) | 1× |
| DMEM medium | 85% |
8. 0.5× TBE
| Reagent | Final concentration |
|---|---|
| Tris base | 0.045 M |
| H3BO3 | 0.045 M |
| EDTA (pH 8.3) | 0.01 M |
9. Denature buffer
| Reagent | Final concentration |
|---|---|
| NaCl | 1.5 M |
| NaOH | 0.05 M |
10. Neutralization buffer
| Reagent | Final concentration |
|---|---|
| NaCl | 1.5 M |
| Tris base (pH 8.5) | 9.5 M |
11. Church and Gilbert’s hybridization buffer (Church buffer), pH 7.2
| Reagent | Final concentration |
|---|---|
| Na2HPO4 | 0.34 M |
| NaH2PO4 | 0.16 M |
| SDS | 7% (w/v) |
| EDTA | 0.001 M |
| BSA | 1% (w/v) |
12. 20× SSC, pH 7.0
| Reagent | Final concentration |
|---|---|
| NaCl | 3 M |
| Na3C6H5O7 | 0.3 M |
A series of SSC solutions used in the paper were diluted from 20× SSC with an appropriate volume of MilliQ water.
13. DNA wash solution
| Reagent | Final concentration |
|---|---|
| Na3C6H5O7 | 0.3 M |
| Ethanol | 10% (v/v) |
14. Hank's balanced salt solution (HBSS), pH 7.4
| Reagent | Final concentration |
|---|---|
| NaCl | 0.14 M |
| KCl | 0.005 M |
| CaCl2 | 0.001 M |
| MgSO4 | 0.0004 M |
| MgCl2 | 0.0005 M |
| Na2HPO4 | 0.0003 M |
| KH2PO4 | 0.0004 M |
| D-Glucose | 0.004 M |
| Na2CO3 | 0.004 M |
15. PBS, pH 7.2–7.4
| Reagent | Final concentration |
|---|---|
| NaCl | 0.137 M |
| KCl | 0.0027 M |
| Na2HPO4 | 0.01 M |
| KH2PO4 | 0.0018 M |
16. Red blood cell (RBC) lysis buffer, pH 7.4
| Reagent | Final concentration |
|---|---|
| NH4Cl | 155 M |
| KHCO3 | 10 M |
| EDTA | 0.1 M |
17. Hybridization buffer
| Reagent | Final concentration |
|---|---|
| Tris-HCl (pH 7.2) | 20 mM |
| Deionized formamide | 60% (v/v) |
| Blocking buffer | 0.5% (v/v) |
18. Wash solution I
| Reagent | Final concentration |
|---|---|
| Tris-HCl (pH 7.2) | 20 mM |
| Deionized formamide | 60% (v/v) |
| BSA | 0.1% (w/v) |
| Tween 20 | 0.1% (v/v) |
19. Wash solution II
| Reagent | Final concentration |
|---|---|
| BSA | 0.1% (w/v) |
| Tween 20 | 0.1% (v/v) |
| PBS | Add to final volume |
20. Maleic acid buffer (pH 7.5)
| Reagent | Final concentration |
|---|---|
| Maleic acid | 0.1 M |
| NaCl | 0.15 M |
Laboratory supplies
1. Cell culture multi-well plates (Greiner Bio-One, 12-well plate, 48-well pate, and 96-well plate)
2. 1.5 mL microcentrifuge tubes (Fisher Scientific, catalog number: 05-408-129)
3. 15 mL conical tubes (VWR, catalog number: 89039-658)
4. 50 mL conical tubes (VWR, catalog number: 21008-089)
5. 1 mL cryovials tubes (VWR, catalog number: I210-000001)
6. 5 mL serological pipettes (Greiner Bio-One, catalog number: 606107)
7. 10 mL serological pipettes (Greiner Bio-One, catalog number: 607107)
8. 0.1–10 μL pipette tips (Fisher Scientific, catalog number: 02-707-454)
9. 5–300 μL pipette tips (Fisher Scientific, catalog number: 02-707-447)
10. 100–1,250 μL pipette tips (Fisher Scientific, catalog number: 02-707-400)
11. 0.2 mL PCR tube (Corning, catalog number: PCR-02-A)
12. qRT-PCR plates (Thermo Fisher, catalog number: 4346907)
Equipment
1. PhosphorImager system (GE Healthcare)
2. CO2 incubator (Eppendorf, model: Galaxy 170S)
3. Incubator (VWR)
4. Incubator shaker (New Brunswick, model: I2400)
5. Water bath (Fisher Scientific, model: Isotemp215)
6. Centrifuge (Eppendorf, model: 5424R)
7. Laboratory centrifuge with rotors for 15- and 50-mL conical tubes (Eppendorf, model: 5804R)
8. Thermal cycler machine (Bio-Rad, model: C1000Touch)
9. Applied BiosystemsTM StepOnePlusTM Real-Time PCR System
10. CytoFLEX (Bechman Coulter)
11. Tissue cell culture hood
12. Liquid nitrogen (N2) tank
13. Freezer (-20 °C)
14. Refrigerator (2–8 °C)
15. Thermomixer (Eppendorf, model: 5436)
16. Hybridization incubator (Fisherbiotech)
17. UV-crossing link machine (Spectrolink, model: XL-1000)
18. pH meter (VWR, model: B10P)
19. Vortex (Fisher Scientific, catalog number: 12812)
Procedure
A. Single guide RNA (sgRNA) design
To design sgRNAs, we used the Synthego CRISPR Design Tool (https://www.synthego.com/) to identify guide RNA sequences that target regions near the 5′ and 3′ ends of the intended replacement region in the mouse genome (chr13:73769156-73787274), as illustrated in Figure 1. The input sequences for the design tool are provided in the supplemental file SEQ3 and SEQ4. sgRNA 1 (chr13:73770111-73770130) targeted the 5′ intergenic region (5′IR) between mTert and mCrr9 loci, while sgRNA 2 (chr13:73786833-73786852) targeted the distal end of mTert intron 6. Although the TERT locus exhibits conserved synteny between mouse and human, sharing flanking genes and orientation, the non-coding regions, including intergenic sequences, differ significantly. The selected sites were unique within the mouse genome and not conserved in the human genome, minimizing the risk of off-target effects. Upon entering the target sequences, the tool generated a ranked list of sgRNAs based on predicted on-target efficiency and off-target potential. Three potential sgRNAs with the highest scores are presented in Table S1. We selected sgRNA oligonucleotides after careful evaluation of these scores to ensure both specificity and efficiency.
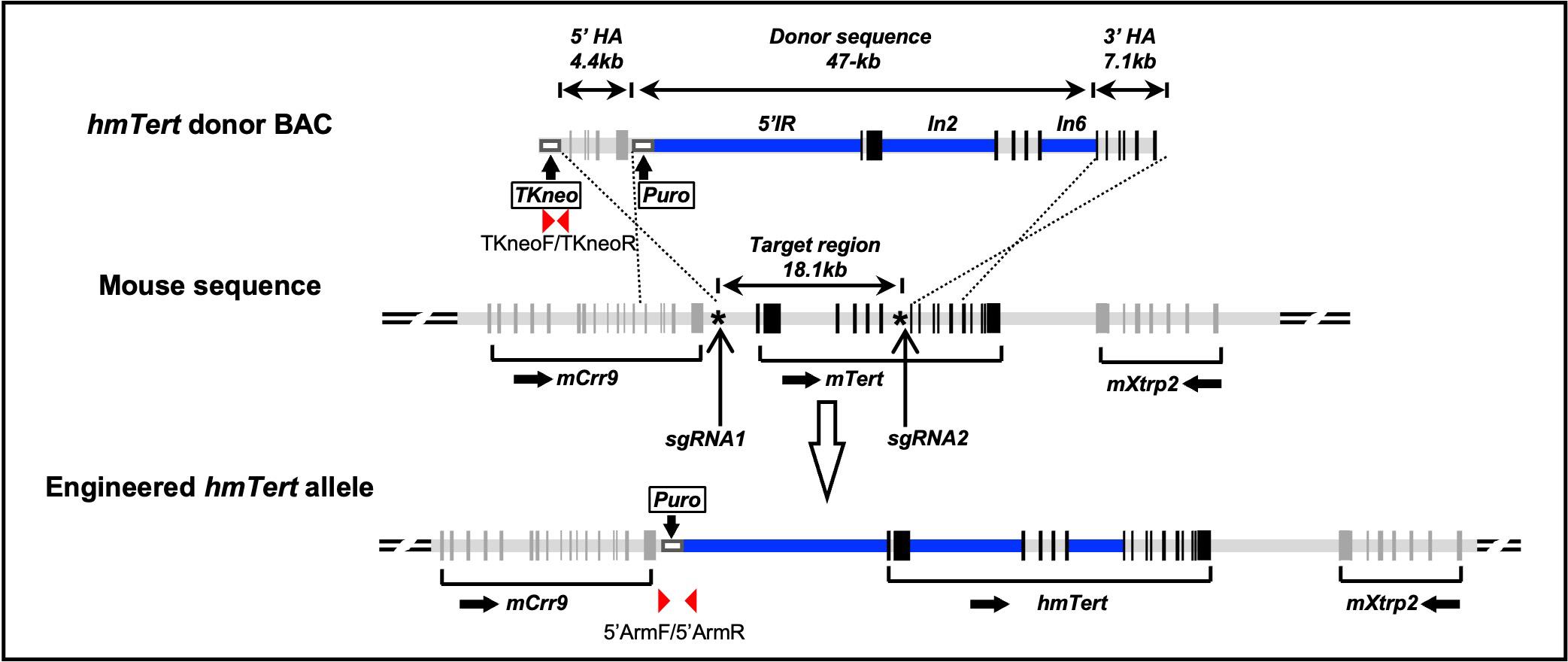
Figure 1. Schematic representation of the donor bacterial artificial chromosome (BAC) construct and engineering strategy for the hmTert allele. The donor BAC contained the mTert gene components (light grey bars). Regions within mTert locus, the 5′ intergenic region (IR), intron 2 (In2), and intron 6 (In6), were substituted with corresponding sequences from the human TERT gene, shown as blue bars. The donor sequence spanned 47 kb and was flanked by 5′ and 3′ homologous arms (HAs). The 5′ HA is a 4.4 kb sequence from the 3′ end of mCrr9 gene, whereas the 3′ HA is a 7.1 kb region spanning exons 7–12 of the mTert gene. In addition, a TKneo and a puromycin-resistant cassette (white boxes) were inserted upstream and downstream of 5′ HA, respectively. The puromycin cassette was surrounded by a pair of lox511 sites and was removed via transient transfection of a Cre expression plasmid in the mouse embryonic stem cells (mESCs) after homologous recombination. The target region was an 18.1 kb region from 5′IR to intron 6 of the mTert gene. Asterisk (*) represents Crispr/Cas9-sgRNAs targeting sites. Black vertical bars represent exons. PCR primers are indicated by red arrowheads.
B. sgRNA plasmid cloning
1. Digest plasmid PX458 (Figure S1) [10] as shown in Table 1 (time: 6 h):
Table 1. Restriction enzyme reaction
| Components | Amount |
|---|---|
| 10× NEB buffer 2.1 | 5.0 μL |
| PX458 | 2.0 μg |
| BbsI | 2.5 μL |
| ddH2O | volume to 50 μL |
| Total | 50 μL |
Perform digestion at 37 °C for 4 h. A correctly digested PX458 backbone has two fragments of 9.3 kb and 22 bp. The 9.3 kb fragment is isolated using QIAEX II Gel Extraction kit and eluted with 20 μL of water for a target concentration of 50 ng/μL.
2. Double-stranded oligos were produced by annealing forward and reverse oligos for sgRNA1 or sgRNA 2, as listed in Table 2. Oligos sequences are listed in Table S2 (time: 40 min).
Table 2. sgRNA targeting site sequences
| sgRNA targeting site | Sequence (5′-3′) | Oligonucleotides |
|---|---|---|
| sgRNA1 | AAGGATGAGGTTGGGCCAAT | sgRNA1-U/ sgRNA1-B |
| sgRNA2 | TCTGCAATGGCGTGGTCCCA | sgRNA2-U/ sgRNA2-B |
Annealing reactions were set up in 0.2 mL PCR tubes as shown in Table 3.
Table 3. sgRNA annealing reaction mix
| Components | Amount |
|---|---|
| Upper oligonucleotide (100 μM) | 1.0 μL |
| Bottom oligonucleotide (100 μM) | 1.0 μL |
| 10× T4 DNA ligation buffer | 1.0 μL |
| T4 polynucleotide kinase | 0.5 μL |
| ddH2O | 6.5 μL |
| Total | 8 μL |
Annealing was done using the following program (Table 4) in a thermocycler:
Table 4. sgRNA annealing reaction
| Temperature | Time |
|---|---|
| 37 °C | 30 min |
| 95 °C | 5 min |
| ramping down to 25 °C at 5 °C/min | |
| 4 °C | Hold |
3. Assemble PX458-sgRNA: set up assembling reaction in a 0.2 mL PCR tube as shown in Table 5 (time: 16 h):
Table 5. Fragment assembling reaction
| Components | Amount |
|---|---|
| Digested PX458 (50 ng/μL) | 0.5 μL |
| Annealed oligonucleotide (1:250 dilution) | 0.5 μL |
| 10× T4 DNA ligation buffer | 1.0 μL |
| T4 DNA ligase | 0.25 μL |
| ddH2O | 7.75 μL |
| Total | 10 μL |
Run the reaction at 16 °C overnight in the thermocycler.
4. Transform 5 μL of the ligated product from step B3 using NEB Chemical Competent DH5α E. coli according to the manufacturer’s protocol. Upon transformation, spread all competent cells onto LB plates containing 100 μg/mL ampicillin (time: 1 h).
5. Bacterial colony identification: Culture three colonies in 3 mL of LB medium with 100 μg/mL of ampicillin. Shake overnight at 37 °C. Isolate plasmid DNAs from the cultures using the GenEluteTM Plasmid Miniprep kit according to the manufacturer’s instructions. Plasmids were digested with BbsI and AgeI to identify and confirm the correct constructs (Figure S1) (time: 3 h). Set up the digestion reaction as shown in Table 6.
Table 6. Restriction enzyme digestion reaction
| Components | Amount |
|---|---|
| 10× NEB buffer 1.1 | 1 μL |
| BbsI | 0.4 μL |
| AgeI | 0.2 μL |
| PX485-sgRNA plasmid DNA | 1.0 μL |
| ddH2O | 7.4 μL |
| Total | 10 μL |
Incubate at 37 °C for 2 h in a water bath. PX458-sgRNA plasmids were digested to a 9.3 kb band, while the parental PX458 plasmid was digested into 8.3 kb and 971 bp bands (Figure 2).
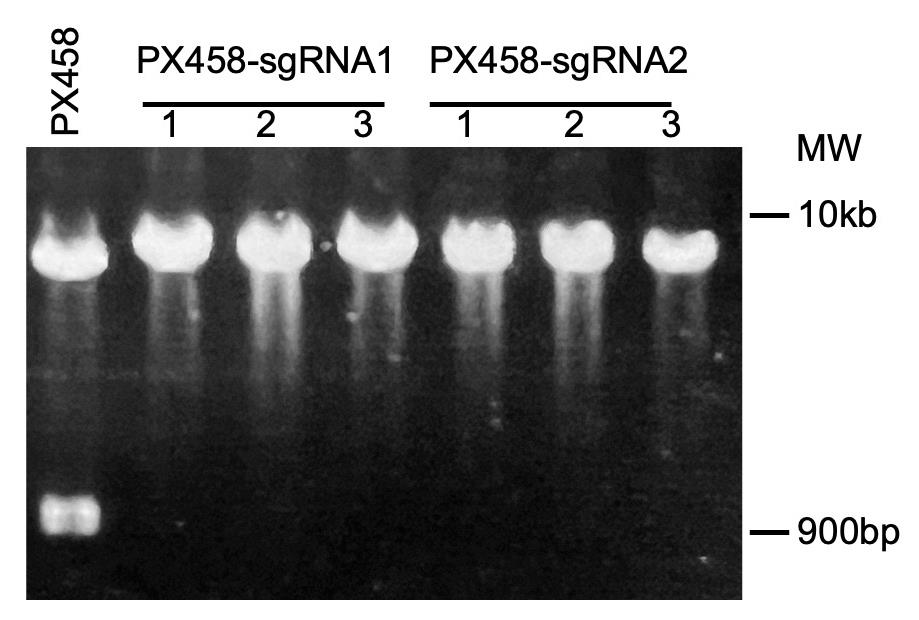
Figure 2. Identification of sgRNA plasmids. Correct PX485-sgRNA plasmids were identified and digested with BbsI and AgeI. MW: molecular weight markers.
6. PX458-sgRNA plasmids were validated by Sanger sequencing using the U6-Fwd primer: 5′-GACTATCATATGCTTACCGT-3′.
C. Homologous recombination in mouse ESCs (total time: 24 d)
hmTert donor BAC was co-transfected with two sgRNA-expressing plasmids into mESCs (G4) [9]. The construction of the hmTert donor BAC has been previously described [5]. Briefly, in addition to incorporating the human TERT regulatory sequences specified above, we introduced the following modifications: (1) insertion of a puromycin resistance (Puro) cassette at the 3′ end of mCrr9; (2) replacement of mCrr9 exons 1–12 with a thymidine kinase-neomycin resistance (TK-neo) selection cassette; and (3) deletion of sequences downstream of mTert exon 12. These modifications collectively generated extended homologous recombination arms, as indicated in Figure 1, with their sequences provided in the supplemental file SEQ1 and SEQ2.
1. Day 1: Seed 3.5 × 106 mESCs into each well of a 12-well-plate with DR4 feeder cells. Transfect cells with 0.25 μg of PX458-sgRNA1 and PX458-sgRNA2 plasmids, together with 1.0 μg of donor BAC construct using lipofectamine 2000 according to the manufacturer’s instructions. Eight hours later, passage transfected mESCs into 6-well plates. A brief protocol of cell passaging is as follows:
a. For a well in a 12-well plate, aspirate the medium and wash the cells twice with 2 mL of PBS.
b. Then, add 0.25 mL of prewarmed 0.025% Trypsin solution and incubate for approximately 2 min or until the cells are uniformly dispersed into small clumps.
c. Next, add 2.5 mL of mESCs culture medium to neutralize the trypsin, and transfer the cells to a new 6-well plate pre-coated with fresh DR4 feeder cells.
Note: DR4 feeder cells are mouse embryonic fibroblasts (MEFs) derived from 13.5-day-old mouse embryos and inactivated by X-ray irradiation. These cells are resistant to multiple antibiotics, including puromycin, making them suitable for selection-based experiments. In our setup, DR4 feeder cells were seeded at a density of 5 × 104 cells per well in a 12-well plate. The optimal feeder cell density corresponds to approximately 50% confluence of the plate surface area, which provides sufficient support for mESC growth without overcrowding. Feeder cells are maintained in standard MEF culture medium and should be used within a few days post-irradiation to ensure viability and support function.
2. Day 2: Select mESCs with 1.5 μg/mL puromycin for two days.
3. Day 4: Wash mESCs once with PBS and culture with fresh mESCs culture medium.
4. Day 6: Select mESCs with 1.5 μg/mL puromycin for two days again.
5. Day 8: Wash mESCs once with PBS and culture with fresh mESCs culture medium.
6. Day 10: Select 800 puromycin resistant colonies and seed them individually into 96‐well plates with fresh DR4-feeder cells.
7. Day 11: Culture mESCs in culture medium containing 1.5 μg/mL puromycin.
8. Day 13: Wash cells once with PBS and culture them in fresh mESCs culture medium.
9. Day 14: Culture puromycin-resistant colonies in mESCs culture medium containing 50 μM GCV for two days.
Note: Following GCV selection, many colonies were eliminated. GCV served as a negative selection agent by killing cells that retained the TK-neo cassette, indicating random integration of the donor BAC at non-targeted chromosomal sites.
10. Day 16: Wash surviving mESC colonies (approximately 5,000–10,000 mESCs of each colony) once with PBS and passage them into 96‐well plates coated with fresh DR4-feeder cells.
11. Expand surviving colonies for further analysis. The duration of colony expansion depends on the initial size of the colony when picked from the primary screening dish, typically requiring 4–6 days to reach sufficient confluence.
Note: Cells were split into 48-well plates (for frozen) and 24-well plates (for genomic DNA).
D. Identification of recombinants
1. PCR identification: extract genomic DNAs from puromycin- and GCV-selected mESC colonies using Wizard® Genomic DNA Purification kit (time: 9 h).
a. 5′ end recombination was identified by PCR using primers (5′ArmF/5′ArmR) designed out of 5′ homology arm (Figures 1 and 3A). Colonies with correct 5′ end recombination had a 4.9 kb PCR band. Primer sequences are listed in Table S2. Set up PCR reactions as shown in Table 7 and run reactions following the program in Table 8.
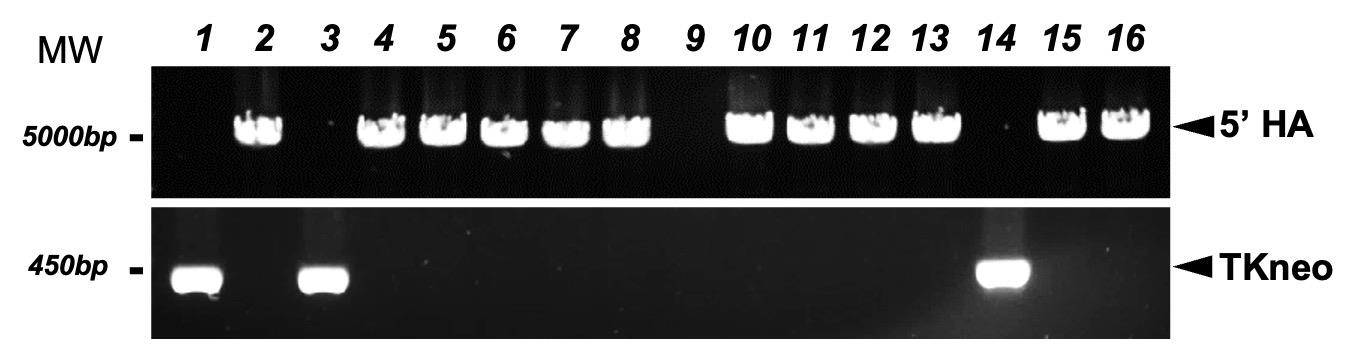
Figure 3. Characterization of mouse embryonic stem cell (mESC) clones with hmTert locus. PCR-based identification of candidate mESC clones. Primers designed outside of 5′ HA detected a 4,888 bp fragment, indicating successful homologous recombination at the 5′ end. Primers targeting the TKneo cassette amplified a 473 bp band, indicating random integration of the donor BAC.
Table 7. PCR reaction
| Components | Amount |
|---|---|
| 10× LA PCR buffer (Mg2+ PLUS) | 1 μL |
| LA Taq polymerase | 0.1 μL |
| dNTP (2.5 mM) | 1.6 μL |
| Primers (10 μM) | 0.2 μL |
| mESCs genomic DNA (10ng/μL) | 1.0 μL |
| ddH2O | 6.1 μL |
| Total | 10 μL |
Table 8. PCR cycling conditions
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 94 °C | 1 min | 1 |
| Denaturation | 98 °C | 10 s | 31 cycles |
| Annealing | 68 °C | 5 min | |
| Final extension | 72 °C | 10 min | 1 |
| Hold | 4 °C |
b. Detect TK-neo sequence by PCR using primers TKneoF and TKneoR. Amplification of a 473 bp PCR product suggests that the mESC clone contained randomly integrated donor BAC constructs (Figure 3A). Primer sequences were listed in Table S2. Set up PCR reactions as shown in Table 9 and run reactions following the program in Table 10.
Table 9. PCR reaction
| Components | Amount |
|---|---|
| 10× ThermoPol® buffer | 1 μL |
| Taq polymerase | 0.1 μL |
| dNTP (10 mM) | 0.2 μL |
| Primers (10 μM) | 0.2 μL |
| mESCs genomic DNA (10 ng/μL) | 1.0 μL |
| ddH2O | 7.5 μL |
| Total | 10 μL |
Table 10. PCR cycling conditions
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 94 °C | 5 min | 1 |
| Denaturation | 95 °C | 30 s | 35 cycles |
| Annealing | 60 °C | 30 s | |
| Extension | 72 °C | 45 s | |
| Final extension | 72 °C | 5 min | 1 |
| Hold | 4 °C |
Among 110 puromycin- and GCV-resistant clones, 84 yielded a 4.9 kb PCR fragment, indicating that these clones had undergone homologous recombination at the 5′ end.
2. For further verification, verify correct recombination with Southern blot analysis of the entire chimeric hmTert region (time: 4 d).
a. Day 1: Digest 2.0 μg mESCs genomic DNAs with BamHI, HindIII, or EcoRV, followed by hybridization with different probes. Correctly recombined hmTert sequences and wildtype mTert alleles were identified by comparing the size of restriction fragments (Figure 4A). Set up restriction enzyme digestion reactions as shown in Table 11.
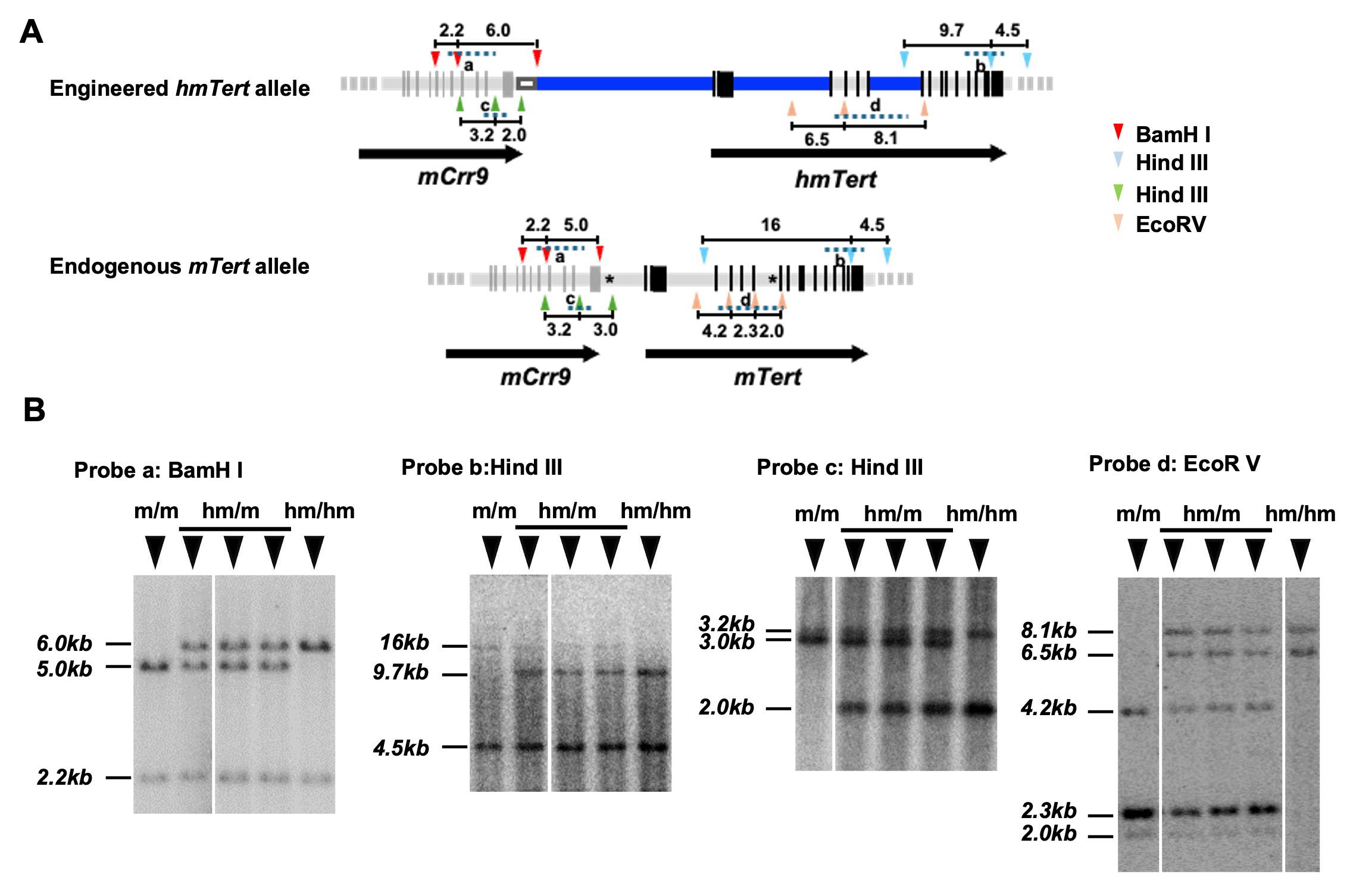
Figure 4. Characterization of the hmTert alleles by Southern blot analysis. (A) Schematic representation of the hmTert and mTert alleles. Restriction enzyme sites (BamH1, HindIII, and EcoRV) used in the analysis are indicated by arrowheads om corresponding colors. Distance between restriction sites are labeled in kilobases (kb). Probes used for hybridization are shown as dashed lines. Black vertical bars represent exons, and asterisks (*) mark the sgRNA cleavage sites. (B) Representative Southern blot images using four different DNA probes. Genomic DNA (2 μg) from individual mouse embryonic stem cell (mESC) clones were digested with the indicated enzymes and hybridized with 32P-labeled DNA probes. Genotypes are indicated as follows: m/m, homozygous mTert alleles (Tert+/+); hm/m, heterozygous hmTert/mTert alleles (Terth/+); hm/hm, homozygous hmTert alleles (Terth/h).
Table 11. Restriction enzyme reaction
| Components | Amount (μL) |
|---|---|
| 10× NEB buffer | 2.5 μL |
| Restriction enzyme | 0.4 μL |
| mESCs genomic DNA | 2.0 μg |
| ddH2O | up to 25 μL |
| Total | 25 |
Incubate at 37 °C overnight in a water bath.
b. Day 2: Analyze digested genomic DNAs on a 0.7% agarose gel with 0.5× TBE for 4 h. Wash agarose gels with denature buffer twice for 20 min each, followed by washing with neutralization buffer twice for 20 min each at room temperature. Rinse the agarose gels with 10× SSC buffer and transfer DNAs from gel to nylon membranes through the capillary transfer method at room temperature overnight.
c. Day 3: Wash the nylon membranes with 2× SSC once and UV-crosslink. Rinse the membranes with 2× SSC and pre-hybridize with Church buffer at 60 °C in rotating tubes for 4 h. Meanwhile, label the probes with 32P-dCTP using the Ladderman DNA Labeling kit according to the manufacturer’s instructions. Hybridize the membranes with 5 mL of fresh Church buffer containing 2 μL of 32P-labeled DNA probes in rotating tubes overnight at 60 °C.
d. Day 4: Wash membranes three times with 1× SSC (+0.1% SDS) for 10 min each at room temperature and in 0.2× SSC (+0.1% SDS) twice for 15 min each at 60 °C. Detect signals using the PhosphorImager system (GE Healthcare) (Figure 4B).
Note: Our data showed that approximately 80% (130 out of 156) of puromycin- and GCV-resistant clones underwent successful homologous recombination following CRISPR/Cas9-mediated double-stranded breaks. Among these, 29% (45 out of 156) harbored the correctly targeted hmTert locus.
E. Removal of the puromycin cassette
Four mESC clones with the correctly targeted hmTert locus were selected for removal of the puromycin resistance cassette.
1. Transient transfection with pCBM plasmid (Cre recombinase expression vector) (time: 6 d).
a. Day 1: Seed 1.8 × 106 hmTert knocked-in mESCs [9] into each well of a 24-well plate containing DR4 feeder cells. Cells were transfected with 0.2 μg of the pCBM plasmids using Lipofectamine 2000. Eight hours post-transfection, passage cells into a 6-well plate.
b. Day 2: Culture cells with fresh mESCs culture medium.
c. Day 4–6: Seed individual mESC colonies per well in a 96-well plate for expansion and further analysis.
2. Identification of puromycin-negative colonies (time: 1 d).
Colonies that had successfully excised the puromycin cassette were identified by PCR amplification of a 333-bp fragment using primer pair RePuroF1/RePuroR1, which flanks the puromycin cassette (Figure 5). Primer sequences are listed in Table S2. Set up PCR reactions as shown in Table 12 and run the reactions following the program in Table 13.
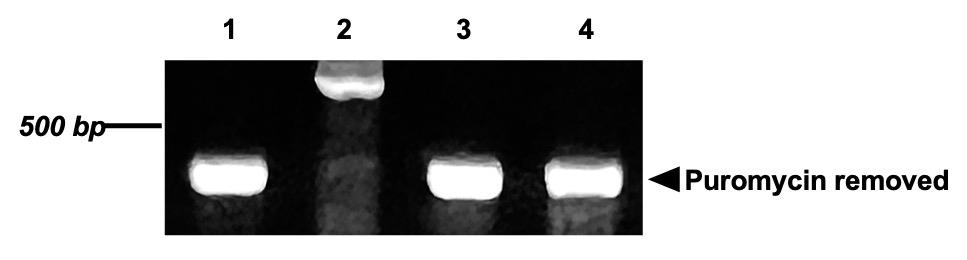
Figure 5. Identification of puromycin cassette removal in hmTert knock-in mouse embryonic stem cell (mESC) clones. Genomic DNA was amplified using the primer pair RePuroF1/RePuroR1. Clones 1, 3, and 4 produced a 333 bp PCR fragment, confirming successful excision of the puromycin resistance cassette.
Table 12. PCR reaction
| Components | Amount |
|---|---|
| 10× ThermoPol® buffer | 1 μL |
| Taq polymerase | 0.1 μL |
| dNTP (10 mM) | 0.2 μL |
| Primers (10 μM) | 0.2 μL |
| Genomic DNA (10 ng/μL) | 1.0 μL |
| ddH2O | 7.5 μL |
| Total | 10 μL |
Table 13. PCR cycling conditions
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 94 °C | 5 min | 1 |
| Denaturation | 95 °C | 20 s | 36 cycles |
| Annealing | 60 °C | 30 s | |
| Extension | 72 °C | 45 s | |
| Final Extension | 72 °C | 5 min | 1 |
| Hold | 4 °C |
F. Generation of chimeric mice
1. Inject G4 mESCs (Terth/+), carrying one hmTert and one wildtype mTert allele, into C57BL/6J embryos (Transgenic Core, Cornell University, US). Six chimeric mice were obtained as indicated by coat coloration (time: 18 d).
2. Detect hmTert or mTert mRNA expression (time: 6 h):
a. Isolate total RNAs from various tissues of chimeric mice using TRI reagent according to the manufacturer’s instructions.
b. Synthesize first-strand cDNAs using SuperScriptTM III reverse transcriptase.
c. hmTert or mTert mRNA expression levels were determined by quantitative RT-PCR using PowerUpTM SYBRTM Green Master Mix (Figure 6A). Primer sets are listed in Table 14. Tert mRNAs from hmTert or mTert allele were distinguished by nine silent nucleotide substitutions near the 3′ end of exon 2 of the hmTert locus, which overlapped with the forward primers [5]. Primer sequences are listed in Table S2.
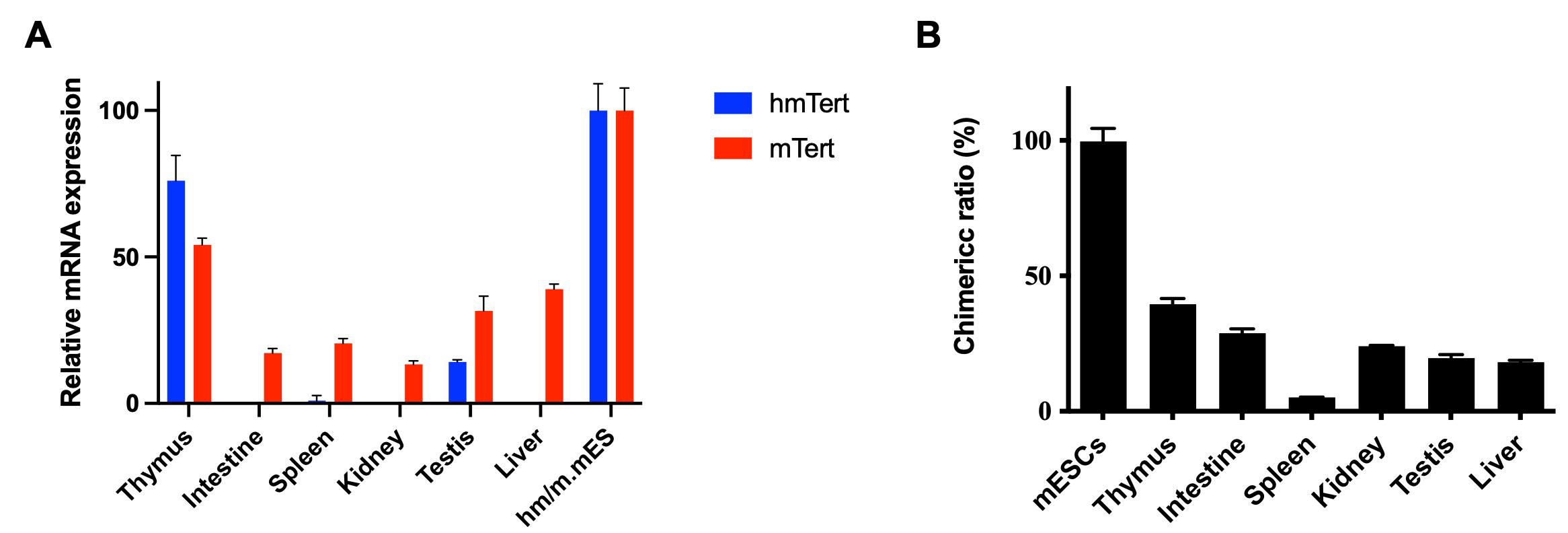
Figure 6. Analysis of chimera mice. (A) Detection of hmTert mRNA in tissues of a chimera mouse. Total RNAs were isolated from tissues, and hmTert and mTert mRNA levels were quantified by qRT-PCR. Expression levels were normalized to the chimeric ratio in the corresponding tissues (see panel B). Terth/+ mouse embryonic stem cells (mESCs) were used as the control, and mRNA levels were normalized to 18S rRNA. (B) Chimeric ratio in mouse tissues. The chimeric contribution of hmTert/mTert in various tissues was determined by quantitative genomic PCR targeting hmTert and mTert sequences. The ratio of mESCs (Terth/+) was defined as 100%. Data are presented as mean values with SD.
Table 14. Primer sets used for quantitative RT-PCR
| Allele | Primer sets |
|---|---|
| hmTert | mTert.Mut F/ mTert.Endo R |
| mTert | mTert.Endo F/ mTert.Endo R |
| 18s | 18s RNA control F/18s RNA control R |
d. The chimeric ratios of various tissues were determined by quantitative PCR of 5′ end recombination site. Isolate genomic DNAs from the phenol phase and interphase of the Trizol/chloroform step of total RNA isolation. Add 1/3 volume of 100% ethanol per TRI reagent and mix solution by inversion. Incubate the samples at room temperature for 5 min and then centrifuge at 2,000× g for 5 min at 4 °C to precipitate DNA. Wash the DNA pellets twice with DNA wash solution for 30 min at room temperature, and enrich the DNAs again by centrifuging samples at 2,000× g for 5 min at 4 °C. Wash the DNA twice with 75% ethanol. Dry DNA pellets at room temperature, and dissolve pellets in 8 mM NaOH. Adjust the pH of the solution with 0.1 M HEPES to pH 8.0. Add EDTA to the final concentration of 1 mM to stabilize DNA.
e. The ratios of hmTert and mTert alleles were assessed by quantitative PCR using PowerUpTM SYBRTM Green Master Mix and the primer sets listed in Table 15 (Figure 6B). Primer sequences are listed in Table S2.
Table 15. Primer sets used for quantitative PCR
| Allele | Primer sets | Chromosome position |
|---|---|---|
| hmTert | hmTert up5k F/ hmTert up5k R | chr5: 1300555-1300574/chr5: 1300435-1300454 |
| mTert | mTert up5k F/ mTert up5k R | chr13: 73770040-73770059/chr13: 73621998-93622018 |
G. Reducing telomere length in mice with the hmTert allele
1. Cross male chimeric mice with C57BL/6J female mice, resulting in progeny with a germline hmTert allele (F0). Cross again the mice with C57BL/6J mice to generate F1 Terthm/+ mice with 88% C57BL/6J genetic background. Mouse genotypes were determined by PCR using primers specific for hmTert or mTert alleles. Isolate genomic DNAs from the toes of 7-day-old mice using the Wizard® Genomic DNA Purification kit and the primer sets listed in Table 16. Primer sequences are listed in Table S2 (time: 6 months).
Table 16. Primer sets used for mouse genotyping
| Allele | Primer sets | PCR amplicon length (bp) |
|---|---|---|
| hmTert | hmTert F/ hmTert R | 363 |
| mTert | mTert F/ mTert R | 467 |
Set up PCR reactions as shown in Table 17 and run the reaction following the PCR program in Table 18.
Table 17. PCR reaction
| Components | Amount |
|---|---|
| 10× ThermoPol® buffer | 1 μL |
| Taq polymerase | 0.1 μL |
| dNTP (10 mM) | 0.2 μL |
| Primers (10 μM) | 0.2 μL |
| Mouse genomic DNA (10 ng/μL) | 1.0 μL |
| ddH2O | 7.5 μL |
| Total | 10 μL |
Table 18. PCR cycling conditions
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 94 °C | 5 min | 1 |
| Denaturation | 94 °C | 30 s | 35 |
| Annealing | 60 °C | 30 s | |
| Extension | 72 °C | 30 s | |
| Final extension | 72 °C | 5 min | 1 |
| Hold | 4 °C |
2. Terth/- mice were generated by crossing F1 Terth/+ mice with Tert+/- mice (C57BL/6J background, a gift of Dr. Lea Herrington). The resulting (Terth/-) mice were subsequently bred with Tert-knockout (Tert-/-, C57BL/6J) mice for successive generations. The Tert-KO alleles were detected by PCR using primers (mTert-KO F/ mTert-KO R), which were designed to span the mTert knockout region as previously reported [11], producing a 467 bp amplicon. Primer sequences are listed in Table S2. Set up PCR reactions following Table 19 and run the reactions following Table 20.
Table 19. PCR reaction
| Components | Amount |
|---|---|
| 10× ThermoPol® buffer | 1 μL |
| Taq polymerase | 0.1 μL |
| dNTP (10 mM) | 0.2 μL |
| Primers (10 μM) | 0.2 μL |
| Mouse genomic DNA (10 ng/μL) | 1.0 μL |
| ddH2O | 7.5 μL |
| Total | 10 μL |
Table 20. PCR cycling conditions
| Steps | Temperature | Time | Cycles |
| Initial denaturation | 94 °C | 5 min | 1 |
| Denaturation | 94 °C | 30 s | 32 |
| Annealing | 60 °C | 30 s | |
| Extension | 72 °C | 30 s | |
| Final extension | 72 °C | 5 min | 1 |
| Hold | 4 °C |
3. The Terth/- mice from the fourth-generation Tert-KO breeding were intercrossed for 20 generations, from G4.1 to G4.20 (Figure 7A) (time: 5 years).
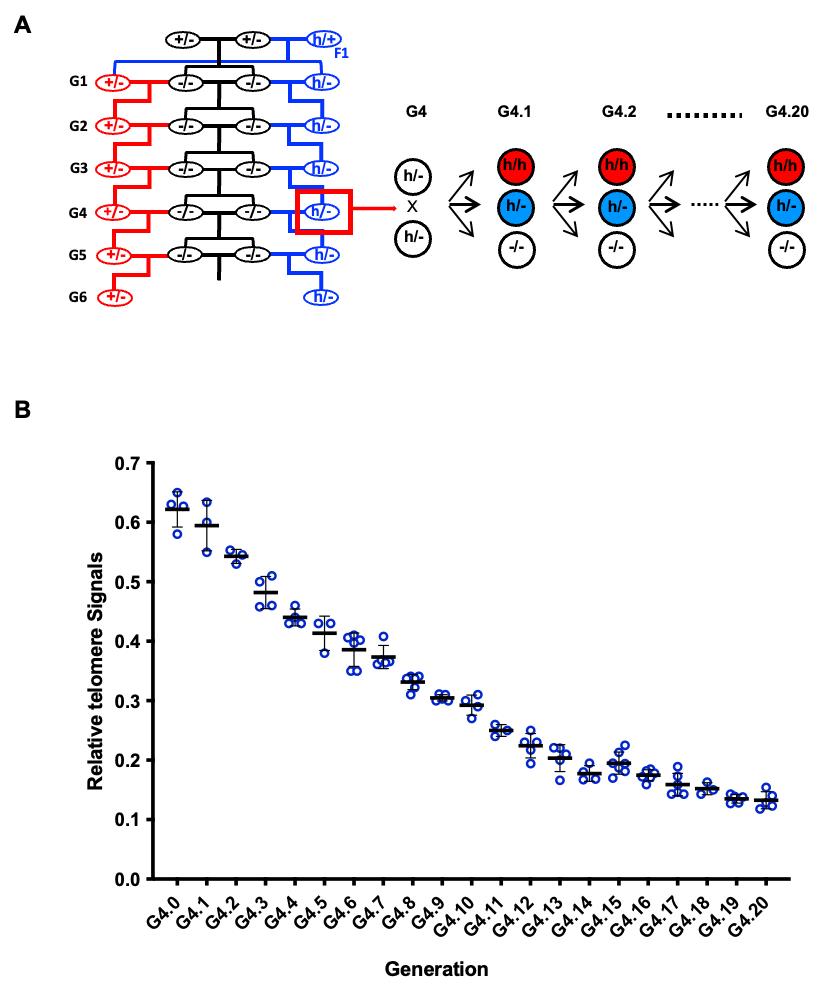
Figure 7. Telomere lengths in mice carrying the hmTert gene. (A) Breeding strategy. Tert-knockout mice (Tert-/-) were initially generated by intercrossing mTert heterozygous (Tert+/-) parents. In each subsequent generation, Tert-/- mice were bred with the corresponding generation of either Tert+/- or Terth/- mice. This strategy established and maintained two parallel heterozygous lines, Tert+/- and Terth/-, through six generations (G1–G6). Beginning at generation 4 (G4), Terth/- mice were intercrossed over successive generations, resulting in progressive telomere shortening and the eventual establishment of human-like telomere lengths. (B) Average telomere lengths across generations of Terth/- intercrosses. Telomere lengths in splenocyte were measured by Flow-FISH. Hybridization signals were normalized with those of wildtype C57BL/6J mice (Tert+/+), which have an average telomere length of approximately 50 kb. Data are presented as mean values with SD.
4. Average telomere lengths in Terth/- mice were determined by Flow-FISH (time: 2 d).
a. Collect spleens from adult mice in 1.5 mL tubes with 0.5 mL of HBSS or RPMI medium (+2% FBS).
b. Smash spleens on a 70 μm strainer on top of a 50 mL tube using a stick. Resuspend splenocytes in 1.5 mL of HBSS or RPMI medium (+2% FBS) and centrifuge tubes at 800× g for 10 min.
c. Resuspend cell pellets in 5 mL of RBC lysis buffer for 5 min at room temperature.
d. Halt the lysis process by adding 5 mL of complete RPMI medium (+10% FBS). Centrifuge samples at 800× g for 5 min. Resuspend the pellets in 10 mL of PBS (+2% FBS) and centrifuge again at 800× g for 5 min to enrich the cells.
e. Filter cell suspensions through a 70 μm strainer. Count cell numbers and move 4 million cells into 1.5 mL tubes. Add PBS (+0.1% BSA) to a final volume of 1 mL. Centrifuge tubes at 800× g for 5 min.
Note: The cell count typically ranges from 1 to 4 million. However, the loss and clogging of splenocytes during the experimental procedure can reduce the final yield of single cells, which are used for probe signal quantification. To ensure reliable data, it is recommended to start with a higher number of cells for hybridization.
f. Resuspend cell pellets by vortexing for 10 s and then mix with 500 μL of hybridization buffer containing the FISH probe (TelC-FAM, 0.14 μg/mL). Three samples without the probe are included as negative controls. Incubate tubes at 80 °C for 10 min with continuous shaking, then allow to cool to room temperature and leave overnight in the dark.
g. Centrifuge the samples at 800× g for 10 min at 16 °C. Vortex the pellets for 20 s and resuspend cells with 1 mL of wash solution I.
Note: Approximately 20 μL of residual solution is left in the tube to aid in resuspending the cell pellets by vortexing, helping to reduce cell clogging.
h. Repeat step G4g.
i. Centrifuge the samples at 800× g for 10 min at 16 °C. Vortex the pellets for 20 s and resuspend cells with 1 mL of wash solution II.
j. Centrifuge the samples at 800× g for 10 min at 16 °C. Vortex the pellets and incubate cells with 400 μL of PBS with 5 μL of 7-AAD.
k. Filter the cells through a 40 μm strainer and analyze samples using the flow cytometry machine Cytoflex. Capture the telomere and DNA signals using the appropriate channels: B525 for FAM and Y675 for 7-AAD. Analyze the data using the CytExpert software (Figure S2). Telomere signals of Terth/- mice were normalized with those of wildtype C57BL/6J mice (Figure 7B, Figure S2).
Validation of protocol
A. Validation of hmTert allele engineering in mESC cells
The protocol has been validated and successfully applied to engineer the hmTert allele in an additional mESC cell line, TC1, as described in the following publications:
Zhang et al. [8]. Crispr/Cas9-mediated cleavages facilitate homologous recombination during genetic engineering of a large chromosomal region. Biotechnology Bioengineer (Table 2)
Cheng et al. [5]. Engineering a humanized telomerase reverse transcriptase gene in mouse embryonic stem cells. Scientific Report (Figure 3, Figure S1, and Table S1)
B. Validation of telomere length shortening in mice with the hmTert allele
This protocol has also been validated and applied in the following publication to establish a parallel breeding line, successfully generating Terth/-heterozygous mice with human-like telomere lengths:
Zhang et al. [12]. Modification of the telomerase gene with human regulatory sequences resets mouse telomeres to human length. Nature Communications (Figure 8)
General notes and troubleshooting
General notes
1. Since the larger DNA fragment (about 47 kb) was used in this protocol, Southern blotting and PCR analyses were conducted to ensure the accuracy of engineered sequences.
2. Cells are more prone to clogging after hybridization with a FISH probe, primarily due to the high temperature involved in the process. To minimize background noise and prevent cell aggregation, it is crucial to thoroughly wash and resuspend the cells. This includes vortexing the cell pellets before adding the washing buffer and mixing the suspension thoroughly by pipetting up and down.
Supplementary information
The following supporting information can be downloaded here:
1. Table S1. Scores of potential sgRNAs
2. Table S2. Sequences of oligo primers
3. Figure S1. Illustration map of pSpCa9 (BB)-2A-GFP
4. Figure S2. Mice telomere length was quantified using flow cytometry analysis of TelC-FAM fluorescence intensity
5. SEQ 1. Sequence of 5′ end homologous recombination arm (4,361 bp, chr.13: 73764794-73769155)
6. SEQ 2. Sequence of 3′ end homologous recombination arm (7,152 bp, chr13: 73787275-73794428)
7. SEQ 3. Input sequence for sgRNA1 design (1,000 bp, chr13: 73769582-73770581, 5′ intergenic region between mTert and mCrr9)
8. SEQ 4. Input sequence for sgRNA2 design (1,000 bp, chr13: 73786261-73787260, intron 6 of mTert)
Acknowledgments
This work was supported in part by NIH Grants R21OD021432, R01AG073423, and R35GM149529 to J.Z., a Team Science Award (ME220261) from Department of Defense to J.Z., and Health Sciences & Services Authority (HSSA) of Spokane County. We would like to express our gratitude to Drs. Yie Liu and Lea Harrington for their generous gift of mTert knockout mice. We thank the Program of Laboratory Animal Resources (PLAR) and Histology Core of Washington State University Spokane.
Conception and design: J. Zhu. Data acquisition: F. Zhang, D. Cheng, K.I. Porter. Data analysis and interpretation: F. Zhang, S. Wang, J. Zhu. Manuscript writing and revision: J. Zhu, F. Zhang. Study supervision: J. Zhu.
Competing interests
The authors declare no competing interests.
Ethical considerations
All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Animals and were approved under protocols ASAF#6659 and ASAF#6693 by the Institutional Animal Care and Use Committee of Washington State University.
References
- Zhang, F., Cheng, D., Wang, S. and Zhu, J. (2016). Human Specific Regulation of the Telomerase Reverse Transcriptase Gene. Genes (Basel). 7(7): 30. https://doi.org/10.3390/genes7070030
- Zhang, J., Zhang, F., Porter, K. I., Dakup, P. P., Wang, S., Robertson, G. P., Gaddameedhi, S. and Zhu, J. (2023). Telomere dysfunction in Tert knockout mice delays BrafV600E‐induced melanoma development. Int J Cancer. 154(3): 548–560. https://doi.org/10.1002/ijc.34713
- Blasco, M. A., Lee, H. W., Hande, M., Samper, E., Lansdorp, P. M., DePinho, R. A. and Greider, C. W. (1997). Telomere Shortening and Tumor Formation by Mouse Cells Lacking Telomerase RNA. Cell. 91(1): 25–34. https://doi.org/10.1016/s0092-8674(01)80006-4
- Zhu, J., Zhao, Y. and Wang, S. (2010). Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell. 1(1): 22–32. https://doi.org/10.1007/s13238-010-0014-1
- Cheng, D., Zhao, Y., Zhang, F., Zhang, J., Wang, S. and Zhu, J. (2019). Engineering a humanized telomerase reverse transcriptase gene in mouse embryonic stem cells. Sci Rep. 9(1): 9683. https://doi.org/10.1038/s41598-019-46160-5
- Wang, S., Zhao, Y., Leiby, M. and Zhu, J. (2009). A New Positive/Negative Selection Scheme for Precise BAC Recombineering. Mol Biotechnol. 42(1): 110–116. https://doi.org/10.1007/s12033-009-9142-3
- Cheng, D., Wang, S., Jia, W., Zhao, Y., Zhang, F., Kang, J. and Zhu, J. (2017). Regulation of human and mouse telomerase genes by genomic contexts and transcription factors during embryonic stem cell differentiation. Sci Rep. 7(1): 16444. https://doi.org/10.1038/s41598-017-16764-w
- Zhang, F., Cheng, D., Wang, S. and Zhu, J. (2020). Crispr/Cas9‐mediated cleavages facilitate homologous recombination during genetic engineering of a large chromosomal region. Biotechnol Bioeng. 117(9): 2816–2826. https://doi.org/10.1002/bit.27441
- George, S. H. L., Gertsenstein, M., Vintersten, K., Korets-Smith, E., Murphy, J., Stevens, M. E., Haigh, J. J. and Nagy, A. (2007). Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci USA. 104(11): 4455–4460. https://doi.org/10.1073/pnas.0609277104
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 8(11): 2281–2308. https://doi.org/10.1038/nprot.2013.143
- Liu, Y., Snow, B. E., Hande, M. P., Yeung, D., Erdmann, N. J., Wakeham, A., Itie, A., Siderovski, D. P., Lansdorp, P. M., Robinson, M. O. et al. (2001). The telomerase reverse transcriptase is limiting and necessary for telomerase function in vivo. Curr Biol. 11(11): 907. https://doi.org/10.1016/s0960-9822(01)00264-0
- Zhang, F., Cheng, D., Porter, K. I., Heck, E. A., Wang, S., Zhang, H., Davis, C. J., Robertson, G. P. and Zhu, J. (2025). Modification of the telomerase gene with human regulatory sequences resets mouse telomeres to human length. Nat Commun. 16(1): 1211. https://doi.org/10.1038/s41467-025-56559-6
Article Information
Publication history
Received: May 18, 2025
Accepted: Aug 3, 2025
Available online: Aug 28, 2025
Published: Sep 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Zhang, F., Cheng, D., Porter, K. I., Wang, S. and Zhu, J. (2025). Genetic Engineering of Humanized Telomere Mice. Bio-protocol 15(18): e5445. DOI: 10.21769/BioProtoc.5445.
Category
Developmental Biology > Genome editing > Targeted integration
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.