- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient Gene Knockdown in Adult Zebrafish Retina by Intravitreal Injection
Published: Vol 15, Iss 17, Sep 5, 2025 DOI: 10.21769/BioProtoc.5436 Views: 1392
Reviewed by: Alessandro DidonnaAnonymous reviewer(s)
Original research article
The authors used this protocol in:
Jan 2024

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
High-throughput sequencing has created a tremendous amount of information about the genes expressed in various cell types and tissues throughout the body. As such, there is a need for a quick and effective method to knock down genes of interest in order to investigate their roles. While there are many approaches for this in mammalian models, there are limited ways to knock down genes of interest in adult zebrafish. Unlike mammals, zebrafish have the natural ability to regenerate their neurons after injury or disease is detected, making them a staple in regenerative studies. Unfortunately, current approaches for gene knockdown in the retina of adult zebrafish are costly and provide a barrier for many scientists. We provide two cost-effective approaches for targeted gene knockdowns in adult zebrafish retinas. We describe this approach through the use of Vivo-morpholinos and lipid-encapsulated siRNAs that target the expression of the proliferating cell nuclear antigen (PCNA) gene in adult zebrafish. We also describe how to collect and process retina samples for downstream immunohistochemistry, imaging, and quantification. Overall, this protocol will provide researchers with a straightforward, cheap, and effective method to perform targeted gene knockdowns in adult zebrafish retinas.
Key features
• This protocol provides researchers with an approach to knock down genes of interest in adult zebrafish retina without the use of electroporation.
• This protocol can be performed without causing an acute damage response in the retina.
• This protocol allows targeting of genes in both proliferating cells and terminally differentiated cells.
• This protocol allows the retina to be collected and processed for further downstream analysis.
Keywords: Gene knockdownsGraphical overview
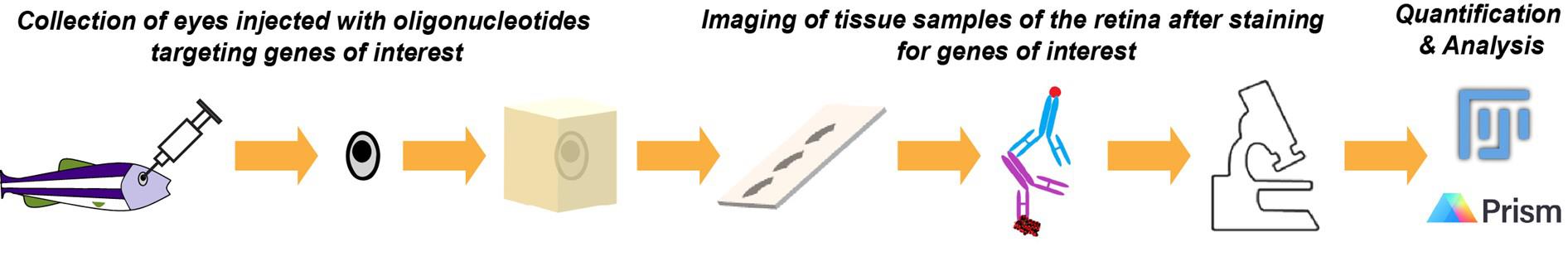
Graphical abstract illustrating the procedures required to knock down and validate the knockdown of genes of interest
Background
Ease of access to high-throughput sequencing in recent years has provided researchers with a plethora of data to mine. Whether they are investigating genes in relation to chronic disease, developmental defects, or even evolution, cell atlases have come to provide researchers with a multitude of genes to investigate. Unfortunately, limited resources and funding for most labs requires a quick and efficient method to screen genes of interest through gene knockdowns. While researchers can readily utilize a diverse array of viral vectors, CRISPR-Cas systems, and nanoparticles to target and knock down genes of interest in mammalian model systems, adult zebrafish models still lack a quick and effective method for targeted gene knockdown [1–3]. Unlike mammalian models, zebrafish have the remarkable ability to regenerate their fins, cardiomyocytes, and even neurons throughout their entire lifespans. As such, many researchers are trying to identify the network of genes responsible for regeneration so they may translate these findings to mammalian models. In adult zebrafish retina, targeted gene knockdowns have been achieved through the electroporation of morpholino oligonucleotides into the retina [4–6]. Many zebrafish labs do not have an electroporation device readily available, which may cost in excess of US$16,000, and must rely on alternative strategies. One alternative approach is the use of Vivo-morpholino oligonucleotides. Vivo-morpholinos are designed so that a morpholino oligomer is linked to a molecular transporter with eight guanidinium head groups and does not require electroporation for successful penetrance of cells [7]. While several studies have successfully used Vivo-morpholinos for targeted gene knockdowns in adult zebrafish, none of these studies worked on the retina [8–13]. Another popular strategy for targeted gene knockdowns is through the use of siRNAs. SiRNAs degrade mRNA strands of interest by utilizing the endogenous RISC cascade [14,15]. One limitation of siRNAs for in vivo experiments is their successful delivery to the cells of interest. One strategy to overcome this limitation is through encapsulation in lipid particles; however, the efficiency of these particles varies from species to species and tissue to tissue. In this study, we describe a quick and easy method to effectively knock down genes of interest in adult zebrafish retina through the use of either Vivo-morpholinos or lipid-encapsulated siRNAs. We provide detailed reagents and a step-by-step protocol on how to prepare the solutions needed to knock down PCNA in adult zebrafish retina. The model system used in this study is a transgenic zebrafish expressing a Flag-tagged mouse rhodopsin harboring the P23H mutation in rod photoreceptors. This results in rapid degeneration of rods, producing a model of Retinitis Pigmentosa [16]. Because of the robust regenerative capabilities of the zebrafish, this model displays continuous regeneration of degenerated rods, with rapid proliferation of retinal progenitor cells that express PCNA. In this protocol, we will explain how to process samples of the retina and quantify the expression of PCNA between treatment and control conditions. We show cost-effective strategies that will allow researchers to quickly and efficiently target and knock down genes in the retina of adult zebrafish.
Materials and reagents
Biological materials
1. Adult P23H rhodopsin transgenic zebrafish [generated in-house, RRID: uth4tg(AB); ZFIN ID: ZDB-FISH-220323-6]
Reagents
1. 0.01 M phosphate-buffered saline (PBS) (Millipore-Sigma, catalog number: P3813)
2. UltraPure DNase/RNase-free distilled water (Invitrogen, catalog number: 10977015)
3. DDH2O (made in-house)
4. Tricaine/MS222 (Syndel, catalog number: SYNC-M-GR-US02)
5. Tris (Fisher, catalog number: BP152-500)
6. 32% Formaldehyde (PFA) (Electron Microscopy Sciences, catalog number: 15714-S)
7. 190 Proof (95%) ethanol (ETOH) (Fisher, catalog number: 04-355-454)
8. Sucrose (Millipore-Sigma, catalog number: S0389-500G)
9. Tissue-Tek CRYO-O.C.T. compound (Fisher Scientific, catalog number: 14-373-65)
10. Dry ice
11. Triton X-100 (Sigma-Aldrich, catalog number: X100-1L)
12. Normal donkey serum (NDS) (Jackson ImmunoResearch, catalog number: 017-000-121)
13. Flag-DDK primary antibody (Origene, catalog number: TA50011; RRID: AB_2622345)
14. PCNA primary antibody (Abcam, catalog number: Ab18197; RRID: AB_444313)
15. Cy3 secondary antibody (Jackson ImmunoResearch, catalog number: 115-165-206; RRID: AB_2338695)
16. Alexa Fluor 488 secondary antibody (Jackson ImmunoResearch, catalog number: 711-545-152; RRID: AB_2313584)
17. Vectashield with DAPI (Vector Laboratories, catalog number: H-1000)
18. Altogen Nanoparticle In Vivo Transfection kit (Altogen, catalog number: 5030)
19. Zebrafish water (reverse osmosis-purified water adjusted to 700 nS conductivity with Instant Ocean sea salt mix and pH 7.0 with sodium bicarbonate)
20. Custom translation blocking Vivo-morpholino targeting PCNA (Gene Tools, sequence: TTTCTTAGTTTGGAGTAGGAGGAAC)
21. Standard negative control Vivo-morpholino (Gene Tools, sequence: CCTCTTACCTCAGTTACAATTTATA)
22. Custom siRNA targeting PCNA #1 (IDT, target sequence: GTCCAAGACGGTCACACTTAGCATG)
23. Custom siRNA targeting PCNA #2 (IDT, target sequence: AAGACGGTCACACTTAGCATGTCCG)
24. Custom siRNA targeting EGFP (negative control) (IDT, target sequence: GCATGCATCTCAATTAGTCAGCAAC)
Solutions
1. Tricaine stock solution (4%) (see Recipes)
2. Anesthesia solution (see Recipes)
3. Euthanasia solution (see Recipes)
4. Fixation solution (see Recipes)
5. Blocking buffer (see Recipes)
6. Primary antibody solution (see Recipes)
7. Secondary antibody solution (see Recipes)
8. Vivo-morpholino solution (see Recipes)
9. siRNA solution (see Recipes)
10. 25% sucrose solution (see Recipes)
11. 10% Triton X-100 (see Recipes)
Recipes
1. Tricaine stock solution (4%)
Add 4.0 g of Tricaine/MS222 to 97 mL of ddH2O. Adjust pH to 7.0 with 1 M Tris base. Top off to 100 mL and store in 10 mL aliquots frozen at -20 °C.
2. Anesthesia solution
Each adult zebrafish must be anesthetized with Tricaine/MS222 before performing any injections. To check that the zebrafish is fully anesthetized, the experimenter should be able to use a plastic spoon to pick up the zebrafish without any response. This typically takes about 3–5 min. Anesthesia solution should be made fresh for each experiment.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tricaine stock solution | 0.02% | 0.5 mL |
| Zebrafish water | - | 100 mL |
3. Euthanasia solution
Zebrafish should be euthanized immediately prior to the collection of samples for downstream analysis. Euthanasia solution should be made fresh for each experiment.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tricaine stock solution | 0.04% | 1 mL |
| Zebrafish water | - | 100 mL |
4. Fixation solution
Fixation solution should be made fresh for each experiment. It consists of a 9:1 95% EtOH to 32% PFA ratio.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Ethanol | 85.5% | 9 mL |
| PFA | 3.2% | 1 mL |
5. Blocking buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 10% Triton X-100 | 0.3% | 9 μL |
| NDS | 5% | 15 μL |
| 1× PBS | - | 276 μL |
6. Primary antibody solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 10% Triton X-100 | 0.3% | 9 μL |
| NDS | 5% | 15 μL |
| Flag-DDK primary antibody | 1:200 dilution | 1.5 μL |
| PCNA primary antibody | 1:100 dilution | 3 μL |
| 1× PBS | - | 271.5 μL |
7. Secondary antibody solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 10% Triton X-100 | 0.3% | 9 μL |
| NDS | 5% | 15 μL |
| Cy3 secondary antibody | 1:500 dilution | 0.6 μL |
| Alexa 488 fluor secondary antibody | 1:500 dilution | 0.6 μL |
| 1× PBS | - | 274.8 μL |
8. Vivo-morpholino solution
Vivo-morpholinos are delivered as powders and should be stored in the dark and at room temperature until ready to use. When making the stock solution, dissolve the Vivo-morpholino in ultraclean water to 400 μM. This concentration may need to be adjusted depending on oligo properties; follow the manufacturer’s instructions. The manufacturer recommends storing the stock solution at room temperature. To make the working solution, dilute the stock solution with sterile-filtered PBS. Typically, Vivo-morpholinos precipitate out of the stock solution and lose efficacy within 1 month. This may vary depending on the Vivo-morpholino. Always check the stock solution for precipitates before making the working solution. The working solution should be made fresh on the day of injections. Each Vivo-morpholino will have its own unique efficacy. Be sure to use freshly prepared Vivo-morpholino when evaluating efficacy.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Vivo-morpholino | 40 μM | 2 μL |
| 0.01 M PBS | - | 18 μL |
9. siRNA solution
Dissolve siRNA in 10 mM Tris, pH 8.0, following manufacturer’s instructions. siRNA stock is generally 225 μM, and should be stored at -80 °C. Each siRNA will have a different efficacy when optimally transfected. It is often more effective to combine two or more siRNAs to the desired target gene. The Altogen Nanoparticle reagents typically have a shelf life of 3 months when stored at 4 °C. Always check for precipitates before using.
| Reagent | Final concentration | Quantity or Volume |
| siRNA #1 | 37.5 μM | 1.5 μL |
| siRNA #2 | 37.5 μM | 1.5 μL |
| Altogen Nanoparticle In Vivo transfection reagent | - | 5 μL |
| Incubate at room temperature for 20 min. Then, add 1 μL of Altogen Nanoparticle In Vivo transfection enhancer. Incubate for 5 min at room temperature; then, it is ready to use. | ||
10. 25% sucrose solution
2.5 g of sucrose in a final volume of 10 mL of ddH2O; made fresh for each experiment.
11. 10% Triton X-100
1 mL of Triton X-100 and 9 mL of ddH2O.
Laboratory supplies
1. Petri dish (Fisher, catalog number: 08-757-100B)
2. Disposable pipettes (Fisher, catalog number: 13-711-7M)
3. 50 mL Falcon tubes (Fisher, catalog number: 14-959-49A)
4. 1.5 mL Eppendorf tubes (Fisher, catalog number: 14-222-158)
5. Paper towels (Fisher, catalog number: 06-666-32B)
6. Saphire blade (World Precision Instruments, catalog number: 504072)
7. Sterile 32-gauge blunt 5 μL NEUROS Syringes (Stoelting, catalog number: 53496)
8. 24-well plate (Fisher, catalog number: 09-761-146)
9. Embedding molds (Fisher, catalog number: 2219)
10. Microtome blades (Fisher, catalog number: 4280L)
11. Superfrost Plus Gold slides (Fisher, catalog number: 1518848)
12. Hydrophobic barrier PAP pen (Cole-Parmer, catalog number: 7595553)
13. Coplin jars (Electron Microscopy Sciences, catalog number: 70315)
14. Coverslips (Fisher, catalog number: 15-541-000)
15. Nail polish (Fisher, catalog number: 50-949-071)
16. Kimwipes (Fisher, catalog number: 06-666)
Equipment
1. Mechanical micromanipulator (Sutter Instruments, model: MM-33)
2. Stereomicroscope (Olympus, model: SZ51)
3. Cryostat (Leica, model: CM1950)
4. Confocal microscope (Zeiss, model: LSM 800)
5. PC (Windows 11)
Software and datasets
1. FIJI (1.54F; free): https://imagej.net/software/fiji/
2. Zen (Black; free): https://www.zeiss.com/microscopy/us/products/software/zeiss-zen.htmL
3. Excel (2016; requires a license but Google Sheets can be used as a free alternative)
4. GraphPad Prism 9 (9.5.0; requires a license)
Procedure
A. Setting up the injection station
1. Position the syringe in the micromanipulator so it is at a 45° angle with the flat surface and microscope.
2. Wet a paper towel with zebrafish water and fold in half. The fish will be placed within the folds so that only the site of injection is exposed.
3. Place a folded paper towel in the lid of a Petri dish.
4. Have the sapphire blade ready to use.
5. Make the anesthesia solution.
6. Set up the recovery tank for zebrafish to be placed in after injections are complete.
B. Injecting zebrafish
1. Anesthetize zebrafish. Make sure the zebrafish is no longer responsive when lifting it out of the anesthesia solution with a plastic spoon before proceeding to the next step.
2. Place the zebrafish in the moist paper towel from step A3. Make sure only the head is exposed, with the eye to be injected facing upward (Figure 1A).
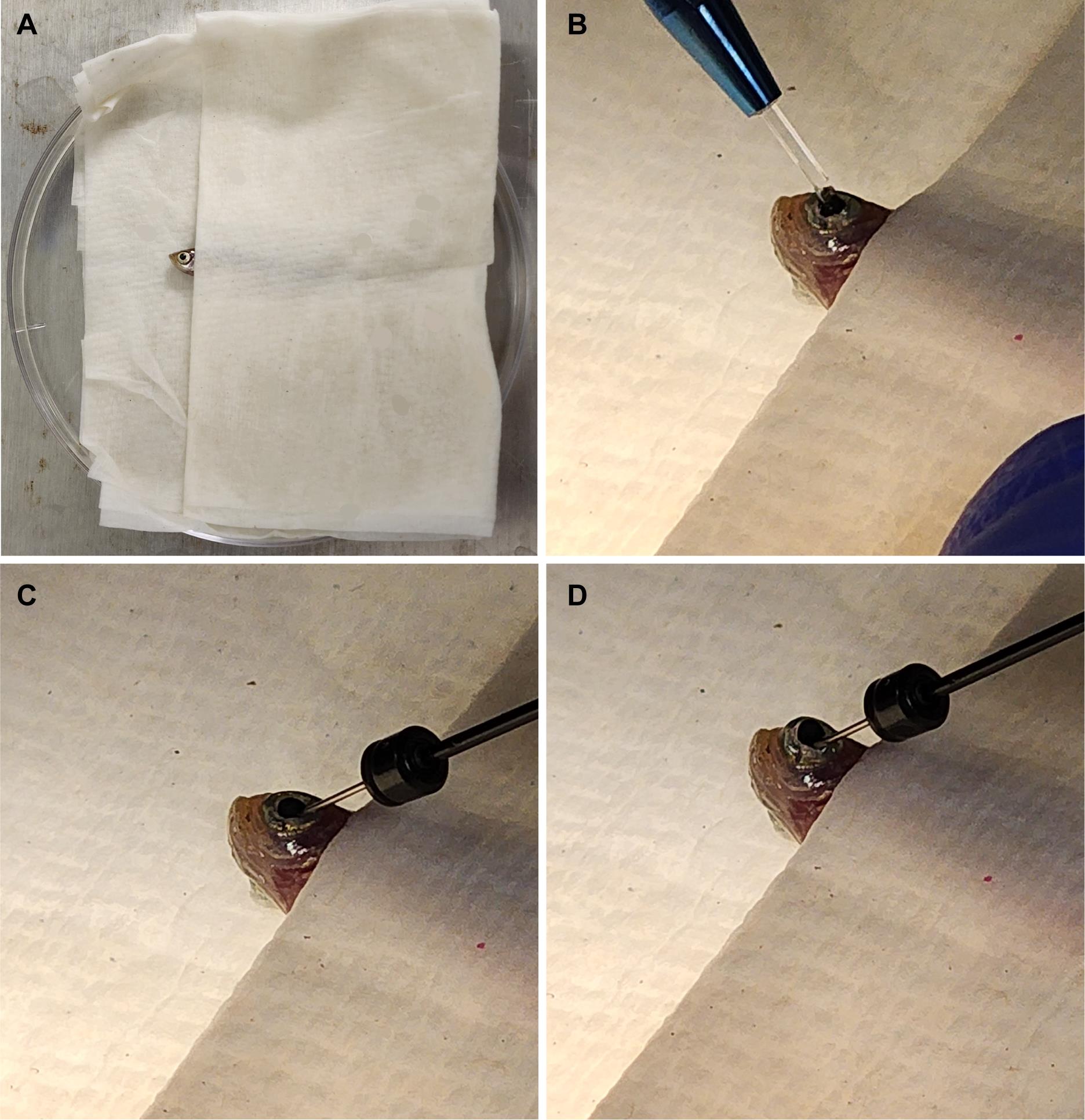
Figure 1. Fish setup for intravitreal injections. (A) Fish are placed in a moist towel so that only their heads are exposed for intravitreal injections after being anesthetized. (B) A sapphire blade is used to make an incision between the pupil and the outer edge of the iris. (C) A blunt syringe is used to remove 1.0 μL of vitreous humor from the eye. (D) 1.0 μL of the solution of interest is injected at the incision site. This image was adapted from Shihabeddin et al. [17].
3. Place the Petri dish lid with zebrafish under a microscope, viewing at approximately 10×.
4. Use the sapphire blade to make an incision between the pupil and the outer edge of the iris (Figure 1B).
5. Insert the syringe at the site of incision and remove 1.0 μL of vitreous humor. It is best to angle the needle toward the lens to avoid damaging the retina (Figure 1C). Important: Removal of the vitreous humor is critical for the oligonucleotides to reach and penetrate the retina.
6. Remove the syringe from the incision site and expel the vitreous humor collected.
7. Use a syringe to collect 1.0 μL of either Vivo-morpholino or siRNA solution.
8. Re-insert the syringe at the site of incision and inject solution into the eye (Figure 1D), being careful to avoid touching the retina.
9. Remove the syringe and place the zebrafish in the recovery tank.
10. Use a disposable pipette to aerate the tank until the zebrafish awakens.
A video of the injection procedure can be found in Shihabeddin et al. [17]: https://www.frontiersin.org/journals/cellular-neuroscience/articles/10.3389/fncel.2023.1321337/full#supplementary-material
C. Collecting zebrafish
1. Euthanize zebrafish immediately prior to dissection.
2. Excise the eyeball and place it in 1 mL of fixation solution for 30 min at room temperature. It can be placed in a 12-well plate.
3. Give the eyeball 3 × 5 min washes with 1 mL of 1× PBS solution at room temperature.
4. Place eyeballs in 1 mL of 25% sucrose solution overnight at 4 °C.
D. Embedding eyeball
1. Place dry ice in a container. Make sure that dry ice is very fine (can use a mallet to crush dry ice until fine).
2. Fill the embedding mold with O.C.T. compound.
3. Place the mold in dry ice until the bottom layer of O.C.T. compound is completely frozen. This step is optional but often helps keep the eyeball from misorienting during the embedding process.
4. Take the eyeball out from the sucrose solution and dab gently with a towel.
5. Place the mold under the microscope and insert the eyeball in the orientation of interest.
6. Place the mold with the eyeball in dry ice and let freeze for 30 min.
7. Move the mold into a -80 °C freezer until ready to cryosection.
E. Cryosectioning
1. Place the mold in the cryostat chamber for 15–20 min. This allows the mold to come up to the same temperature as the chamber. Typically, this is kept at -20 °C.
2. Remove the O.C.T. block with the embedded eyeball from the mold.
3. Embed the O.C.T. block onto the chuck.
4. Set up coarse and fine sectioning thicknesses. Typically, coarse thickness is used to cut away unembedded parts of the O.C.T block, while fine thickness is used for cutting tissue slices of interest. For this protocol, coarse thickness is set to 40 µm, and fine thickness is set to 12 µm.
5. Begin cryosectioning with coarse thickness until you reach the retina.
6. Switch to fine thickness and begin collecting tissue slices on the Superfrost Plus slides.
7. Collected slides can be stored at -20 °C until ready for immunohistochemistry.
F. Immunohistochemistry
1. Select the slides of interest and let them sit at room temperature for about 5 min.
2. Wash slides in a Coplin jar filled with 1× PBS, 3 × 10 min at room temperature.
3. Dab slides dry around the tissue sections with a Kimwipe, being careful not to touch the sections. Circle each section with a PAP pen.
4. Apply 30 μL of blocking solution to each sample. Incubate in the humidity chamber for 1 h at room temperature.
5. Remove blocking solution, apply 30 μL of primary antibody solution, and leave in the humidity chamber overnight at room temperature.
6. Wash slides in a Coplin jar with 1× PBS, 3 × 10 min at room temperature.
7. Apply 30 μL of secondary antibody solution and leave in the humidity chamber in the dark for 1 h at room temperature.
8. Wash slides in a Coplin jar with 1× PBS, 3 × 10 min at room temperature in the dark.
9. Dry slides as much as possible without drying out tissue samples.
10. Apply Vectashield mounting medium with DAPI.
11. Add coverslips to each slide and seal with nail polish. Keep slides in the dark. Slides can be stored at 4 °C until ready to image.
Note: Mounting medium needs a few hours to solidify, so it is best to wait that long before imaging.
G. Confocal imaging and image analysis
1. Image samples using a laser scanning confocal microscope and a 40× oil objective. Make sure to select lasers that can detect DAPI, Alexa 488, and Cy3 fluorescence. Take a z-stack that will sufficiently represent the entire depth of the retina tissue. Make sure that depth and laser settings are reused between each image acquisition. This is important for quantifications.
2. Export a maximum intensity projection of each z-stack taken (Figure 2A, B, D, and E).
3. Import the image into FIJI. Here, you can crop the size of the image to a specific region of interest that encompasses the total area covered by the gene of interest or keep it whole. However, make sure all images are cropped the same way. Downstream analysis will measure the expression levels for the protein of interest within the total area of the image.
4. Split the image by each channel. Under the Image tab at the top, select color, followed by split channels.
5. Under the Image tab at the top, select adjust, followed by Threshold… Keep the threshold the same for all images. After applying a threshold, a binary image mask will show the signal as white and the background as black.
6. Under the Analyze tab at the top, select histogram. Copy and paste the data into Excel.
7. Take the ratio of white pixels (signal) to total pixels and multiply by 100 to get the percentage of the region of interest that expresses the signal.
8. In GraphPad Prism 9, perform a two-tailed t-test to compare signals between the control and injected fish groups. A p-value <0.05 is considered significantly different (Figure 2C and F).
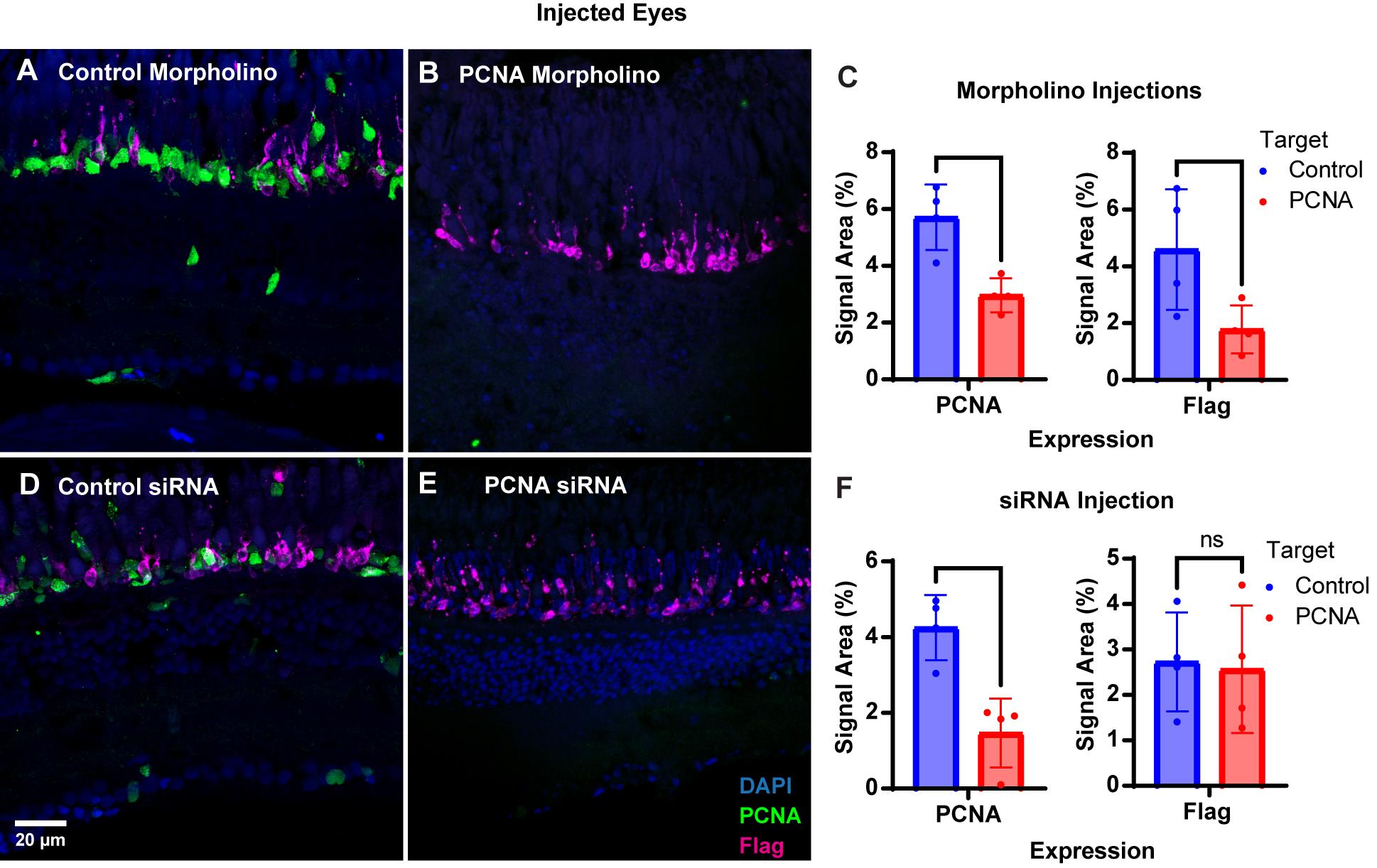
Figure 2. PCNA knockdown in retinas of injected eyes. (A) PCNA and Flag tag expression in retinas of eyes injected with control Vivo-morpholino. (B) PCNA and Flag tag expression in retinas of eyes injected with PCNA Vivo-morpholino. (C) Quantitative analysis comparing PCNA and Flag tag expression between conditions. (D) PCNA and Flag tag expression in retinas of eyes injected with control siRNA encapsulated through Altogen lipid nanoparticles. (E) PCNA and Flag tag expression in retinas of eyes injected with PCNA siRNA encapsulated through Altogen lipid nanoparticles. (F) Quantitative analysis comparing PCNA and Flag tag expression between conditions. Data in C and F are means ± 1SD. Two-tailed T-test results: ns = not significant; * p < 0.05; ** p < 0.01. This image was adapted from Shihabeddin et al. [17].
Validation of protocol
This protocol or parts of it has been used and validated in the following manuscripts:
Shihabeddin et al. [17]. Cost-effective strategies to knock down genes of interest in the retinas of adult zebrafish. Frontiers in Cellular Neuroscience (Figure 1A–D, Figure 3A–D, Figure 4A–E, Figure 5E–H, Figure 6A–F, and Figure 7A–D).
Shihabeddin et al. [18]. Master control genes in the regeneration of rod photoreceptors from endogenous progenitor cells in zebrafish retina. eLife (Figure 6A–I, Figure 7A–B, Figure 8A–D, and Figure 9A–G).
General notes and troubleshooting
1. It is very important that the vitreous humor be removed from the eye prior to injecting the siRNA or Vivo-morpholino solution. In this protocol, 1.0 μL of vitreous was removed, and 1.0 μL of solution was injected. Depending on the size of the zebrafish, this volume can be increased or decreased. This protocol has been optimized for zebrafish with a length of about 30 mm and a weight of 50–150 mg.
2. To increase the efficiency of gene knockdown through siRNAs, we combined siRNAs targeting different sites of the mRNA. This is not always necessary and can be adjusted so that only one site is targeted. Conversely, sometimes it will be necessary to utilize multiple siRNAs, as some may be ineffective.
3. With Vivo-morpholinos, the target sequence is very important. We have noticed that even a shift by about 3 nucleotides greatly affects efficacy. Furthermore, some genes may have multiple transcripts with different translation start sites. The design service from Gene Tools, LLC can provide recommendations for the best target sequences. Multiple Vivo-morpholinos can be mixed together and injected into the eye together.
4. When using PCNA Vivo-morpholinos, it was found that the non-injected contralateral eye would show a significant knockdown of PCNA expression. This indicates that the Vivo-morpholino is able to successfully exit the injected eye and eventually enter the contralateral eye. As this phenomenon has been seen with other Vivo-morpholinos in the lab, further investigation is needed to understand the mechanism at hand.
5. Both Vivo-morpholinos and lipid encapsulation delivery of siRNAs have given us issues with long-term storage. Vivo-morpholinos progressively lose efficacy as the stock solution ages and precipitates out of solution within a month. The Altogen transfection reagent has to be replaced after about 3 months. Always check reagents before starting to ensure that no precipitation is visible.
6. In our hands, translation-blocking Vivo-morpholino oligonucleotides had slightly higher efficacy than siRNAs. This is likely to result from a better penetration into cell types of interest. As we reported in [17], in vivo transfection reagents from some manufacturers did not work at all, indicating that the nanoparticle/liposome formulation needs to be optimized for the species and cell type studied.
7. The method used to fix eyeballs can vary depending on the antibody used or the downstream analysis of interest. For example, some antibodies require fixation with 2% or 4% PFA in 0.1 M phosphate buffer with no ethanol [16,19], while other downstream applications may require different fixation buffers altogether [20]. Table 1 lists antibodies that we have validated to work in zebrafish retina, along with fixation conditions that have worked for immunofluorescence experiments.
8. In this protocol, we validated knockdown through immunohistochemistry. For siRNAs, qPCR can also be performed to validate knockdown of the gene of interest, as well as genes downstream of the target. For Vivo-morpholinos, mass spec analysis can be performed to validate protein knockdown. Both approaches have been shown in [17,18].
Table 1. Fixation conditions for antibodies validated to work in the zebrafish retina
| Antibody | Host | Source | Catalog number | RRID | Fixative | References |
|---|---|---|---|---|---|---|
| BrdU (BU-1) | Ms | Invitrogen | MA3-071 | AB_10986341 | 4% PFA, works in EtOH/PFA | [16,18] |
| Connexin 35/36 | Ms | Millipore | MAB3045 | AB_94632 | 2% PFA, 4% carbodiimide | [20] |
| Flag-DDK | Ms | Origene | TA50011 | AB_2622345 | 4% PFA, works in EtOH/PFA | [16–18] |
| Glutamine synthetase (GS6) | Ms | Millipore | MAB302 | AB_2110656 | 4% PFA | [16] |
| PCNA | Ms | Abcam | ab29 | AB_303394 | EtOH/PFA | [16] |
| PCNA | Rb | Abcam | Ab18197 | AB_444313 | EtOH/PFA | [16,18,19,21] |
| PKCα | Rb | Millipore | P4334 | AB_477345 | 4% PFA, works in EtOH/PFA | [16,18,19] |
| Rhodopsin (Retp1) | Ms | Novus Biologicals | NB120-3267 | AB_791922 | 4% PFA, works in EtOH/PFA | [16,18,19] |
| SV2 | Ms | Developmental Studies Hybridoma Bank | SV2 | AB_2315387 | 4% PFA | [16,20] |
| ZO-1 | Ms | ThermoFisher | 33-9100 | AB_2533147 | 4% PFA | [19] |
| Zpr1/Fret43 | Ms | Zebrafish International Resource Center (ZIRC) | ab174435 | AB_10013803 | 4% PFA, works in EtOH/PFA | [16,18,20] |
Acknowledgments
E.S. conceived the project. E.S., S.T., A.A., and A.S. performed experiments and analyzed data. J.O. and E.S. acquired funding. E.S. wrote the original draft. All authors reviewed and edited the manuscript.
The authors would like to thank Dr. Steven W. Wang for helpful discussions in injection technique, quantification, and reagents available to use. This protocol was validated in Shihabeddin et al. [17] and used in Shihabeddin et al. [18].
Competing interests
The authors declare no competing interests.
Ethical considerations
All procedures comply with the U.S. Public Health Service policy on humane care and use of laboratory animals and the NRC Guide for the Care and Use of Laboratory Animals and have been reviewed and approved by the Institutional Animal Care and Use Committees at the University of Texas Health Science Center at Houston under protocol HSC-AWC-21-0040 and at the University of Houston under protocol PROTO202100037.
References
- Turchinovich, A., Zoidl, G. and Dermietzel, R. (2010). Non-viral siRNA delivery into the mouse retina in vivo. BMC Ophthalmol. 10(1): 1–5. https://doi.org/10.1186/1471-2415-10-25
- Hatakeyama, H., Wu, S. Y., Mangala, L. S., Lopez-Berestein, G. and Sood, A. K. (2016). Assessment of In Vivo siRNA Delivery in Cancer Mouse Models. Methods Mol Biol. 189–197. https://doi.org/10.1007/978-1-4939-3378-5_15
- Hana, S., Peterson, M., McLaughlin, H., Marshall, E., Fabian, A. J., McKissick, O., Koszka, K., Marsh, G., Craft, M., Xu, S., et al. (2021). Highly efficient neuronal gene knockout in vivo by CRISPR-Cas9 via neonatal intracerebroventricular injection of AAV in mice. Gene Ther. 28: 646–658. https://doi.org/10.1038/s41434-021-00224-2
- Thummel, R., Bailey, T. J. and Hyde, D. R. (2011). In vivo Electroporation of Morpholinos into the Adult Zebrafish Retina. J Visualized Exp. e3791/3603. https://doi.org/10.3791/3603
- Craig, S. E. L., Thummel, R., Ahmed, H., Vasta, G. R., Hyde, D. R. and Hitchcock, P. F. (2010). The Zebrafish Galectin Drgal1-L2 Is Expressed by Proliferating Müller Glia and Photoreceptor Progenitors and Regulates the Regeneration of Rod Photoreceptors. Invest Ophthalmol Vis Sci. 51(6): 3244. https://doi.org/10.1167/iovs.09-4879
- Thummel, R., Enright, J. M., Kassen, S. C., Montgomery, J. E., Bailey, T. J. and Hyde, D. R. (2010). Pax6a and Pax6b are required at different points in neuronal progenitor cell proliferation during zebrafish photoreceptor regeneration. Exp Eye Res. 90(5): 572–582. https://doi.org/10.1016/j.exer.2010.02.001
- Morcos, P. A., Li, Y. and Jiang, S. (2008). Vivo-Morpholinos: A Non-Peptide Transporter Delivers Morpholinos into a Wide Array of Mouse Tissues. Biotechniques. 45(6): 613–623. https://doi.org/10.2144/000113005
- Kim, S., Radhakrishnan, U. P., Rajpurohit, S. K., Kulkarni, V. and Jagadeeswaran, P. (2010). Vivo-Morpholino knockdown of αIIb: A novel approach to inhibit thrombocyte function in adult zebrafish. Blood Cells Mol Dis. 44(3): 169–174. https://doi.org/10.1016/j.bcmd.2009.12.004
- Kizil, C., Iltzsche, A., Kaslin, J. and Brand, M. (2013). Micromanipulation of Gene Expression in the Adult Zebrafish Brain Using Cerebroventricular Microinjection of Morpholino Oligonucleotides. J Visualized Exp. e3791/50415. https://doi.org/10.3791/50415
- Hughes, C. E., Radhakrishnan, U. P., Lordkipanidzé, M., Egginton, S., Dijkstra, J. M., Jagadeeswaran, P. and Watson, S. P. (2012). G6f-Like Is an ITAM-Containing Collagen Receptor in Thrombocytes. PLoS One. 7(12): e52622. https://doi.org/10.1371/journal.pone.0052622
- Pfefferli, C., Müller, F., Jaźwińska, A. and Wicky, C. (2014). Specific NuRD components are required for fin regeneration in zebrafish. BMC Biol. 12(1): e1186/1741–7007–12–30. https://doi.org/10.1186/1741-7007-12-30
- Kyritsis, N., Kizil, C., Zocher, S., Kroehne, V., Kaslin, J., Freudenreich, D., Iltzsche, A. and Brand, M. (2012). Acute Inflammation Initiates the Regenerative Response in the Adult Zebrafish Brain. Science. 338(6112): 1353–1356. https://doi.org/10.1126/science.1228773
- Chiang, K. Y., Li, Y. W., Li, Y. H., Huang, S. J., Wu, C. L., Gong, H. Y. and Wu, J. L. (2021). Progranulin A Promotes Compensatory Hepatocyte Proliferation via HGF/c-Met Signaling after Partial Hepatectomy in Zebrafish. Int J Mol Sci. 22(20): 11217. https://doi.org/10.3390/ijms222011217
- Meister, G. and Tuschl, T. (2004). Mechanisms of gene silencing by double-stranded RNA. Nature. 431(7006): 343–349. https://doi.org/10.1038/nature02873
- Han, H. (2018). RNA Interference to Knock Down Gene Expression. Methods Mol Biol. : 293–302. https://doi.org/10.1007/978-1-4939-7471-9_16
- Santhanam, A., Shihabeddin, E., Atkinson, J. A., Nguyen, D., Lin, Y. P. and O’Brien, J. (2020). A Zebrafish Model of Retinitis Pigmentosa Shows Continuous Degeneration and Regeneration of Rod Photoreceptors. Cells. 9(10): 2242. https://doi.org/10.3390/cells9102242
- Shihabeddin, E., Santhanam, A., Aronowitz, A. L. and O’Brien, J. (2024). Cost-effective strategies to knock down genes of interest in the retinas of adult zebrafish. Front Cell Neurosci. 17: e1321337. https://doi.org/10.3389/fncel.2023.1321337
- Shihabeddin, E., Santhanam, A., Tetenborg, S., Aronowitz, A. L., Wei, H., Qin, G., Cai, C., Wu, J. and O’Brien, J. (2025). Master control genes in the regeneration of rod photoreceptors from endogenous progenitor cells in zebrafish retina. e105965. https://doi.org/10.7554/elife.105965
- Santhanam, A., Shihabeddin, E., Wei, H., Wu, J. and O’Brien, J. (2023). Molecular basis of retinal remodeling in a zebrafish model of retinitis pigmentosa. Cell Mol Life Sci. 80(12): 362. https://doi.org/10.1007/s00018-023-05021-1
- Li, H., Chuang, A. Z. and O'Brien, J. (2009). Photoreceptor Coupling Is Controlled by Connexin 35 Phosphorylation in Zebrafish Retina. J Neurosci. 29(48): 15178–15186. https://doi.org/10.1523/jneurosci.3517-09.2009
- Thummel, R., Kassen, S. C., Montgomery, J. E., Enright, J. M. and Hyde, D. R. (2007). Inhibition of Müller glial cell division blocks regeneration of the light‐damaged zebrafish retina. Dev Neurobiol. 68(3): 392–408. https://doi.org/10.1002/dneu.20596
Article Information
Publication history
Received: Apr 6, 2025
Accepted: Jul 22, 2025
Available online: Aug 12, 2025
Published: Sep 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Shihabeddin, E., Santhanam, A., Tetenborg, S., Aronowitz, A. L. and O`Brien, J. (2025). Efficient Gene Knockdown in Adult Zebrafish Retina by Intravitreal Injection. Bio-protocol 15(17): e5436. DOI: 10.21769/BioProtoc.5436.
Category
Neuroscience > Sensory and motor systems > Retina
Cell Biology > Cell engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.