- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Simultaneous Capture of Chromatin-Associated RNA and Global RNA–RNA Interactions With Reduced Input Requirements
(*contributed equally to this work) Published: Vol 15, Iss 17, Sep 5, 2025 DOI: 10.21769/BioProtoc.5430 Views: 3600
Reviewed by: Wei DaiAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2025

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Chromatin-associated RNAs (caRNAs) have been increasingly recognized as key regulators of gene expression and genome architecture. A few technologies, such as ChRD-PET and RedChIP, have emerged to assess protein-mediated RNA–chromatin interactions, but each has limitations. Here, we describe the TaDRIM-seq (targeted DNA-associated RNA and RNA–RNA interaction mapping by sequencing) technique, which combines Protein G (PG)-Tn5-targeted DNA tagmentation with in situ proximity ligation to simultaneously profile caRNAs across genomic regions and capture global RNA–RNA interactions within intact nuclei. This approach reduces the required cell input, shortens the experimental duration compared to existing protocols, and is applicable to both mammalian and plant systems.
Key features
• A multi-omics sequencing strategy.
• Compatible with mammalian and plant systems.
• Profiling of epigenome, RNA–DNA, and RNA–RNA interactomes.
Keywords: Chromatin-associated RNAs (caRNAs)Graphical overview
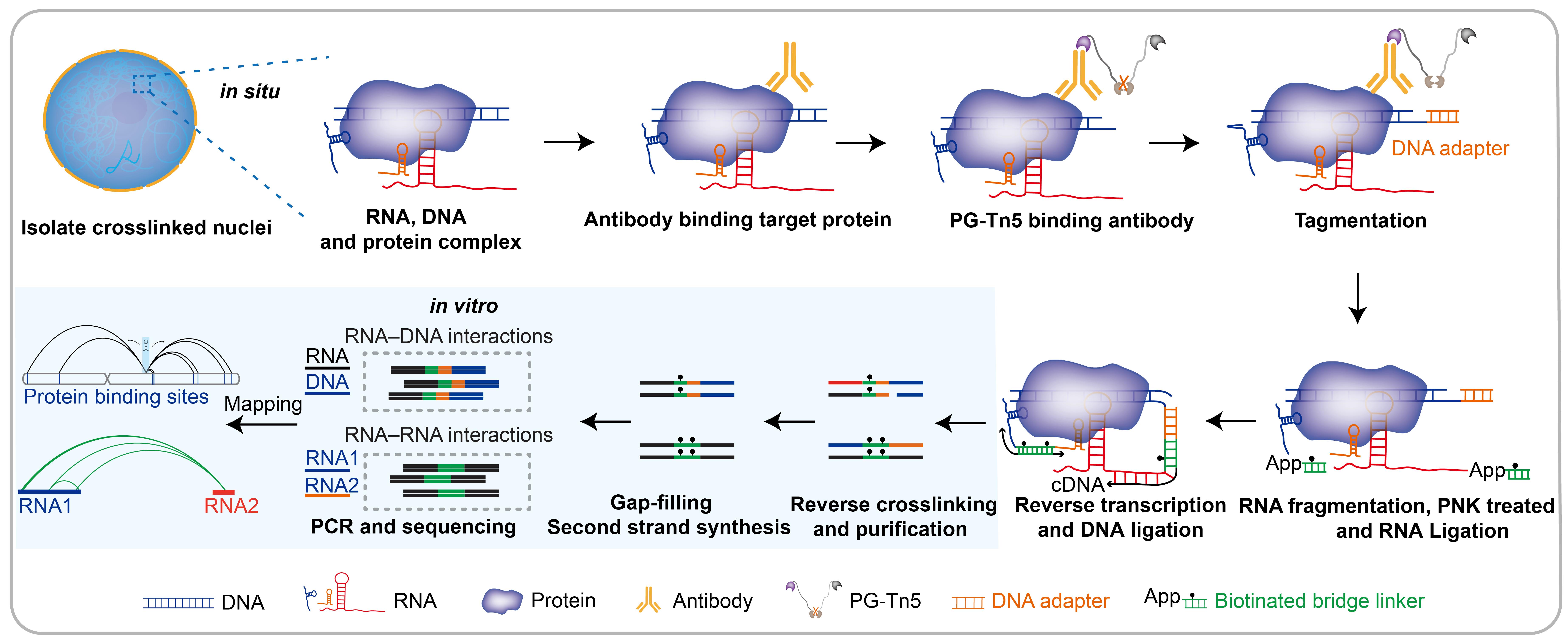
Targeted DNA-associated RNA and RNA–RNA interaction mapping by sequencing (TaDRIM-seq) technique schematic
Background
Chromatin-associated RNAs (caRNAs) have been proposed to represent an additional regulatory layer of the epigenome [1]. Some caRNAs act in trans, being recruited to distant genomic loci (trans-acting RNAs), while others function in cis, remaining enriched at their transcription sites (cis-acting RNAs) [2]. Increasing evidence has demonstrated that caRNAs play crucial roles in regulating gene transcription [3–5], X-chromosome inactivation [6], and the three-dimensional organization of the genome [7,8].
Current methods, such as ChRD-PET [9] and RedChIP [10], enable the identification of DNA elements targeted by specific caRNAs. However, these approaches are resource-intensive, time-consuming, and inefficient due to in vitro ligation limitations, and they are unable to capture RNA–RNA interactions. To overcome these limitations, we recently developed a method named TaDRIM-seq [11]. In this protocol, we provide a step-by-step guide for performing TaDRIM-seq in rice and human cells. The assay is also compatible with other cell lines, as well as plant and animal tissues, making it broadly applicable to the study of caRNAs-mediated chromatin regulation.
Materials and reagents
Biological materials
1. Human K562 (ATCC, catalog number: CCL-243)
2. Rice leaf tissue [Xian/indica cv. Minghui 63 (MH63)]
Reagents
1. Formaldehyde (37%) (Sigma-Aldrich, catalog number: F8775)
2. Glycine (Sigma-Aldrich, catalog number: G7126-1kg)
3. PBS (10×) (Invitrogen, catalog number: AM9625)
4. HEPES (1 M) (Coolaber, catalog number: SL6056-500mL)
5. NaCl (5 M) (Invitrogen, catalog number: AM9759)
6. Spermidine (Sigma-Aldrich, catalog number: S2626-1g)
7. Protease inhibitor cocktail tablets (Roche, catalog number: 58914000)
8. RiboLock RNase inhibitor (40 U/μL) (Invitrogen, catalog number: EO0381)
9. Nuclease-free water (NF-H2O) (Thermo Fisher Scientific, catalog number: EF0651)
10. Digitonin (5%) (Thermo Fisher Scientific, catalog number: BN2006)
11. EDTA (0.5 M) (Invitrogen, catalog number: AM9260G)
12. BSA (20 mg/mL) (NEB, catalog number: B9000S)
13. MgCl2 (1 M) (Invitrogen, catalog number: AM9530G)
14. SDS (10%) (Invitrogen, catalog number: AM9822)
15. dNTPs (10 mM) (Invitrogen, catalog number: 18427088)
16. Triton X-100 (Sigma-Aldrich, catalog number: 9036-19-5)
17. H3K4me3 antibody (50 μg) (Abcam, catalog number: ab8580)
18. H3K27me3 antibody (100 μg) (Abcam, catalog number: ab6002)
19. Tris-HCl (1 M, pH 7.5) (Invitrogen, catalog number: 15567027)
20. rCutSmartTM buffer (10×) (NEB, catalog number: B6004V)
21. T4 polynucleotide kinase (T4 PNK, 10 U/μL) (Thermo Fisher Scientific, catalog number: EK0032)
22. T4 polynucleotide kinase reaction buffer (10×) (Thermo Fisher Scientific, catalog number: EK0032)
23. 5′ DNA Adenylation kit (NEB, catalog number: E2610L)
24. T4 RNA ligase 2, truncated KQ (200 U/μL) (NEB, catalog number: M0373L)
25. T4 RNA ligase reaction buffer (10×) (NEB, catalog number: B0216S)
26. PEG 8000, 50% (w/v) (NEB, catalog number: B1004S)
27. T4 DNA Ligase (400 U/μL) (NEB, catalog number: M0202S)
28. DNA ligase reaction buffer (10×) (NEB, catalog number: B0202S)
29. Phenol/chloroform/isoamyl alcohol (phenol/ChCl3/IAA, 25:24:1, pH 7.9) (Ambion, catalog number: AM9730)
30. Sodium acetate (3 M, pH 5.2) (VWR, catalog number: E521-100mL)
31. Ethanol absolute (pure) (Sangon Biotech, catalog number: A500737)
32. T4 DNA polymerase (3 U/μL) (NEB, catalog number: M0203L)
33. NEBufferTM r2.1 (10×) (NEB, catalog number: B6002S)
34. Tween 20 (Sigma-Aldrich, catalog number: P9416-100ml)
35. Isopropanol (Sigma-Aldrich, catalog number: I-9516-500ml)
36. Buffer EB (QIAGEN, catalog number: 19086-250ml)
37. ATP (10 mM) (NEB, catalog number: P0756L)
38. RNase remover and nucleic acid decontamination reagent (Vazyme, catalog number: R504)
39. Hyperactive pG-Tn5 Transposase (500 ng/μL) (Vazyme, catalog number: S602)
40. TruePrep DNA Library Prep Kit V2 for Illumina (Vazyme, catalog number: TD501)
41. DNA Clean Beads (Vazyme, catalog number: N411-03)
42. DynabeadsTM M-280 (10 mg/mL) (Invitrogen, catalog number: 11205D)
43. I-Block protein-based blocking reagent (Thermo Fisher, catalog number: T2015)
44. M-MuLV reverse transcriptase (200 U/μL) (NEB, catalog number: M0253L)
45. M-MuLV reverse transcriptase reaction buffer (10×) (NEB, catalog number: B0253L)
46. RNase IF (NEB, catalog number: M0243S)
47. NEBNext® second strand synthesis enzyme mix (NEB, catalog number: E6111L)
48. DNA Clean & Concentrator-5 (Zymo, catalog number: D4013)
49. Oligo Clean & Concentrator (Zymo, catalog number: D4060)
50. Primers (synthesized by Sangon Biotech with HPLC purification)
Solutions
1. Buffer S (see Recipes)
2. Buffer F (see Recipes)
3. 1× wash buffer A (see Recipes)
4. Antibody buffer (see Recipes)
5. 1× transposase incubation buffer (see Recipes)
6. 1× wash buffer B (see Recipes)
7. Lysis buffer (see Recipes)
8. Proteinase K solution buffer (see Recipes)
9. iBlock buffer (see Recipes)
10. 2× binding and washing buffer (see Recipes)
11. 1× binding and washing buffer (see Recipes)
12. 2× SSC/0.5% (w/v) SDS (see Recipes)
13. Sheared DNA solution (see Recipes)
Recipes
1. Buffer S
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M HEPES | 50 mM | 0.5 mL |
| 5 M NaCl | 150 mM | 0.3 mL |
| 0.5M EDTA | 1 mM | 0.02 mL |
| Triton X-100 | 1% | 0.1 mL |
| 10% sodium deoxycholate | 0.1% | 0.1 mL |
| 10% SDS | 1% | 1 mL |
| 50× protease inhibitors | 1× | 0.2 mL |
| RiboLock RNase (40 U/μL) | 0.2 U/μL | 0.05 mL |
| H2O | n/a | 7.73 mL |
| Total | n/a | 10 mL |
Note: This buffer should be prepared fresh before use.
2. Buffer F
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M HEPES | 50 mM | 0.5 mL |
| 5 M NaCl | 150 mM | 0.3 mL |
| 2 M Spermidine | 1 mM | 0.005 mL |
| Triton X-100 | 1% | 0.1 mL |
| 10% sodium deoxycholate | 0.1% | 0.1 mL |
| 50× protease inhibitors | 1× | 0.2 mL |
| RiboLock RNase (40 U/μL) | 0.2 U/μL | 0.05 mL |
| H2O | n/a | 8.745 mL |
| Total | n/a | 10 mL |
Note: This buffer should be prepared fresh before use.
3. 1× wash buffer A
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M HEPES (pH 7.6) | 20 mM | 100 μL |
| 5 M NaCl | 150 mM | 150 μL |
| 2 M Spermidine | 0.5 mM | 1.25 μL |
| 50× protease inhibitors | 1× | 100 μL |
| RiboLock RNase (40 U/μL) | 0.2 U/μL | 25 μL |
| H2O | n/a | 4.623 mL |
| Total | n/a | 5 mL |
Note: This buffer should be prepared fresh before use.
4. Antibody buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1× wash buffer A | n/a | 495 μL |
| 0.5M EDTA | 2 mM | 2 μL |
| 30% BSA | 0.1% | 1.6 μL |
| 5% Digitonin | 0.05% | 5 μL |
| Total | n/a | 503.6 μL |
Note: This buffer should be prepared fresh before use.
5. 1× transposase incubation buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M HEPES (pH 7.6) | 20 mM | 100 μL |
| 5 M NaCl | 300 mM | 300 μL |
| 2 M Spermidine | 0.5 mM | 1.25 μL |
| 5% Digitonin | 0.01% | 10 μL |
| 50× protease inhibitors | 1× | 100 μL |
| RiboLock RNase (40 U/μL) | 0.2 U/μL | 25 μL |
| H2O | n/a | 4.467 mL |
| Total | n/a | 5 mL |
Note: This buffer should be prepared fresh before use.
6. 1× wash buffer B
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl (pH 7.5) | 20 mM | 1 mL |
| 1 M MgCl2 | 10 mM | 0.5 mL |
| 10% Tween 20 | 0.2% | 1 mL |
| RiboLock RNase (40 U/μL) | 0.2 U/μL | 0.25 mL |
| H2O | n/a | 47.25 mL |
| Total | n/a | 50 mL |
Note: This buffer should be prepared fresh before use.
7. Lysis buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl (pH 7.5) | 10 mM | 10 μL |
| 5 M NaCl | 10 mM | 2 μL |
| 10% NP40 | 0.2% | 20 μL |
| 50× protease inhibitors | 1× | 20 μL |
| RiboLock RNase (40 U/μL) | 0.2 U/μL | 5 μL |
| H2O | n/a | 943 μL |
| Total | n/a | 1 mL |
Note: This buffer should be prepared fresh before use.
8. Proteinase K solution buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl (pH 7.5) | 50 mM | 50 μL |
| 10% SDS | 1% | 100 μL |
| 5 M NaCl | 100 mM | 20 μL |
| 0.5 M EDTA | 1 mM | 2 μL |
| Proteinase K (20mg/mL) | 1 mg/mL | 50 μL |
| H2O | n/a | 778 μL |
| Total | n/a | 1 mL |
Note: This buffer should be prepared fresh before use.
9. iBlock buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| iBlock | 2% (w/v) | 0.2 g |
| 10% SDS | 0.5% | 0.5 mL |
| H2O | n/a | 9.5 mL |
| Total | n/a | 10 mL |
Note: Incubate the reagent in a 65 °C water bath to facilitate dissolution. The buffer is stable for up to 6 months when stored at room temperature.
10. 2× binding and washing buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl (pH 8) | 10 mM | 0.5 mL |
| 5 M NaCl | 2 M | 20 mL |
| 0.5 M EDTA | 1 mM | 0.1 mL |
| H2O | n/a | 29.4 mL |
| Total | n/a | 50 mL |
Note: This buffer is stable for up to 6 months when stored at 4 °C.
11. 1× binding and washing buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl (pH 8) | 10 mM | 0.5 mL |
| 5 M NaCl | 1 M | 10 mL |
| 0.5 M EDTA | 1 mM | 0.1 mL |
| H2O | n/a | 39.4 mL |
| Total | n/a | 50 mL |
Note: This buffer is stable for up to 6 months when stored at 4 °C.
12. 2× SSC/0.5% (w/v) SDS
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 20× SSC | 2× | 5 mL |
| 10% SDS | 0.5% | 2.5 mL |
| H2O | n/a | 42.5 mL |
| Total | n/a | 50 mL |
Note: This buffer can be stored at room temperature for up to 3 months; however, lower room temperatures may cause SDS to precipitate. If this occurs, place the buffer in a 25 °C water bath to fully dissolve the SDS before use.
13. Sheared DNA solution
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Sheared DNA | 0.5 ng/μL | x μL |
| 2× binding and washing buffer | 1× | 50 μL |
| H2O | n/a | 50 – x μL |
| Total | n/a | 100 μL |
Note: The genomic DNA isolated from plant or animal was fragmented into 300–500 bp fragments by sonication using a Bioruptor (Diagenode) set to high intensity with 30 cycles (30 s ON, 50 s OFF) at 4 °C. The sheared DNA solution is stable for up to 6 months when stored at -20 °C.
Equipment
1. Eppendorf Thermo Mixer® C (Eppendorf, model: ThermoMixer C)
2. Centrifuge (Eppendorf, model: 5425R)
3. DynaMag-2 Magnet (Life Technologies, catalog number: 12321D)
4. Thermal Cycler (Bio-Rad, model: T100TM)
5. Vortex-Genie Mixers (VWR, catalog number: 58816-121)
6. Low-temperature incubator (Being, catalog number: BC-60L)
7. Qubit 2.0 Fluorometer (Invitrogen, catalog number: Q32866)
8. Intelli-mixer RM-2L (Rose Scientific, catalog number: MX1000)
9. Bioruptor® Plus sonication device (Diagenode)
Software and datasets
1. All data and code have been deposited to GitHub: https://github.com/304243504/HiR
Procedure
A. Crosslinking (1–1.5 h)
1. Cells
a. Prepare the cell suspension and wash the cells twice with 1× PBS. Pin down the cells by centrifugation at 1,000× g for 5 min.
b. Resuspend the cells with 1 mL of 1% formaldehyde in 1× PBS for 10 min with rotation at room temperature.
Critical step: Using a formaldehyde concentration lower than 1% can result in a low signal-to-noise ratio.
c. Add glycine to a final concentration of 0.2 M for 5 min at room temperature with rotation to quench the crosslinking reaction. Wash cells three times with 1× PBS and store at -80 °C.
2. Plant tissue
a. Cross-link mature rice leaves with 1% formaldehyde (add 4 mL of 37% formaldehyde to 144 mL of 1× PBS) and apply vacuum for 20 min.
b. Quench formaldehyde by adding 13.2 mL of 2.5 M glycine to the media to a final concentration of 0.2 M and vacuum for 5 min.
c. Wash the leaves three times with NF-H2O and store at -80 °C.
B. pG-Tn5 transposase assembly (timing: 2.5 h)
1. Dissolve primer A (5′-phos-CTGTCTCTTATACACATCTT-3′) and primer B (5′-phos-AAGATGTGTATAAGAGACAG-3′) to 100 μM in annealing buffer.
2. Mix the equimolar mixture of primer A and B (primer mix) and perform the PCR program as described in Table 1.
Table 1. PCR program for primer annealing
| Temperature (°C) | Time (min) |
|---|---|
| 75 | 15 |
| 60 | 10 |
| 50 | 10 |
| 40 | 10 |
| 25 | 30 |
3. Prepare the assembled mixture as in Table 2.
Table 2. Assembly of pG-Tn5 transposase reaction mix (TTE mix)
| Reagent | Volume (μL) |
|---|---|
| Hyperactive pG-Tn5 Transposase (500 ng/μL) | 20 |
| Primer mix | 3.5 |
| Coupling buffer | 14 |
4. Thoroughly mix the components and incubate for 1 h at 30 °C using a thermal cycler. Label as TTE Mix.
C. Preparation of bridge linker (timing: ~2 h)
1. Dissolve the bridge linker forward and reverse oligonucleotides in NF-H2O to a final concentration of 100 μM.
The forward strand: 5′-[5Phos]CCTCGATAC/iBIOdT/TGAGCTGAC-3′
Reverse strand: 5′-[5Phos]GTCAGC/iBIOdT/CAAGTATCGAG-3′
Note: The iBIOdT-modified primers were custom-synthesized by Sangon Biotech with HPLC purification.
2. Prepare the adenylation reaction mixture (Table 3).
Table 3. Adenylation reaction mix for bridge linker preparation
| Reagent | Volume (μL) |
|---|---|
| 10× 5ʹ DNA adenylation reaction buffer | 12 |
| 1 mM ATP | 12 |
| Forward strand oligo (100 μM) | 6 |
| Mth RNA ligase | 12 |
| Nuclease-free water | 78 |
3. Incubate the mixture at 65 °C for 1 h to allow annealing, then heat at 85 °C for 5 min to inactivate enzymatic activity.
4. Add 6 μL of the reverse strand oligo to the adenylation reaction mixture and incubate by PCR using the following program: 95 °C for 2 min; ramp from 95 °C to 75 °C at 0.1 °C/s, hold at 75 °C for 2 min; ramp from 75 °C to 65 °C at 0.1 °C/s, hold at 65 °C for 2 min; ramp from 65 °C to 50 °C at 0.1 °C/s, hold at 50 °C for 2 min; ramp from 50 °C to 37 °C at 0.1 °C/s, hold at 37 °C for 2 min; ramp from 37 °C to 20 °C at 0.1 °C/s, hold at 20 °C for 2 min and hold at 4 °C.
5. Purify the annealed linker using the Oligo Clean & Concentrator kit (Zymo) and elute in 100 μL of nuclease-free water. Label as the bridge linker.
D. Nuclei preparation and tagmentation (timing: ~1 h)
1. Plant nuclei isolation
a. Grind the crosslinked tissue (0.3–0.5 g) into a fine powder using liquid nitrogen.
b. Add 300 μL of Buffer S to the powdered tissue and incubate at 4 °C for 30 min with gentle rotation.
c. Add 1.2 mL of Buffer F and continue incubation at 4 °C for 15 min with rotation. Centrifuge at 1,000× g for 10 min at 4 °C to collect the nuclei.
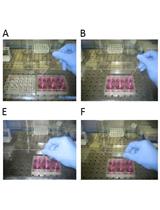
Note: After nuclei isolation, the experiment should be performed immediately. The extraction from mature rice leaves is shown in Figure 1.

Figure 1. Isolated nuclei are stained with DAPI. Crosslinked rice tissue was ground into a fine powder in liquid nitrogen. The powder was suspended in buffer S/F, and the nuclei were isolated by centrifugation. The nuclei were stained with DAPI and visualized using a fluorescence microscope. Scale bar: 10 μm.
2. Cell nuclei isolation
a. Resuspend crosslinked cells (1 × 106 to 1 × 107) in 1 mL of lysis buffer and incubate for 10 min on ice followed by centrifugation at 2,500× g for 3 min at 4 °C.
3. pG-Tn5 transposase binding to the antibody and tagmentation
a. Wash cells or plant nuclei twice with 300 μL of 1× wash buffer A. Collect nuclei by centrifugation at 2,500× g for 3 min at 4 °C.
b. Resuspend nuclei in 500 μL of antibody buffer, add 5–10 μg of antibody, and incubate at 4 °C for 2 h with rotation.
c. Collect nuclei by centrifugation at 2,500× g for 3 min at 4 °C. Wash the nuclei with 300 μL of 1× wash buffer A and perform 3–5 washing steps.
d. Resuspend nuclei in 500 μL of 1×transposase incubation buffer. Add 5 μL of TTE Mix and incubate at 4 °C for 1 h with rotation.
e. Collect the nuclei by centrifugation at 2,500× g for 3 min at 4 °C and wash 3–5 times with 300 μL of 1× transposase incubation buffer.
f. Resuspend nuclei in 500 μL of 1× transposase incubation buffer, add 5 μL of 1 M MgCl2, and incubate at 37 °C for 1 h.
g. Add 500 μL of 40 mM EDTA to stop the reaction.
h. Collect nuclei by centrifugation at 2,500× g for 3 min at 4 °C. Wash the nuclei pellet twice with 500 μL of 1× PBS and then incubate in 200 μL of 0.5% SDS at 62 °C for 10 min.
i. Immediately quench the SDS by adding 580 μL of nuclease-free water and 100 μL of 10% (v/v) Triton X-100. Incubate at 37 °C for 15 min with rotation.
E. RNA fragmentation and PNK treated (timing: ~1.5 h)
1. Collect nuclei by centrifugation at 2,500× g for 3 min and wash twice with 500 μL of 1× wash buffer B.
2. Resuspend nuclei in 500 μL of 1× CutSmart buffer, add 1.6 μL of RNase IF (previously diluted 10-fold in 1× PBS), and incubate at 37 °C for 3 min, followed by an additional 10-min incubation on ice.
3. Prepare the PNK mixture as in Table 4.
Table 4. Reaction mixture for T4 polynucleotide kinase
| Reagent | Volume (μL) |
|---|---|
| 10× T4 PNK reaction buffer | 40 |
| 100 mM ATP | 4 |
| RiboLock RNase | 10 |
| T4 PNK | 20 |
| Nuclease-free water | 326 |
4. Resuspend nuclei and incubate at 37 °C for 40 min in a thermo mixer with shaking at 500 rpm.
F. In situ ligation DNA and RNA (timing: overnight reaction)
1. Wash the isolated nuclei twice with 500 μL of 1× wash buffer B, then perform in situ RNA ligation using the prepared ligation solution (Table 5).
Table 5. T4 RNA polymerase master mix
| Reagent | Volume (μL) |
|---|---|
| 10× T4 RNA ligase reaction buffer | 40 |
| Bridge linker (from section C) | 50 |
| RiboLock RNase | 10 |
| 50% (w/v) PEG 8000 | 120 |
| 10% Triton X-100 | 4 |
| T4 RNA Ligase 2 truncated KQ | 20 |
| Nuclease-free water | 156 |
2. Resuspend nuclei and incubate overnight at 16 °C using a Intelli-mixer RM-2L with rotation.
3. Collect the nuclei by centrifugation at 2,500× g for 3 min at 4 °C. Wash the pellet three times with 500 μL of 1× wash buffer B and add the reverse transcription solution. Perform reverse transcription as in Table 6.
Table 6. Reverse transcription mixture
| Reagent | Volume (μL) |
|---|---|
| 10× MMLV buffer | 40 |
| 10mM dNTP | 20 |
| RiboLock RNase | 10 |
| MMLV (RNase H-) | 20 |
| Nuclease-free water | 310 |
4. Resuspend nuclei and then incubate at 42 °C for 1 h in a ThermoMixer with shaking at 500 rpm.
5. Wash isolated nuclei three times with 500 μL of 1× wash buffer B and perform in situ DNA ligation solution (Table 7).
Table 7. T4 DNA polymerase master mix
| Reagent | Volume (μL) |
|---|---|
| 10× DNA ligase reaction buffer with 10 mM ATP | 120 |
| BSA (20 mg/mL) | 6 |
| 10% Triton X-100 | 12 |
| T4 DNA ligase | 12 |
| Nuclease-free water | 1050 |
6. Resuspend the nuclei and incubate overnight at 16 °C with rotation.
G. Reversal crosslinking and RNA/DNA purification (timing: ~4.5 h)
1. Centrifuge at 2,500× g for 3 min at 4 °C and resuspend the nuclei in 500 μL of proteinase K solution buffer at 65 °C for 3 h with shaking at 800 rpm using a ThermoMixer.
2. Add an equivalent volume of phenol:chloroform:IAA (25:24:1) (pH 7.9).
3. Centrifuge a MaxTract high density tube at 13,000× g for 1 min and add the mixture from step G1 to the tube. Then centrifuge the tube at 13,000× g for 5 min.
4. Transfer the upper aqueous phase into a new 1.5 mL tube and add 2 μL of glycoblue, 1/10th volume of 3 M sodium acetate (pH 5.2), and an equivalent volume of ice-cold 100% (v/v) isopropanol. Mix well and incubate at -80 °C for 30 min.
5. Centrifuge at 13,000× g for 15 min at 4 °C to precipitate the nucleic acid pellet and wash twice with 75% cold ethanol. Air-dry the pellet and dissolve the nucleic acid pellet in 50 μL of nuclease-free water.
H. Gap-filling and second-strand cDNA synthesis (timing: ~3 h)
1. Prepare the Gap-filling mixture as in Table 8.
Table 8. Gap-filling reaction mixture
| Reagent | Volume (μL) |
|---|---|
| 10× NEBuffer r2.1 | 10 |
| 10 mM dNTP | 8 |
| DNA (from section G) | 50 |
| T4 DNA polymerase | 6 |
| Nuclease-free water | 26 |
2. Mix and incubate at room temperature for 40 min, and then purify nucleic acid using AMPure XP beads. Dissolve the nucleic acid in 50 μL of nuclease-free water.
3. Prepare second-strand cDNA synthesis mixture (Table 9).
Table 9. Second-strand cDNA synthesis mixture
| Reagent | Volume(μL) |
|---|---|
| NEBNext second strand synthesis reaction buffer | 8 |
| NEBNext second strand synthesis enzyme mix | 4 |
| DNA (from step H2) | 50 |
| Nuclease-free water | 18 |
4. Mix and incubate at 16 °C for 1 h in a ThermalCycler and purify nucleic acid using AMPure XP beads. Dissolve the nucleic acid in 50 μL of QIAGEN Buffer EB. Measure the concentration of the product with Qubit.
I. DNA library preparation, PCR enrichment, and sequencing (timing: ~6 h)
1. Fragment the proximity-ligated nucleic acid (from section H) using the Tn5 transposome (from the TruePrep DNA Library Prep Kit for Illumina) according to the manufacturer’s instructions (Table 10).
Table 10. Proximity ligation nucleic acid fragmentation mixture with Tn5 transposome
| Reagent | Volume (μL) |
|---|---|
| 50 ng proximity ligation nucleic acid | x |
| 5× TTBL | 10 |
| Tn5 Tagmentation enzyme | 5 |
| Nuclease-free water | 35 – x |
2. Incubate at 55 °C for 10 min in a ThermalCycler and purify nucleic acid using DNA Clean & Concentrator-5. Dissolve the proximity ligation nucleic acid in 50 μL of nuclease-free water.
3. Mix the M280 Streptavidin–Dynabeads suspension thoroughly and transfer 30 μL of the M280 beads to a 1.5-mL LoBind tube.
4. Place the tube on the magnetic stand, discard the supernatant, and wash the beads twice with 200 μL of 2× B&W buffer.
5. Resuspend the M280 beads in 100 μL of iBlock buffer and incubate at room temperature for 45 min with shaking at 50 rpm on an Intelli-Mixer (UU, 50 rpm).
6. Place the tube on the magnetic rack, discard the iBlock buffer, and wash the M280 beads twice with 200 μL of 1× B&W buffer.
7. Resuspend the M280 beads in 100 μL of sheared DNA solution and incubate at room temperature for 30 min with shaking at 50 rpm on an Intelli-Mixer (UU, 50 rpm).
8. Place the tube on the magnetic rack, discard the sheared DNA solution, and wash the M280 beads twice with 200 μL of 1× B&W buffer.
9. Resuspend the M280 beads in 100 μL of proximity ligation nucleic acid and incubate at room temperature for 45 min with shaking at 50 rpm on an Intelli-Mixer (UU, 50 rpm).
10. Place the tube on the magnetic rack, discard the supernatant, and wash the M280 beads five times using 500 μL of 2× SSC/0.5% SDS. Then, wash the M280 beads two times using 500 μL of 1× B&W buffer.
11. Resuspend the M280 beads in 24 μL of nuclease-free water. Use the resuspended beads as the template for PCR amplification of the library, as in Table 11. Run the PCR amplification with the program described in Table 12.
Table 11. M280 beads as a template for PCR amplification
| Reagent | Volume (μL) |
|---|---|
| Bead solution (from step I11) | 24 |
| 5× TAB | 10 |
| Index primer (i7) | 5 |
| Index primer (i5) | 5 |
| PPM | 5 |
| TAE | 1 |
Table 12. Thermal cycling conditions for library amplification
| Temperature (°C) | Time (s) | |
| 72 | 180 | |
| 98 | 30 | |
| 98 | 10 | 10–12 cycles |
| 60 | 30 | |
| 72 | 180 | |
| 72 | 300 | |
| 4 | ∞ |
12. Perform double-sided size selection using DNA Clean Beads as follows:
a. Add 27.5 μL (0.55×) of DNA clean beads to the 50 μL sample. Mix thoroughly by pipetting and incubate at room temperature for 5 min.
b. Place the tube on a magnetic rack and carefully transfer the supernatant to a new tube.
c. Add 10 μL (0.2×) of DNA Clean Beads to the supernatant. Mix well and incubate at room temperature for 5 min. Place the tube on the magnetic rack and discard the supernatant.
d. Wash the beads twice with 80% ethanol (freshly prepared), keeping the tube on the magnetic rack during washing.
e. Carefully remove all residual ethanol. Air-dry the beads at room temperature, avoiding over-drying.
f. Resuspend the beads in 16 μL of EB buffer and incubate for 5 min.
g. Place the tube on the magnetic rack and transfer the supernatant to a new tube. This is the final TaDRIM-seq library (as shown in Figure 2).
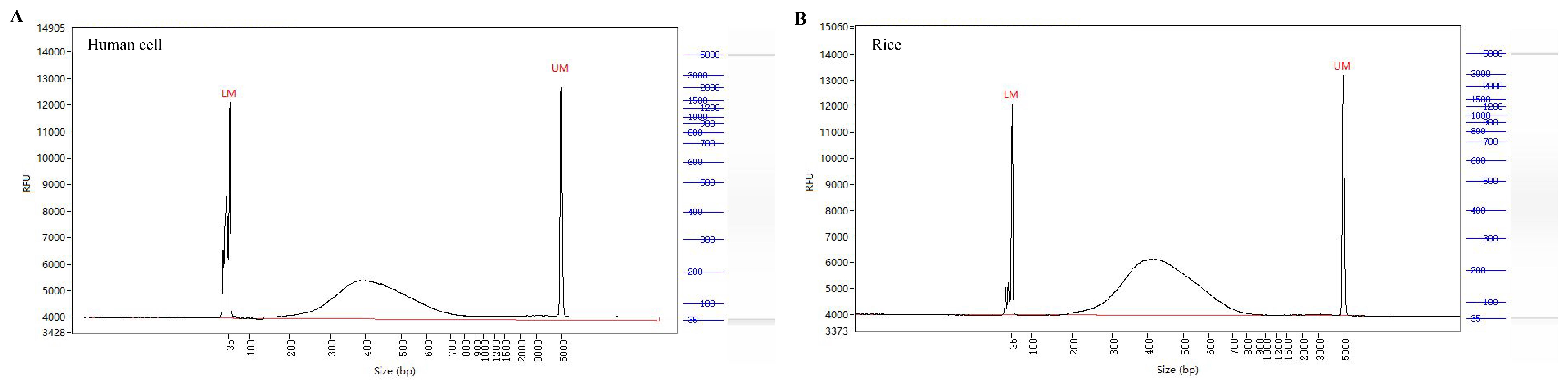
Figure 2. Size distribution of final TaDRIM-seq libraries.
(A) Human cell. (B) Rice.
Data analysis
1. Use flash (version 1.2.11) and cutadapt (version 2.3) to split data into combined or independent reads and find the linker.
• flash --threads 20 -M 150 -O --output-prefix PREFIX.flash --output-directory PREFIX_R1.fq.gz PREFIX_R1.fq.gz• cutadapt -b file:$Linker_file -n 14 --no-indels -o PREFIX.combine.noLinker --info-file PREFIX.combine.Linker_info --discard -O 35 PREFIX.flash.extendedFrags.fastq > PREFIX.combine.stat• cutadapt -b file:$Linker_file -n 8 --no-indels -o PREFIX.notCombined.1.noLinker --info-file PREFIX.notCombined.1.Linker_info --discard -O 35 PREFIX.flash.notCombined_1.fastq > PREFIX.notCombined.1.stat• cutadapt -b file:$Linker_file -n 8 --no-indels -o PREFIX.notCombined.2.noLinker --info-file PREFIX.notCombined.2.Linker_info --discard -O 35 PREFIX.flash.notCombined_2.fastq > PREFIX.notCombined.2.statThe Linker_file file should be in the following format:
A. CCTCGATACTTGAGCTGACAAGATGTGTATAAGAGACAG
B. CTGTCTCTTATACACATCTTGTCAGCTCAAGTATCGAGG
• perl ts_cutadapt2oneline.pl PREFIX.combine.Linker_info PREFIX.combine.cut.info > PREFIX.combine.cut.info.log• perl ts_cutadapt2oneline.pl PREFIX.notCombined.1.Linker_info PREFIX.notCombined.1.cut.info > PREFIX.notCombined.1.cut.info.log• perl ts_cutadapt2oneline.pl PREFIX.notCombined.2.Linker_info PREFIX.notCombined.2.cut.info > PREFIX.notCombined.2.cut.info.log2. Separate RNA and DNA sequences based on the linker sequence.
• perl split.combine.pl PREFIX.combine.cut.info PREFIX 1> PREFIX.combine.fq.stat 2 > PREFIX.combine.fq.stat.log• perl split.only.pl PREFIX.notCombined.1.cut.info PREFIX.notCombined.2.cut.info PREFIX.only 1> PREFIX.only.fq.stat 2>PREFIX.only.fq.stat.log• cat PREFIX.combine.RNA.fq PREFIX.only.RNA.fq > PREFIX.RNA.fastq• cat PREFIX.combine.DNA.fq PREFIX.only.DNA.fq > PREFIX.DNA.fastq3. Use bwa (version 0.7.15-r1140), hisat2 (version 2.1.0), and bowtie2 (version 2.3.5.1) to map reads to a reference genome, and make multi-align to only match. Use samtools (version 1.9) to convert a sm file to a bam file.
• perl split.pl PREFIX.DNA.fastq $OUTPUT_PREFIX 70• bwa aln -t 20 -f PREFIX.DNA.short.sai $BWA_GENOME_INDEX PREFIX.DNA.short.fastq && bwa samse -f PREFIX.DNA.short.sam $BWA_GENOME_INDEX PREFIX.DNA.short.sai PREFIX.DNA.short.fastq• bwa mem -t 20 $BWA_GENOME_INDEX PREFIX.DNA.long.fastq > PREFIX.DNA.long.sam• hisat2 --known-splicesite-infile $knownspl -p 20 -x $HISAT_GENOME_INDEX --rna-strandness F -U PREFIX.RNA.fastq -S PREFIX.RNA.sam && python flt_bam.py PREFIX.RNA.sam PREFIX.RNA && samtools sort -@ 20 -o PREFIX.RNA.hisat.bam PREFIX.RNA.uniqmap.bam• samtools view -Sb -F 256 PREFIX.RNA.multimap.bam | bamToFastq -i - -fq PREFIX.tmp.multi.fq• bamToFastq -i PREFIX.RNA.unmap.bam -fq PREFIX.tmp.unmap.fq• cat PREFIX.tmp.multi.fq PREFIX.tmp.unmap.fq > PREFIX.remap.fq• bowtie2 -p 20 --local --very-sensitive-local -x $BOWTIE2_GENOME_INDEX -U PREFIX.remap.fq -S PREFIX.remap.sam && samtools view -Sb -q 2 PREFIX.remap.sam | samtools sort -@ 20 -o PREFIX.remap.bam• samtools merge -f PREFIX.RNA.bam PREFIX.RNA.hisat.bam PREFIX.remap.bam• samtools index PREFIX.RNA.bam4. Use samtools (version 1.9) to get the unique mapped reads.
• java -cp LGL.jar LGL.util.UniqueSam PREFIX.DNA.long.sam PREFIX.DNA.long.rmdup.sam• python combine_DNA_file.py PREFIX.DNA.short.sam PREFIX.DNA.long.rmdup.sam PREFIX.DNA.sam• samtools view -Sb PREFIX.DNA.sam | samtools sort -@ 20 -o PREFIX.DNA.bam• samtools index PREFIX.DNA.bam5. Use samtools and deeptools (version 3.2.1) to convert the bam file to a BigWig file.
• samtools index PREFIX.DNA.bam• samtools index PREFIX.RNA.bam• bamCoverage -o PREFIX.DNA.bw --binSize 5 -b PREFIX.DNA.bam --numberOfProcessors 20 --minMappingQuality 20• bamCoverage -o PREFIX.RNA.bw --binSize 5 -b PREFIX.RNA.bam --numberOfProcessors 20 --minMappingQuality 206. Merge DNA–RNA interaction according to read ID and extraction of high-confidence DNA–RNA interactions using the hypergeometric distribution test.
• bamToBed -cigar -i PREFIX.DNA.bam | awk -v OFS="\t" '{if($6=="+"){print $1,$2,$2+500,$4,$5,$6,$7}else{start=$3-500;if(start<0){start=0;}print $1,start,$3,$4,$5,$6,$7}}' | intersectBed -a - -b $DNA_ANCHOR -wao | python uniq_DNA.py > PREFIX.DNA.bed &• htseq-count -o PREFIX.RNA.yes.gene.sam -f bam -r name -s yes -t gene -i ID -m intersection-nonempty PREFIX.RNA.bam $gene_file > PREFIX.RNA.yes.gene.count• samtools view -H PREFIX.RNA.bam | cat - PREFIX.RNA.yes.gene.sam > PREFIX.RNA.yes.gene.cg.sam• htseq-count -o PREFIX.RNA.yes.exon.sam -f sam -r name -s yes -t exon -i ID -m intersection-nonempty REFIX.RNA.yes.gene.cg.sam $exon_file > PREFIX.RNA.yes.exon.count• samtools view -H PREFIX.RNA.bam | cat - PREFIX.RNA.yes.exon.sam | perl -lane 'if(/XF:Z/){$_=~s/XF:Z/GE:Z/;print $_}else{print $_}' > PREFIX.RNA.yes.sam• python sam2bed2.py PREFIX.RNA.yes.sam 1> PREFIX.RNA.yes.bed 2>PREFIX.RNA.yes.bed.log• python combineDNAandRNA_withanchor.py PREFIX.DNA.bed PREFIX.RNA.yes.bed PREFIX.DNARNA.bedpe• awk -v qu=$MAPPING_CUTOFF '$8>=qu' PREFIX.DNARNA.bedpe | sort --parallel=$NTHREADS -k14,14 -k13,13 -k1,1 -k2,2n -k3,3n -k4,4 -k5,5n -k6,6n -k8,8n -k9,9 -k10,10 -k11,11 -k12,12 > PREFIX.DNARNA.sorted.bedpe• python uniqread.py PREFIX.DNARNA.sorted.bedpe > PREFIX.DNARNA.uniq.bedpe• python PetClusterWithGivenAnchors.py $DNA_ANCHOR $BED_file PREFIX.DNARNA.uniq.bedpe | sort --parallel=$NTHREADS -k1,1 -k4,4 -k2,2n -k5,5n -k3,3n -k6,6n > PREFIX.DNARNA.givenanchor.cluster• awk '$9>1' PREFIX.DNARNA.givenanchor.cluster > PREFIX.DNARNA.givenanchor.gt1.clustertmp=`wc -l PREFIX.DNARNA.uniq.bedpe | awk '{print $1}'`• Rscript $PROGRAM_DIRECTORY/hypergeometric5.r PREFIX.DNARNA.givenanchor.gt1.cluster PREFIX.DNARNA.givenanchor.gt1.cluster.withpvalue• awk '$13+0.0<=0.05' PREFIX.DNARNA.givenanchor.gt1.cluster.withpvalue > PREFIX.DNARNA.givenanchor.FDRfiltered.txt• awk '$9>2'PREFIX.DNARNA.givenanchor.FDRfiltered.txt > PREFIX.DNARNA.givenanchor.FDRfiltered.gt2.txt• cat PREFIX.DNARNA.givenanchor.FDRfiltered.gt2.txt | awk '{if($1==$4 && ($5+$6)>=($2+$3)) print $1"\t"$2"\t"$3"\t"$4"\t"$5"\t"$6"\t"$9"\t"$12"\t"$13}' > PREFIX.givenanchor.cluster.intra.DNA-RNA.curve• cat PREFIX.DNARNA.givenanchor.FDRfiltered.gt2.txt |awk '{if($1==$4 && ($5+$6)<($2+$3)) print $4"\t"$5"\t"$6"\t"$1"\t"$2"\t"$3"\t"$9"\t"$12"\t"$13}' > PREFIX.givenanchor.cluster.intra.RNA-DNA.curveThe main code files used in the process of RNA–DNA data processing are as follows:
https://github.com/fengchuiguo1994/ChRDPETpipeline/blob/main/run.sh
https://github.com/fengchuiguo1994/ChRDPETpipeline/blob/main/run.coverage.sh
https://github.com/fengchuiguo1994/ChRDPETpipeline/blob/main/run.interaction.sh
TaDRIM data process pipeline to get RNA–RNA interaction information
1. First, merge fragments shorter than or equal to 290 bp in sequenced files (*.fq.gz) with FLASH (version 1.2.11).
• flash -M 145 -O --output-prefix PREFIX.flash --output-directory PREFIX_R1.fq PREFIX_R1.fq2. Use cutadapt (version 2.3) to identify the RNA–RNA bridge linker for merged (≤290 bp) and unmerged (>290 bp) fragments.
• Use cutadapt to identify the RNA-RNA bridge linker for merged (≤290 bp) and unmerged (>290 bp) fragments.• cutadapt -b file:$Linker_file -n 8 -e 0.2 --no-indels -o PREFIX.combine.noLinker --info-file PREFIX.combine.Linker_info --discard -O 34 PREFIX.flash.extendedFrags.fastq > PREFIX.combine.stat• cutadapt -b file:$Linker_file -n 8 -e 0.2 --no-indels -o PREFIX.notCombined.1.noLinker --info-file PREFIX.notCombined.1.Linker_info --discard -O 34 PREFIX.flash.notCombined_1.fastq > PREFIX.notCombined.1.stat• cutadapt -b file:$Linker_file -n 8 -e 0.2 --no-indels -o PREFIX.notCombined.2.noLinker --info-file PREFIX.notCombined.2.Linker_info --discard -O 34 PREFIX.flash.notCombined_2.fastq > PREFIX.notCombined.2.statThe Linker_file file should be in the following format:
A. CCTCGATACTTGAGCTGACGTCAGCTCAAGTATCGAGG
B. CCTCGATACTTGAGCTGACGTCAGCTCAAGTATCGAGG
3. Quantitative profiling of RNA-RNA linker distributions in merged and non-merged paired-end reads
• perl linker_info_sta.pl PREFIX.combine.Linker_info PREFIX.combine.cut.info > PREFIX.combine.cut.info.log• perl linker_info_sta.pl PREFIX.notCombined.1.Linker_info PREFIX.notCombined.1.cut.info > PREFIX.notCombined.1.cut.info.log• perl linker_info_sta.pl PREFIX.notCombined.2.Linker_info PREFIX.notCombined.2.cut.info > PREFIX.notCombined.2.cut.info.log4. Extract tags from dual-end sequencing results with only one linker, where the lengths of both ends of the linker are greater than the threshold (13 bp).
• sh scriptdir/Extract_valid_data.sh PREFIX 135. Reverse complementary sequencing of the 3′ end of the linker to obtain sequence-specific interaction information:
• python sequence_RC.py PREFIX.total-PET_SE_3nd.txt PREFIX.total-PET_SE_3nd_FXHBcorrected.fq6. Remove PCR replicates with identical PETs.
• python remove_duplication.py -r1 PREFIX.total-PET_SE_5nd.fq.gz -r2 PREFIX.total-PET_SE_3nd_FXHBcorrected.fq.gz -o1 PREFIX.part1.fq -o2 PREFIX.part2.fq7. Use hista2 (version 2.1.0) to align the sequences interacting at both ends of the fragments to the reference genome indexed in advance.
• hisat2 -x ${HISAT_GENOME_INDEX} -U PREFIX.part1.fq -S PREFIX.part1.sam --summary-file PREFIX.part1_hisat2_aln_out.txt• hisat2 -x ${HISAT_GENOME_INDEX} -U PREFIX.part2.fq -S PREFIX.part2.sam --summary-file PREFIX.part2_hisat2_aln_out.txt8. Filter the interaction information: only sequencing records with high-quality (Q > 20) and uniquely mapped pair-end tags are retained.
• cat PREFIX.part1.sam | grep 'NH:i:1$' > PREFIX.part1_unique.sam• cat PREFIX.part2.sam | grep 'NH:i:1$' > PREFIX.part2_unique.sam• awk '{if($5>=20) print $0}' PREFIX.part1_unique.sam > PREFIX.part1_unique.q20.sam• awk '{if($5>=20) print $0}' PREFIX.part2_unique.sam > PREFIX.part2_unique.q20.sam• awk '{print $1}' PREFIX.part1_unique.q20.sam > IDtags1.txt• awk '{print $1}' PREFIX.part2_unique.q20.sam > IDtags2.txt• grep -Fwf IDtags2.txt PREFIX.part1_unique.sam > PREFIX.part1_PET_unique_aln.sam• grep -Fwf IDtags1.txt PREFIX.part2_unique.sam > PREFIX.part2_PET_unique_aln.sam9. Convert sam files to a bam format and perform name-based sorting.
• samtools view -H PREFIX.part1.sam > PREFIX.header.sam• cat PREFIX.part1_PET_unique_aln.sam PREFIX.part2_PET_unique_aln.sam > PREFIX.part_PET_unique_aln.sam• cat PREFIX.header.sam PREFIX.part_PET_unique_aln.sam > PREFIX.PET_unique_aln.sam• samtools view -bS PREFIX.PET_unique_aln.sam > PREFIX.PET_unique_aln.bam• samtools sort -n -o PREFIX.PET_unique_aln_sort.bam PREFIX.PET_unique_aln.bam10. Distinguish the interaction information between intra- and intermolecules.
• python class_bam_PairTag.py -g genome.element.bed -i PREFIX.PET_unique_aln_sort.bam -o PREFIX.genome > PREFIX.genome.class.log11. Convert the format of interaction information (from .bam to .bedpe).
• python IntraSam2bedpe.py -i PREFIX.genome.interaction.intraMolecular.bam -o PREFIX.genome.interaction.intraMolecular.bedpe• LANG=C sort -k1,1 -k8,8 -k2,2n -k3,3n -k5,5n -k6,6 PREFIX.genome.interaction.intraMolecular.bedpe > PREFIX.genome.interaction.intraMolecular.sorted.bedpe• python Interbam2bedpe.py -i PREFIX.genome.interaction.interMolecular.bam -o PREFIX.genome.interaction.interMolecular.bedpe12. Calculate and obtain intramolecular RNA–RNA interaction clusters.
• java -jar ClusterCalling.jar call PREFIX.genome.interaction.intraMolecular.sorted.bedpe PREFIX.genome.interaction.intraMolecular.cluster13. Calculate the strength and significance of intramolecular RNA–RNA interaction clusters.
• java -jar ClusterCalling.jar count PREFIX.genome.interaction.intraMolecular.sorted.bedpe PREFIX.genome.interaction.intraMolecular.cluster PREFIX.genome.interaction.intraMolecular.cluster.count• Rscript Hypergeometric.r PREFIX.genome.interaction.intraMolecular.cluster.count PREFIX.genome.interaction.intraMolecular.cluster.count.pvalue14. Calculate and obtain intermolecular RNA–RNA interaction clusters.
• python getInteraction.py -i PREFIX.genome.interaction.interMolecular.bedpe.gz -o PREFIX.total.bedpe.interaction.txt -l genome.element.bed15. Randomly simulate intermolecular interaction information and use the randomly simulated interaction information to calculate the randomly simulated intermolecular RNA–RNA clusters as described previously in Cai et al. [12].
• python generateSim.py -i PREFIX.genome.interaction.interMolecular.bedpe.gz -o sim/PREFIX.total.sim > PREFIX.total.sim.log• for pp in $(seq ${Nsimu}); do pigz sim/PREFIX.total.sim.sim.$pp.tmp.bedpe; done• for pp in $(seq ${Nsimu}); dopython getInteraction.py -i sim/PREFIX.total.sim.sim.${pp}.tmp.bedpe.gz -o sim/PREFIX.total.sim.sim.${pp}.tmp.bedpe.interaction.txt -l genome.element.beddone16. Calculate the significance of real intermolecular interaction clusters based on randomly simulated intermolecular interaction clusters.
• python calpvalue.py -i PREFIX.total.bedpe.interaction.txt.gz -ip sim/PREFIX.total.sim.sim -is tmp.bedpe.interaction.txt.gz -o PREFIX.total.rna-rna.interaction.withpvalue.txtThe main code files used in the process of RNA–RNA data processing are as follows:
https://github.com/304243504/HiR/tree/main/scriptdir
https://github.com/304243504/HiR/blob/main/getGeneElement.py
https://github.com/304243504/HiR/blob/main/Run.sh
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
• Ding et al. [11]. Simultaneous profiling of chromatin-associated RNA at targeted DNA loci and RNA-RNA Interactions through TaDRIM-seq.
General notes and troubleshooting
General notes
1. Before starting the experiment, wipe down the bench surface thoroughly with RNase remover and nucleic acid decontamination reagent to ensure an RNase-free environment.
2. For TaDRIM-seq library preparation, it is recommended to perform 4–6 tagmentation reactions, each using 200–300ng of proximity-ligated nucleic acids.
3. The 50% (w/v) PEG-8000 solution is highly viscous. Mix the bridge linker and enzyme thoroughly first and add PEG-8000 last to ensure proper reagent distribution and reaction efficiency.
4. If the experiment focuses on RNA–DNA interactions, the primer and bridge linker can be pre-designed as follows:
Bridge linker:
Forward strand: 5′-[5Phos]CCTCGATAC/iBIOdT/TGAGCTGACT-3′
Reverse strand: 5′-[5Phos]GTCAGC/iBIOdT/CAAGTATCGAG-3′
Primer: primer A (5′-phos-CTGTCTCTTATACACATCTA-3′)
primer B (5′-phos-AGATGTGTATAAGAGACAG-3′)
5. The ratio of RNA–DNA to DNA–DNA interactions may vary significantly across different species and tissue types.
6. RNA and DNA end specificity are two important characteristics for evaluating the library and the data. The DNA fragment data should align with the corresponding protein ChIP-seq or CUT&Tag sites, while RNA can reflect the transcriptional direction (as shown in Figures 3 and 4).
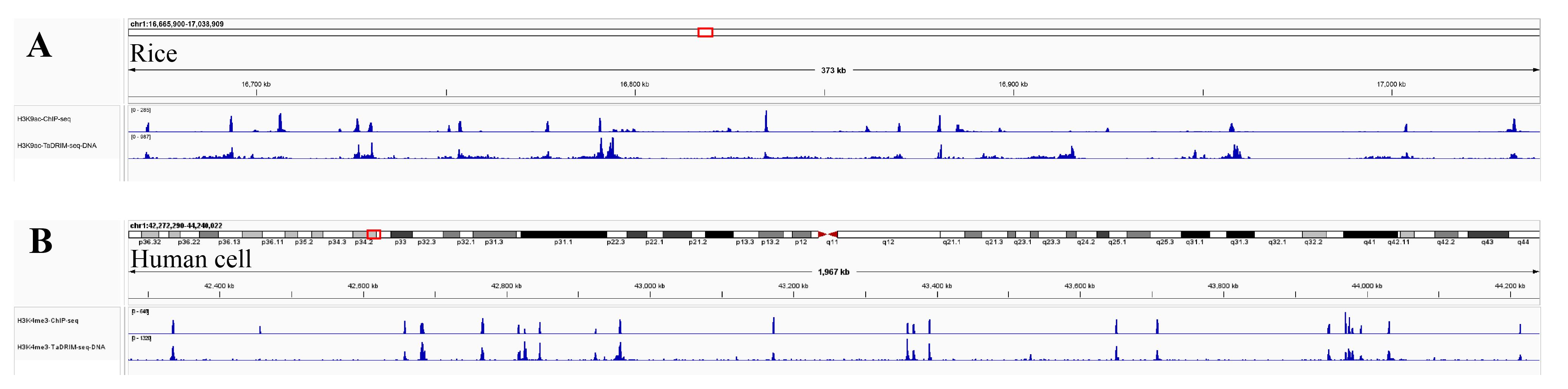
Figure 3. Genome browser tracks showing the distribution of DNA from H3K4me3 and H3K9ac TaDRIM-seq data in (A) rice and (B) human cells
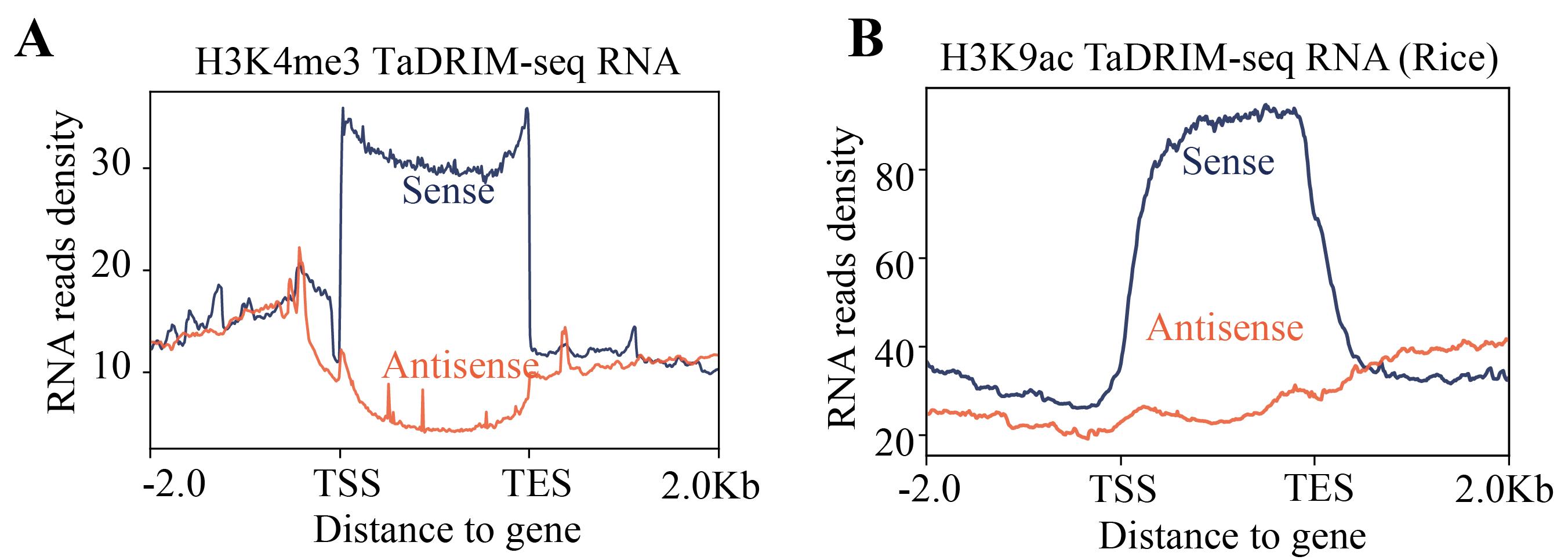
Figure 4. Strand orientation of TaDRIM-seq sense and antisense RNA. (A) H3K4me3 TaDRIM-seq RNA in human cell. (B) H3K9ac TaDRIM-seq RNA in rice.
Troubleshooting
1. Low complexity of the library
Potential solution: Reduce the number of PCR cycles and increase the amount of input DNA during library preparation.
2. Poor DNA-end specificity
Solution: Replace the antibody if necessary or increase the number of washes in step D3 during pG-Tn5 transposase binding and tagmentation to improve specificity.
Acknowledgments
This work was supported by the Biological Breeding-National Science and Technology Major Project (2023ZD04073 to X.L.), the Fundamental Research Funds for the Central Universities (2662023PY002 to X.L., G.L., and J.Y., 2662024SKPY002 to J.Y., and 2662023SKQD002 to M.F.F), the Foundation of Hubei Hongshan Laboratory (2021HSZD010 to C.W. and X.L., 2023HSQD002 to J.Y.), the Postdoctoral Fellowship Program of CPSF (GZB20250589 to C.D. and GZC20240554 to G.C.), the National Natural Science Foundation of China (32070612 to X.L. and 3247040618 to J.Y.), and the National Key Research and Development Program of Hubei Province (2022BBA54 to X.L.). We thank the computing platform of the National Key Laboratory of Crop Genetic Improvement at Huazhong Agricultural University for providing computational resources. This protocol was used in [11].
Authors’ contribution
Conceptualization, C.D., X.L.; Investigation, G.Y., C.G., C.Y., Z.X., J.D.; Writing—Original Draft, C.D., G.C., S.L; Writing—Review & Editing, C.D., G.C., M.F.F; Funding acquisition, J.Y., X.L.; Supervision, J.Y., X.L.
Competing interests
Authors declare that they have no competing interests.
References
- Nguyen, T. C., Zaleta-Rivera, K., Huang, X., Dai, X. and Zhong, S. (2018). RNA, Action through Interactions. Trends Genet. 34(11): 867–882. https://doi.org/10.1016/j.tig.2018.08.001
- Li, X. and Fu, X. D. (2019). Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat Rev Genet. 20(9): 503–519. https://doi.org/10.1038/s41576-019-0135-1
- Morris, K. V., Chan, S. L., Jacobsen, S. E. and Looney, D. J. (2004). Small Interfering RNA-Induced Transcriptional Gene Silencing in Human Cells. Science. 305(5688): 1289–1292. https://doi.org/10.1126/science.1101372
- Kim, D. H. and Sung, S. (2017). Vernalization-Triggered Intragenic Chromatin Loop Formation by Long Noncoding RNAs. Dev Cell. 40(3): 302–312.e4. https://doi.org/10.1016/j.devcel.2016.12.021
- West, J. A., Davis, C. P., Sunwoo, H., Simon, M. D., Sadreyev, R. I., Wang, P. I., Tolstorukov, M. Y. and Kingston, R. E. (2014). The Long Noncoding RNAs NEAT1 and MALAT1 Bind Active Chromatin Sites. Mol Cell. 55(5): 791–802. https://doi.org/10.1016/j.molcel.2014.07.012
- Penny, G. D., Kay, G. F., Sheardown, S. A., Rastan, S. and Brockdorff, N. (1996). Requirement for Xist in X chromosome inactivation. Nature. 379(6561): 131–137. https://doi.org/10.1038/379131a0
- Hsieh, C. L., Fei, T., Chen, Y., Li, T., Gao, Y., Wang, X., Sun, T., Sweeney, C. J., Lee, G. S., Chen, S., et al. (2014). Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci USA. 111(20): 7319–7324. https://doi.org/10.1073/pnas.1324151111
- Kuang, S. and Pollard, K. S. (2024). Exploring the roles of RNAs in chromatin architecture using deep learning. Nat Commun. 15(1): 6373. https://doi.org/10.1038/s41467-024-50573-w
- Xiao, Q., Huang, X., Zhang, Y., Xu, W., Yang, Y., Zhang, Q., Hu, Z., Xing, F., Sun, Q., Li, G., et al. (2022). The landscape of promoter-centred RNA–DNA interactions in rice. Nat Plants. 8(2): 157–170. https://doi.org/10.1038/s41477-021-01089-4
- Gavrilov, A. A., Sultanov, R. I., Magnitov, M. D., Galitsyna, A. A., Dashinimaev, E. B., Lieberman Aiden, E. and Razin, S. V. (2021). RedChIP identifies noncoding RNAs associated with genomic sites occupied by Polycomb and CTCF proteins. Proc Natl Acad Sci USA. 119(1): e2116222119. https://doi.org/10.1073/pnas.2116222119
- Ding, C., Chen, G., Luan, S., Gao, R., Fan, Y., Zhang, Y., Wang, X., Li, G., Foda, M. F., Yan, J., et al. (2025). Simultaneous profiling of chromatin-associated RNA at targeted DNA loci and RNA-RNA Interactions through TaDRIM-seq. Nat Commun. 16(1): 1500. https://doi.org/10.1038/s41467-024-53534-5
- Cai, Z., Cao, C., Ji, L., Ye, R., Wang, D., Xia, C., Wang, S., Du, Z., Hu, N., Yu, X., et al. (2020). RIC-seq for global in situ profiling of RNA-RNA spatial interactions. Nature. 582(7812): 432–437. https://doi.org/10.1038/s41586-020-2249-1
Article Information
Publication history
Received: May 28, 2025
Accepted: Jul 22, 2025
Available online: Aug 11, 2025
Published: Sep 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Ding, C., Chen, G., Luan, S., Gong, Y., Gui, C., Yang, C., Xiang, Z., Du, J., Foda, M. F., Yan, J. and Li, X. (2025). Simultaneous Capture of Chromatin-Associated RNA and Global RNA–RNA Interactions With Reduced Input Requirements. Bio-protocol 15(17): e5430. DOI: 10.21769/BioProtoc.5430.
Category
Bioinformatics and Computational Biology
Molecular Biology > RNA > miRNA-mRNA interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.