- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation and Ex Vivo Testing of CD8+ T-Cell Division and Activation Using Mouse Splenocytes
Published: Vol 15, Iss 16, Aug 20, 2025 DOI: 10.21769/BioProtoc.5423 Views: 4016
Reviewed by: Mary Luz UribeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2025

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
This protocol describes an ex vivo co-culture method to assess CD8+ T-cell activation, proliferation, and cytotoxic potential using bulk splenocytes isolated from immunocompetent mice. Mouse splenocytes are stimulated with anti-CD3 and anti-CD28 antibodies to activate CD8+ T cells, which are then co-incubated with either cancer cells or cancer cell–derived conditioned media (CM) to evaluate tumor-driven modulation of immune cell functions. The use of unfractionated splenocytes preserves physiological cell–cell interactions, eliminating the need for exogenous interleukin (IL-2) and bypassing flow sorting, which simplifies the workflow and reduces experimental variability. CD8+ T-cell responses are measured via flow cytometry, using markers of proliferation (CFSE dilution), activation (CD69), and effector function (Granzyme B and IFNγ). Additionally, immune-mediated tumor cell death is evaluated by Annexin-V/7-AAD staining. Together, this experimental platform supports the investigation of both cell contact-dependent and contact-independent mechanisms of immune cell modulation in a cost-effective and reproducible setting.
Key features
• Enables isolation and stimulation of splenocytes to assess CD8+ T-cell responses to cancer cells or their secreted factors.
• Supports evaluation of CD8+ T-cell activation, proliferation, and effector function by flow cytometry.
• Allows functional assessment of tumor-driven suppression of T-cell activation in co-culture or conditioned media.
• Measures cancer cell death resulting from interactions with activated CD8+ T cells in splenocyte co-cultures.
Keywords: Tumor-immune cross-talkGraphical overview
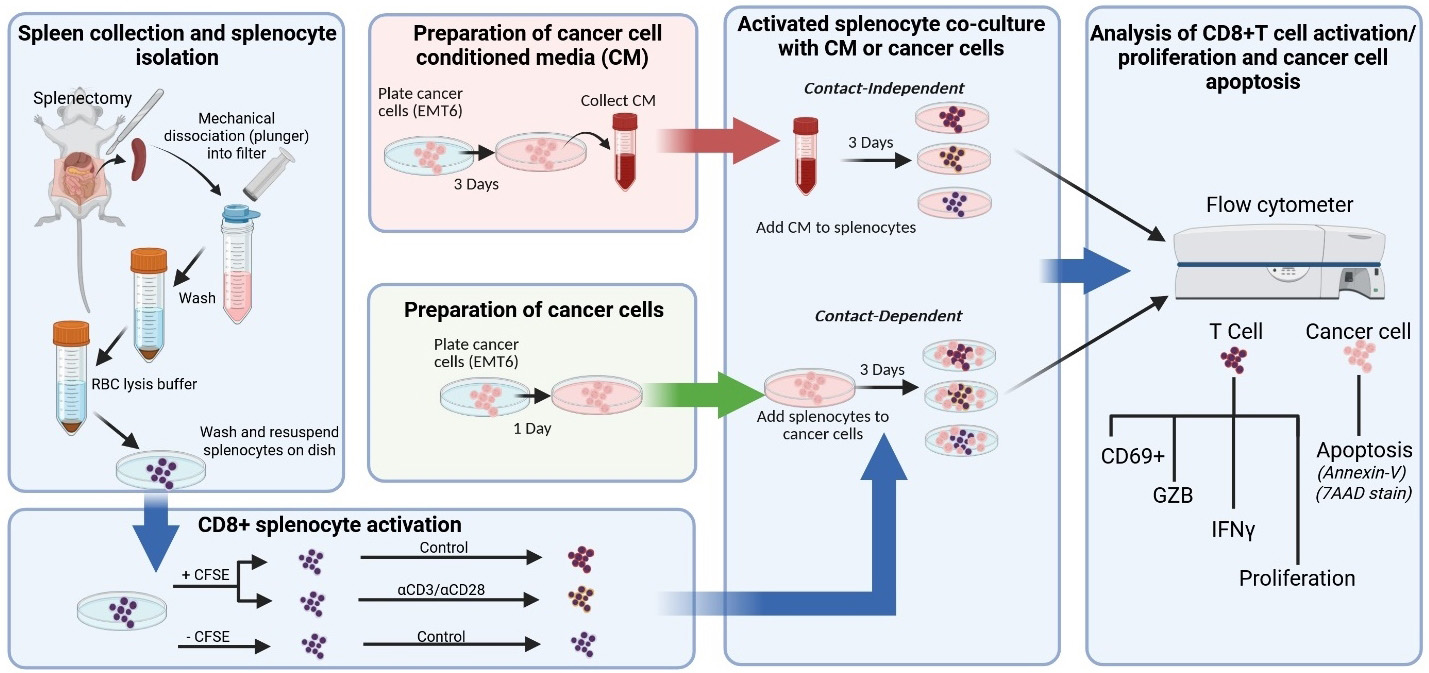
Graphical overview of key protocol steps
Background
Immunotherapies targeting T-cell functions are now approved in several tumor types [1]. This has led to a dramatic increase in the need to develop preclinical methods to identify ways to improve patient responses and evaluate drug resistance [2,3]. In vivo studies with preclinical mouse syngeneic tumor models are most commonly used for gaining functional insight into human cancer progression in an immunocompetent setting, but they are limited by costs and variability. Testing of in vivo–derived immune components in ex vivo settings represents a way to mimic the complexity of tumor and immune cell functions and interactions with improved reproducibility/rigor, reduced cost, and improved control over experimental variables.
Here, we describe a protocol to evaluate the functional interaction between activated CD8+ T cells within mouse splenocyte populations when cultured with cancer cells or cancer cell conditioned media (CM), a technique that allows comparison of cell contact-dependent and contact-independent mechanisms of immune cell modulation [4,5]. The use of bulk mouse splenocytes eliminates the need for stimulation with interleukin-2 (IL-2) and enrichment techniques to isolate pure T-cell populations. IL-2 is a cytokine released by surrounding immune cells (e.g., CD4+ T cells or CD8+ T cells themselves) in secondary lymphoid organs or the tumor microenvironment that promotes expansion of T cells following CD3/T-cell receptor (TCR) and CD28/CD80 activation signaling [6,7]. Endogenous IL-2 produced by bulk splenocytes may better mimic cytokine concentrations present within the secondary lymphoid organs following antigen stimulation by antigen-presenting cells (APCs) [8] and may control for variability across studies that use different IL-2 stimulation concentrations in conventional ex vivo CD8 stimulation/activation protocols [6,7].
Importantly, the protocol described here examines antigen-independent CD8+ T-cell responses that may be advantageous for studying scientific questions involving immune suppressive secreted factors derived from cancer cells and cell surface proteins that can induce T-cell suppression. Ex vivo experiments specifically assessing antigen presentation may also find some aspects of this protocol (e.g., isolation of splenocytes and flow cytometry analysis of T-cell proliferation and activation markers) helpful for guidance in similar steps. We describe the protocol below using EMT6 parental/drug-resistant (DR) murine breast cancer cells and splenocytes derived from the corresponding allogenic BALB/c mice as an example. Alternatively, other mouse cancer cell lines and mouse strains (e.g., commonly used strains including FVB or C57BL/6) can be substituted in this protocol for broader applications. Other critical biological variables, such as age [9] and biological sex [10], which have profound effects on innate and adaptive immune responses and responses to cancer immunotherapy, can be applied to this protocol as well.
Our protocol design allows distinguishing between contact-dependent and contact-independent mechanisms of generalized CD8+ T-cell responses, including the identification of specific soluble secreted factors with strong immune modulating impact [4,5]. While this protocol is broadly applicable to various research contexts, here we compared CD8+ T-cell responses using cancer cells that are naïve or resistant to immunotherapy [4,5]. Together, this method allows elucidation of tumor-immune cell dynamics related to mechanisms of therapeutic resistance in a reproducible and cost-effective setting.
Materials and reagents
Biological materials
1. BALB/cAnNCrl mice (Charles River Laboratory, strain code: 028) as a source for spleens
2. EMT6 murine breast carcinoma cells (gifted by Laboratory of A.Gudkov)
3. EMT6 drug-resistant cell line (generated in [4])
Reagents
1. 1× Dulbecco’s phosphate buffer saline (DPBS) without Ca2+ and Mg2+, pH 7.4 (Corning, catalog number: 21-040-CV)
2. RPMI 1640 media with L-glutamine (Corning, catalog number: 10-040-CV)
3. Heat-inactivated fetal bovine serum (HI-FBS) (Gibco, catalog number: 10437028)
4. Penicillin-Streptomycin (Pen/Strep) (Corning, catalog number: 30-001-CI)
5. Sodium pyruvate (Gibco, catalog number: 11360070)
6. Non-essential amino acids (Gibco, catalog number: 11140050)
7. Beta-mercaptoethanol (β-me) (Gibco, catalog number: 21985023)
8. Red blood cell (RBC) lysis buffer (10×) (BioLegend, catalog number: 420301)
9. Accutase (BioLegend, catalog number: 423201)
10. 0.25% trypsin (Corning, catalog number: 25-053-CI)
11. Trypan blue solution, 0.4% (Corning, catalog number: 25-900-CI)
12. T-cell activation antibodies (see Table 1)
13. Flow cytometry staining antibodies (see Table 1)
Table 1. General antibodies used in the protocol
| Antibody | Purpose | Host | Company | Catalog number |
|---|---|---|---|---|
| Anti-mouse CD3 (clone 17-A2) | Cell activation | Rat | BioLegend | 100202 |
| Anti-mouse CD28 (clone 37.51) | Cell activation | Syrian hamster | BioLegend | 102102 |
| Anti-mouse CD16/CD32 TruStain FcX | Surface cell staining | Rat | BioLegend | 101319 |
| Anti-mouse CD45 (clone 30-F11); Alexa Fluor 700 conjugated | Surface cell staining | Rat | BioLegend | 103128 |
| Anti-mouse CD8b.2 (clone 53-5.8); PE-Cy7 conjugated | Surface cell staining | Rat | BioLegend | 140416 |
| Anti-mouse CD69 (clone H1.2F3); APC conjugated | Surface cell staining | Armenian hamster | BioLegend | 104514 |
| Anti-mouse/human Granzyme B (clone GB11); Pacific Blue conjugated | Intracellular cell staining | Rat | BioLegend | 515408 |
| Anti-mouse IFNγ (clone XMG1.2); PE conjugated | Intracellular cell staining | Rat | BioLegend | 505808 |
14. Fixation buffer (BioLegend, catalog number: 420801)
15. Intracellular staining permeabilization wash buffer (10×) (BioLegend, catalog number: 421002)
16. Carboxyfluorescein succinimidyl ester (CFSE) Cell Division Tracker kit (BioLegend, catalog number: 423801)
17. Sodium azide (NaN3) (Sigma-Aldrich, catalog number: S-2002)
18. Annexin-V APC (BioLegend, catalog number: 640941)
19. 7-AAD viability staining solution (BioLegend, catalog number: 420404)
20. Annexin-V binding buffer (BioLegend, catalog number: 422201)
21. Cell activation cocktail [containing phorbol 12-myristate 13-acetate (PMA) Ionomycin and Brefeldin A1] (BioLegend, catalog number: 423303)
22. Staurosporine (Fisher Scientific, catalog number: AAJ62837MCR)
Solutions
1. Spleen collection solution (see Recipes)
2. Splenocyte wash solution (see Recipes)
3. Splenocyte complete RPMI (see Recipes)
4. RBC lysis buffer (see Recipes)
5. Cancer (EMT6) cell growth media (see Recipes)
6. Flow cytometry staining wash buffer (See Recipes)
7. Intracellular permeabilization and wash buffer (see Recipes)
Recipes
1. Spleen collection solution (5 mL for 1 spleen)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DPBS (1×) | n/a | 4.60 mL |
| HI-FBS | 5% | 50 μL |
| Pen/Strep (100,000 UI/mL) | 1,000 UI/mL | 5 μL |
| Total | 5 mL |
2. Splenocyte wash solution (20 mL for 1 spleen)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DPBS (1×) | n/a | 19.6 mL |
| HI-FBS | 2% | 0.4 mL |
| Total | 20 mL |
3. Splenocyte complete RPMI (500 mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| RPMI 1640 | n/a | 435 mL |
| HI-FBS | 10% | 50 mL |
| Pen/Strep (100,000 UI/mL) | 1,000 UI/mL | 5 mL |
| Non-essential amino acids (100×) | 1× | 5 mL |
| Sodium pyruvate (100×) | 1× | 5 mL |
| β-me (55 mM in DPBS) | 0.1% | 0.5 mL |
| Total | 500.5 mL |
Store at 4 °C for a maximum of 6–8 weeks.
4. RBC lysis buffer (3 mL per 1 spleen)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| RBC lysis buffer (10×) | 1× | 0.3 mL |
| diH2O | n/a | 2.7 mL |
| Total | 3 mL |
Make fresh for every dissociation. Keep sterile and use sterile deionized H2O (diH2O).
5. Cancer (EMT6) cell growth media (500 mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| RPMI 1640 | n/a | 475 mL |
| HI-FBS | 5% | 25 mL |
| Total | 500 mL |
Store at 4 °C for a maximum of 6–8 weeks.
Note: Antibiotics such as Pen/Strep can be optionally added at a final concentration of 1,000 UI/mL when subculturing cancer cells prior to co-culture with isolated splenocytes. Use of antibiotics should be kept consistent with the individual lab’s standard operating procedures. The volume of RPMI 1640 will need to be adjusted accordingly.
6. Flow cytometry staining wash (FACS) buffer (500 mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DPBS (1×) | n/a | 490 mL |
| HI-FBS | 2% | 10 mL |
| NaN3 | 0.1% | 0.5 mL |
| Total | 500.5 mL |
Critical: Wear necessary personal protective equipment (PPE) and adhere to handling precautions as indicated in the material safety data sheet (MSDS) of sodium azide (NaN3).
7. Intracellular permeabilization and wash buffer (100 mL)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Intracellular permeabilization buffer (10×) | 1× | 10 mL |
| diH2O | n/a | 90 mL |
| Total | 100 mL |
Laboratory supplies
1. Pipettes
2. Pipette tips
3. Multichannel pipettes
4. Serological pipettes
5. Serological pipettor
6. 15 mL conical tubes (Fisher Scientific, catalog number: 601052)
7. 50 mL conical tubes (Fisher Scientific, catalog number: 602052)
8. 2 mL microcentrifuge tubes (Laboratory Product Sales Inc., catalog number: L211574)
9. 5 mL Falcon round-bottom polystyrene FACS tubes (Fisher Scientific, catalog number: 14-959-2A)
10. Bio Lite 10 cm cell culture treated plates (Fisher Scientific, catalog number: 12-556-002)
11. Corning 96-well clear round-bottom culture plates (non-treated) (Fisher Scientific, catalog number: 07-200-760)
12. Bio Lite 24-well cell culture treated plates (Fisher Scientific, catalog number: 12-556-006)
13. BD Disposable 3 mL syringes with Luer-Lok tips (Fisher Scientific, catalog number: 14-823-435)
14. Cell strainer 70 μm (Fisher Scientific, catalog number:22-363-548)
15. Corning 0.2 μm syringe filter (Fisher Scientific, catalog number: 09-754-23)
Equipment
1. Surgical scissors
2. Curved forceps
3. Biological safety cabinet (Biosafety Level 2)
4. 37 °C, 5% CO2 cell culture incubator (Thermo, model: Heraeus HeraCell 150)
5. Temperature-regulated water bath
6. -20 °C freezer (Crosley, model: WCF15/F)
7. -80 °C freezer (Sanyo, model: MDF-U76VC)
8. Microscope (Nikon, model: Eclipse TS100)
9. Temperature-controlled centrifuge (Beckman Coulter, model: Allegra 6R)
10. TC20 automated cell counter (Bio-Rad, catalog number: 145-0101)
11. Hemocytometer (Hausser Scientific, catalog number: 1483)
12. Flow cytometer (BD Biosciences, model: LSR II)
Software and datasets
1. BD FACSDiva Software (BD Biosciences)
2. FCS Express 7 (v. 7.24; De Novo Software)
3. GraphPad (v10)
Procedure
Note: All animal studies were approved by the Institutional Animal Care and Use Committee of Roswell Park Comprehensive Cancer Center. All steps in the present protocol should be completed in a sterile hood and using sterile technique and sterile reagents. It is highly recommended that cancer cell lines used be under passage 20, in continuous culture for less than 30 days, and kept under 70% confluence. Labs should also follow common methods to ensure reproducibility guidelines by authenticating cell lines and routinely testing for mycoplasma contamination.
Day 1: Preparation of cancer cell conditioned media (CM)
Note: This step can be performed at any time prior to the splenocyte activation protocol.
1. Seed EMT6 or EMT6 drug-resistant (EMT6-DR) murine breast cancer cells at a density of 7 × 106 cells in a 10 cm tissue culture-treated dish in 10 mL of cancer cell growth media (see Recipe 5).
Critical: Heat-inactivated FBS is used to ensure complement proteins do not impact splenocyte activity in downstream assays. Heat-inactivated FBS can be purchased commercially or inactivated in-house. Heat-inactivating FBS can be performed as follows:
To heat-inactivate FBS, first allow serum to come to room temperature for a minimum of 15 min. Prepare a water bath and bring the temperature to 56 °C. There must be enough water to completely submerge the entire contents of the 50 mL tube, and a control tube containing 50 mL of water should be prepared with a temperature monitor or thermostat to ensure the water bath temperature is at 56 °C. Place the control tube and serum into the 56 °C water bath, and swirl the serum every 3–5 min to ensure the serum remains uniform during heating. Remove the heat-inactivated FBS after 30 min, and gently swirl once more; it is ready for further use.
2. Incubate cancer cells for 3 days at 37 °C with 5% CO2 and then collect CM (generally ~8 mL) into a 15 mL tube on ice.
3. Centrifuge collected CM at 500× g for 5 min at 4 °C. Collect and filter the CM through a 0.2 μm filter to eliminate any floating cancer cells and place in a new sterile 15 mL tube.
4. Cancer cell-derived CM can then be stored at -20 °C for short-term storage (6–8 weeks) or at -70 °C for long-term storage (~6 months).
Day 4: Spleen harvesting and dissociation
Note: Dissociation of one spleen from an 8–12-week-old BALB/c mouse will produce around 8 × 107 to 1.3 × 108 splenocytes [4,5,11].
1. Prepare 5 mL of spleen collection solution and 25 mL of splenocyte wash buffer for processing of one spleen from a BALB/c mouse and keep on ice or at 4 °C (see Recipes 1 and 2, respectively).
2. Euthanize one BALB/c mouse according to institutional guidelines.
3. Using surgical scissors, make an incision to cut open the abdomen and then expose and harvest the spleen using curved forceps (Figure 1A).
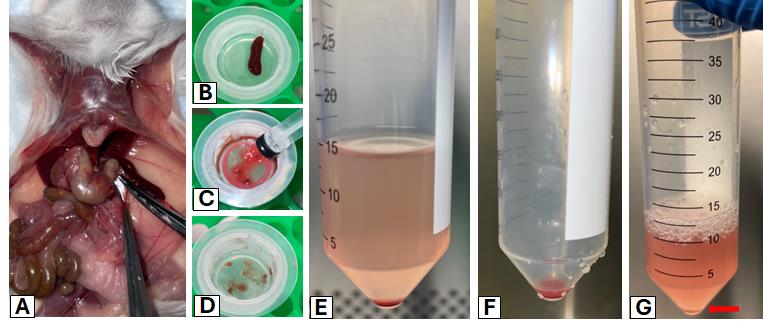
Figure 1. Representative images of key steps for harvesting and dissociating a murine spleen to prepare splenocytes for ex vivo experiments. (A) Spleen harvesting. (B–D) Mechanical spleen dissociation using a syringe plunger, 50 mL tube, and filter. (E, F) Wash and pellet splenocyte cells via centrifugation. (G) Resuspension in red blood cell (RBC) lysis buffer.
4. Place the spleen in the 50 mL tube with the prepared 5 mL of cold spleen collection solution and keep the tube on ice until ready to dissociate.
Note: If collecting multiple spleens, keep the spleens on ice until all spleens are ready to dissociate.
5. In a BSL2 biosafety cabinet under sterile conditions, place a 70 μm filter on a 50 mL tube and pre-wet the filter with 5 mL of cold spleen wash buffer.
6. Place the spleen on the pre-wet filter and, using the rubber plunger of a 3 mL syringe, mechanically crush the spleen through the filter (Figure 1B–D).
7. Wash the filter with 10 mL of cold splenocyte wash buffer.
Note: After this step, it is critical to keep centrifugation steps at 4 °C to maintain splenocyte cell viability.
8. Centrifuge splenocytes at 300× g for 5 min at 4 °C. Carefully aspirate the supernatant and resuspend the splenocyte cell pellet with 10 mL of splenocyte wash buffer (Figure 1E).
9. Centrifuge splenocytes at 300× g for 5 min at 4 °C. Carefully aspirate the supernatant.
Note: The splenocyte cell pellet will be red due to the presence of red blood cells (RBCs), and will need to be treated with RBC lysis buffer to eliminate RBCs (Figure 1F).
10. Resuspend the splenocyte cell pellet with 3 mL of 1× RBC lysis buffer per one spleen (see Recipe 4) for no more than 5 min.
Critical: RBC lysis buffer incubation times longer than 5 min will dramatically impact splenocyte viability. If processing multiple spleens, it is critical to space out incubation times accordingly.
11. Add 5× volume (15 mL) of splenocyte complete RPMI media (see Recipe 3) to inactivate the RBC lysis buffer. This dilution lowers the concentration of ammonium chloride, restoring isotonic conditions and preventing splenocyte death.
12. Centrifuge splenocytes at 300× g for 5 min at 4 °C. Carefully aspirate the supernatant.
Note: After completion of RBC lysis, the red layer (i.e., RBCs) should be gone, leaving a white-yellowish pellet (Figure 1G, red arrow).
13. Resuspend the splenocyte cell pellet in ~10–20 mL of splenocyte complete RPMI media.
14. Collect an aliquot of the splenocyte cell suspension and stain the aliquot with 0.4% trypan blue staining solution to count and assess cellular viability with a hemocytometer or automatic cell counter.
Note: If using a hemocytometer, an accurate count will require a larger resuspension volume to ensure cell density is not too high.
15. Plate splenocytes in a 10 cm dish in 10 mL of splenocyte complete RPMI media and place the dish in a 37 °C, 5% CO2 incubator overnight to allow splenocytes to settle. Proceed with the next steps.
Day 4: Preparation of cancer cells (EMT6) for the cancer cell/splenocyte co-culture assay
Note: All steps are completed under sterile conditions.
1. Remove EMT6 and EMT6-DR cancer cell tissue culture plates from the incubator (37 °C, 5% CO2) and aspirate off the media.
2. Wash the plate with 5 mL of 1× DPBS and aspirate off DPBS.
3. Add 2 mL of 0.25% trypsin and place in the incubator for ~3 min. Visually check cells under a microscope to ensure that cancer cells start to lift off the dish.
4. Collect cancer cells with 8 mL of media and place the cell suspension into a 15 mL tube.
5. Centrifuge cancer cells at 300× g for 5 min at room temperature.
6. Carefully aspirate off the supernatant and resuspend the cancer cells in 10 mL of media.
7. Take an aliquot of the cell suspension and stain the aliquot with a 0.4% trypan blue staining solution to count cancer cells using a cell counter or hemocytometer.
8. Seed 10,000 EMT6 or EMT6-DR cancer cells in a volume of 500 μL of cancer cell growth media in a 24-well treated tissue culture plate and allow cancer cells to settle overnight in a 5% CO2, 37 °C incubator before proceeding to the next step. See Figure 2 for an example of the experimental setup for seeding cancer cells to be co-cultured with splenocytes, including controls, using two 24-well tissue culture-treated plates.
Note: Ensure that any flow cytometry staining controls requiring cancer cells are seeded at this step, including positive and negative controls for inducing cancer cell death, such as untreated cancer cells (e.g., negative control), cancer cells to be heated, and treatment with an apoptosis-inducing agent like Staurosporine (positive controls) [12].
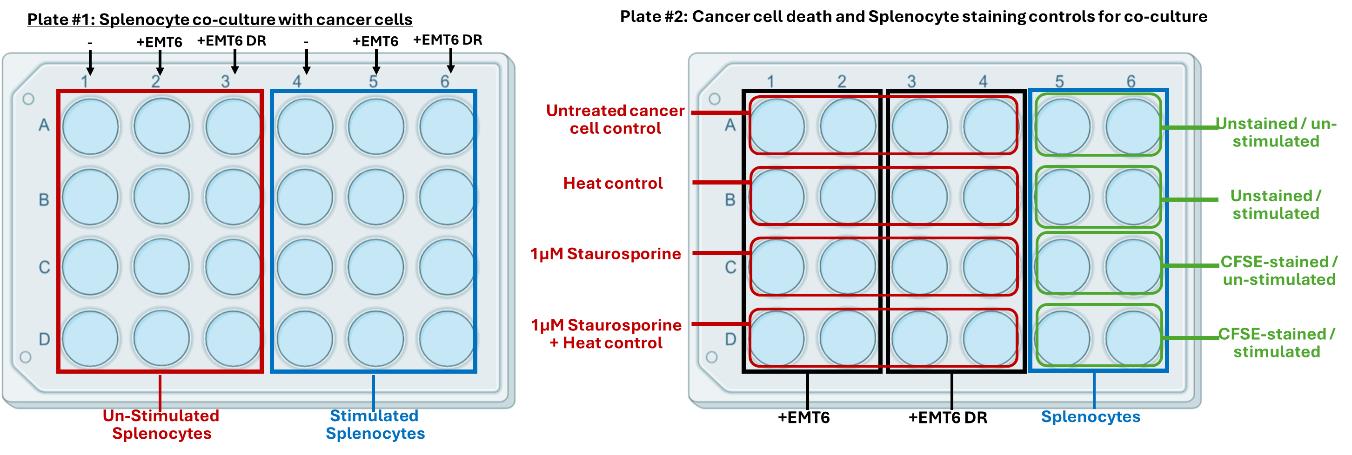
Figure 2. Experimental setup for splenocyte and cancer cell co-culture in a 24-well tissue culture-treated plate. Plate #1 (left): Splenocyte co-culture with cancer cells. Plate #2 (right): Controls to assess cancer cell death and splenocyte staining controls for co-culture.
Day 5: Preparation of splenocytes for co-culture with adherent cancer cells or treatment with cancer-derived CM
1. Grab the 10 cm dish containing isolated splenocytes prepared on Day 4 from the incubator. Using a 10 mL serological pipette, collect the non-adherent splenocytes from the 10 cm plate and place into a 50 mL tube.
2. Add 5 mL of 1× DPBS to wash the plate to collect any remaining splenocytes and add to the same 50 mL tube.
3. Centrifuge collected splenocytes at 300× g for 5 min at 4 °C. Carefully aspirate the supernatant and resuspend the pellet in 10 mL of splenocyte complete RPMI media.
4. Count the splenocytes and divide into two 50 mL tubes:
a. Tube 1 contains splenocytes to be labeled with the live cell division tracker, CFSE, before setting up the co-cultures. 52.8 million CFSE-stained splenocytes are necessary for both co-culture with cancer cells and treatment with CM; therefore, it is recommended that ~80 million splenocytes be used for CFSE staining.
b. Tube 2 contains unstained splenocytes necessary for flow cytometry staining controls. 4.4 million unstained splenocytes are needed for both plating setups; therefore, it is recommended that at least 8 million splenocytes are left unstained.
5. Prepare and label splenocytes with the live cell CFSE cell division tracking dye:
a. Wash the splenocytes in tube 1 two times with 1× DPBS. Following the last wash, resuspend splenocytes at 10 × 106 cells/mL in the 5 μM CFSE working solution according to manufacturer recommendations.
Note: The CFSE working concentration will need to be optimized based on the user’s flow cytometer.
b. Incubate splenocytes for 20 min at 37 °C in the dark.
c. Add 5 times the original staining volume of splenocyte complete RPMI media to quench the CFSE tracking dye.
d. Centrifuge splenocytes at 300× g for 5 min at RT and resuspend in prewarmed splenocyte complete RPMI media.
e. Incubate splenocytes for 10 min at 37 °C in the dark.
f. Collect CFSE-labeled splenocytes from the incubator and count using a hemacytometer or automatic cell counter.
6. Preparation of unlabeled/unstained and CFSE-labeled splenocytes for the 24-well plate co-culture assay with cancer cells:
a. For the 24-well plate setup that includes stimulated splenocytes, prepare a 50 mL tube containing a concentration of 1 × 106 splenocytes per 500 μL in splenocyte complete RPMI media. Add 1 μg/mL of anti-CD3 antibody from a stock concentration of 500 μg/mL and add 1 μg/mL of anti-CD28 antibody from a stock concentration of 500 μg/mL to the tube.
b. For a single 24-well plate setup that includes unstimulated splenocytes, prepare a 50 mL tube containing a concentration of 1 × 106 splenocytes per 500 μL of splenocyte complete RPMI media.
7. Preparation of unlabeled/unstained and CFSE-labeled splenocytes for a 96-well plate setup:
a. For a single 96-well plate setup that includes stimulated splenocytes, prepare a 50 mL tube containing a concentration of 400,000 splenocytes per 100 μL of splenocyte complete RPMI media. Add 2 μg/mL of anti-CD3 antibody from a stock concentration of 500 μg/mL and add 2 μg/mL of anti-CD28 antibody from a stock concentration of 500 μg/mL.
Note: Anti-CD3 and anti-CD28 are added at a 2× concentration to account for a final volume of 200 μL in the co-culture plate (e.g., to account for the volume of added CM).
b. For a single 96-well plate setup that includes unstimulated splenocytes, prepare a 50 mL tube containing a concentration of 400,000 splenocytes per 100 μL of splenocyte complete RPMI media.
8. Splenocytes are ready for the next step and should be used immediately to ensure optimal viability.
Day 5: Addition of prepared splenocytes to co-culture wells for cancer cell or CM incubation
1. Remove 24-well culture plates from the incubator containing seeded EMT6 or EMT6-DR cancer cells and aspirate media.
2. Add 500 μL of stimulated or unstimulated splenocytes to each well containing cancer cells and to splenocyte alone control wells. Each well should have a final volume of 0.5 mL for a total of 1 million splenocytes per well. Incubate co-culture plates at 37 °C in 5% CO2 for 3 days.
3. For 96-well round-bottom plates for cancer cell CM incubation, add prepared splenocytes at a concentration of 400,000 splenocytes per 100 μL. See Figure 3 for an example of the experimental setup for incubating splenocytes with cancer cell-derived CM in a 96-well format.
a. Add 100 μL of cancer cell-derived CM to the 96-well plate, bringing the final volume to 200 μL per well.
b. Incubate splenocytes treated with CM at 37 °C in a 5% CO2 incubator for 3 days.
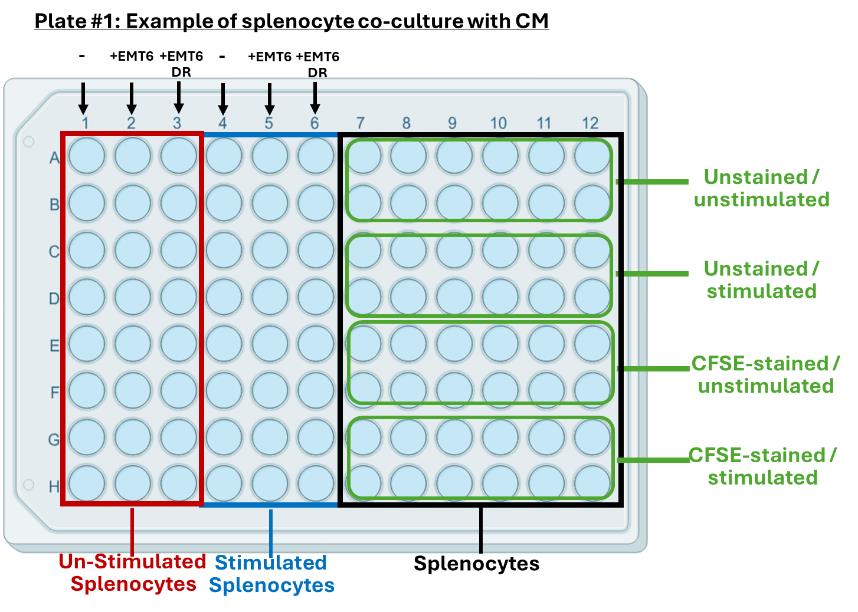
Figure 3. Example of an experimental setup for splenocyte incubation with cancer cell conditioned media (CM) in a 96-well non-tissue-culture-treated U-shaped bottom plate. Experimental groups and splenocyte staining controls can be made to fit on the same plate, depending on experimental design and the number of conditions tested.
Day 8: Sample collection and analysis
Note: It is recommended that enough time is allowed for processing plates for cancer cell-splenocyte co-cultures and splenocytes incubated with cancer cell CM.
In this protocol, processing the 24-well plates containing cancer cells co-cultured with splenocytes is completed first and then left on ice, while processing the 96-well plate containing splenocytes incubated with cancer cell CM. It is expected that processing the 24-well plates will take ~30 min per plate once familiar with the assay. Two separate staining protocols are used to assess cancer cell death and CD8 T-cell activation, as denoted below.
1. Collection and staining of splenocytes and cancer cells from 24-well plates containing co-cultures of cancer cells and splenocytes:
a. Twenty-four hours prior to collection, for cancer cell wells designated for cell death controls (i.e., Staurosporine-alone treated and Staurosporine + Heat treated), add 1 μM of Staurosporine to EMT6 and EMT6-DR control wells and incubate for at least 18 h.
b. Five hours prior to collection, add 2 μL per milliliter of 500× thawed cell activation cocktail containing brefeldin A1 to wells containing stimulated splenocytes and place in the incubator for 5 h. Brefeldin A1 is a protein transport inhibitor that inhibits the release of intracellular cytokines such as interferon (IFN) γ and granzyme B (GZB).
c. Remove plates from the incubator, gently swirl the plate, and collect the non-adherent floating cells with a 1,000 μL pipette into a 2 mL microcentrifuge tube.
Note: For heated cancer cell controls, turn on a heat block set to 55 °C prior to collecting cells to allow time for the heat block to warm up. A heated water bath can also be used.
d. Gently wash wells with 200 μL of 1× DPBS to collect remaining non-adherent splenocytes and pipette into the corresponding 2 mL microcentrifuge tube.
e. Set tubes with non-adherent cells aside on ice and begin preparing for cancer cell collection.
f. Add 200 μL of accutase to wells with cancer cells and place in the incubator for 3 min. Visually check wells to see if cells are lifting off the plate before moving forward.
g. Add 800 μL of cancer cell media to collect cancer cells, place into a 2 mL microcentrifuge tube, and place on ice to begin staining for flow cytometric assessment of annexin-V and 7-AAD staining on cancer cells.
h. Place heated cancer cell controls in the preheated heat block set to 55 °C for 45 min.
i. Once heated cancer cell controls are ready, begin processing cancer cells for cell death flow cytometry staining assessment.
j. Cancer cell death flow cytometry staining protocol:
i. Centrifuge cancer cells at 300× g for 5 min at 4 °C and resuspend in a 96-well round-bottom plate with 200 μL of 1× DPBS.
ii. Wash cells twice with 200 μL of 1× DPBS.
iii. Resuspend cells in 100 μL of annexin-V binding buffer and add 5 μL of annexin-V staining solution to each stained sample, except for indicated single stain controls.
iv. Gently vortex cells and incubate in the dark at room temperature for 15 min.
v. Next, add samples to FACS tubes and bring up to ~400 μL total volume with annexin-V binding buffer (~300 μL of binding buffer).
vi. Add 10 μL of 7-AAD per sample and promptly analyze on cytometer, as 7-AAD is not compatible with fixation.
k. Splenocyte flow cytometry staining protocol:
i. Centrifuge splenocytes at 300× g for 5 min at 4 °C and resuspend in a 96-well round-bottom plate in 200 μL of FACS buffer.
ii. Aspirate supernatant and wash splenocytes twice with 200 μL of FACS buffer.
iii. Centrifuge splenocytes at 300× g for 5 min at 4 °C and resuspend in 100 μL of TruStain FcX (1:100 dilution). Incubate at 4 °C for 15 min in the dark.
iv. Centrifuge splenocytes at 300× g for 5 min at 4 °C, aspirate supernatant, and resuspend samples with 100 μL of the antibody cocktail containing surface markers, single stain controls, or fluorescence minus one (FMO) controls (see Table 2 for single and FMO controls). Incubate for 30 min at 4 °C in the dark.
v. Wash cells twice with 200 μL of FACS buffer.
vi. Centrifuge splenocytes at 300× g for 5 min at 4 °C and resuspend in 100 μL of fixation buffer. Incubate cells at room temperature for 20 min in the dark to fix cells.
vii. Centrifuge splenocytes at 300× g for 5 min at 4 °C and wash with 200 μL of 1× intracellular permeabilization wash buffer (see Recipe 7) three times.
viii. Resuspend samples in 100 μL of intracellular marker antibody cocktail prepared in 1× intracellular permeabilization wash buffer at 4 °C for 30 min in the dark.
ix. Wash cells twice with 200 μL of FACS buffer.
x. Resuspend samples for FACS analysis. Samples can be run on the cytometer in a 96-well plate or in 5 mL FACS tubes.
Table 2. Single color (SC) and fluorescence minus one (FMO) staining controls for flow cytometry analysis of T-cell activation and cancer cell death assessment
| T-cell activation panel for flow cytometry | ||
| Sample/well | Tube | Fluorophore |
| Unstained/unstimulated splenocytes (e.g., Figure 2, plate 2, wells A5, A6; Figure 3, wells A7–12, B7–12) | Unstained | N/A |
| CFSE-stained/unstimulated splenocytes (e.g., Figure 2, plate 2, wells C5, C6; Figure 3, wells E7–12, F7–12) | SC CFSE | CFSE |
| Unstained/unstimulated splenocytes (e.g., Figure 2, plate 2, wells A5, A6; Figure 3, wells A7–12, B7–12) | SC CD45 | Only antibody for CD45 |
| Unstained/unstimulated (e.g., Figure 2, plate 2, wells A5, A6) | SC CD8 | Only antibody for CD8 |
| Unstained/stimulated splenocytes (e.g., Figure 2, plate 2, wells B5, B6; Figure 3, wells C7–12, D7–12) | SC CD69 | Only antibody for CD69 |
| Unstained/stimulated (e.g., Figure 2, plate 2, wells B5, B6) | SC Granzyme B | Only antibody for Granzyme B |
| Unstained/stimulated (e.g., Figure 2, plate 2, wells B5, B6) | SC IFNγ | Only antibody for IFNγ |
| Unstained/stimulated (e.g., Figure 2, plate 2, wells B5, B6) | FMO CFSE | All antibodies |
| CFSE-stained/stimulated splenocytes (e.g., Figure 2, plate 2, wells D5, D6; Figure 3, wells G7–12, H7–12) | FMO CD45 | All antibodies except anti-CD45 |
| FMO CD8 | All antibodies except anti-CD8 | |
| FMO CD69 | All antibodies except anti-CD69 | |
| FMO Granzyme B | All antibodies except anti-Granzyme B | |
| FMO IFNγ | All antibodies except anti-IFNγ | |
| Cancer cell death panel for flow cytometry | ||
| Cancer cells alone (untreated; e.g., Figure 2, plate 2, wells A1–2) | Unstained | N/A |
| Unstained/unstimulated splenocytes (e.g., Figure 2, plate 2, wells A5–6) | SC CD45 | Only antibody for CD45 |
| EMT6 or EMT6-DR + 1 μM Staurosporine (e.g., Figure 2, plate 2, wells B1–2; wells B3–4) | SC annexin V | Only antibody for annexin V |
| EMT6 or EMT6-DR + heat (e.g., Figure 2, plate 2, wells C1–2; wells C3–4) | SC 7AAD | Only 7-AAD staining dye |
| EMT6 or EMT6-DR + 1 μM Staurosporine + heat (e.g., Figure 2, plate 2, wells D1–2; wells D3–4) | FMO CD45 | Antibody for annexin V and 7-AAD staining dye |
| Mix EMT6/EMT6-DR + heat and unstained/unstimulated splenocytes (e.g., Figure 2, plate 2, wells B1–2 + A5–6) | FMO annexin V | Antibody for anti-CD45 and 7-AAD staining dye |
| Mix EMT6/EMT6-DR + 1 μM Staurosporine and unstained/unstimulated splenocytes (e.g., Figure 2, plate 2, wells C1–2 + A5–6) | FMO 7AAD | Antibody for anti-CD45 and annexin V |
2. Collection and staining of splenocytes following incubation with cancer cell CM: Splenocyte staining panel will be used to assess the level of T-cell proliferation and activity as determined by CFSE dilution, surface expression of CD69, and intracellular expression of IFNγ and GZB in CD8+ CD45+ cells.
Note: Splenocytes can be stained in the same 96-well plate, but staining controls must be pooled according to Table 2 and stained in 1.5 mL microcentrifuge tubes or a separate 96-well plate.
a. Five hours prior to collection, add 2 μL per 1 mL volume (for 200 μL, the cell activation cocktail is diluted 10× and then added at a concentration of 2 μL/200 μL) to wells containing stimulated splenocytes and place in an incubator (37 °C, 5% CO2).
b. After 5 h, centrifuge 96-well plates containing splenocytes at 300× g for 5 min at 4 °C.
c. Aspirate supernatant and wash two times with 200 μL of FACS buffer (see Recipe 6).
d. Spin splenocytes down, resuspend in 100 μL of TruStain FcX (1:100 dilution) per sample, and incubate at 4 °C for 15 min in the dark.
e. Spin splenocytes down, aspirate supernatant, and resuspend splenocytes with 100 μL of the antibody cocktail containing surface markers, single stain controls, or FMO controls (see Table 2 for single and FMO controls). Incubate for 30 min at 4 °C in the dark.
f. Wash splenocytes twice with 200 μL of FACS buffer.
g. Spin cells down and resuspend in 100 μL of fixation buffer. Incubate samples at room temperature for 20 min in the dark to fix cells.
h. Spin cells down and wash with 200 μL of 1× intracellular permeabilization wash buffer (see Recipe 7) three times.
i. Resuspend samples in 100 μL of intracellular marker antibody cocktail prepared in 1× intracellular permeabilization wash buffer at 4 °C for 30 min in the dark.
j. Wash cells twice with FACS buffer.
k. Resuspend samples in 200 μL of FACS buffer if using a plate for flow assessment or 300 μL if using FACS tubes for FACS analysis (see Table 3 for an example of flow cytometer settings used for the splenocyte assessment).
Table 3. Example of flow cytometer settings for splenocyte assessment
| Manufacturer | Becton Dickinson | ||
| Model | LSRII | ||
| Data transformation | FSC 3.0 | ||
| Configuration | 2UV-6V-2B-5YG-3R | ||
| Parameter | Laser | Bandpass filter | Voltage setting |
| FSC (forward scatter) | 488 nm (B) | N/A | 615 |
| SSC (side scatter) | 488 nm (B) | B488/10 | 257 |
| Pacific Blue | 405 nm (V) | V450/50 | 490 |
| FITC | 488 nm (B) | B530/30 | 350 |
| PE | 561 nm (YG) | Y582/15 | 500 |
| PE-Cy7 | 561 nm (YG) | Y780/60 | 500 |
| APC | 638 nm (R) | R660/20 | 505 |
| Alexa Fluor 700 | 638 nm (R) | R730/45 | 467 |
Data analysis
Using experimental data comparing a cancer cell (EMT6) to a drug-resistant variant (EMT6-DR), the following figures show flow analysis of data obtained from experiments to evaluate stimulated/unstimulated splenocyte controls (Figure 4) and cancer cell CM (Figure 5). Overall, we show that an EMT6-DR variant can suppress levels of activated CD8+ T cells ex vivo. In addition, we show that differences in cell death between cancer cell variants can be observed following co-culture with stimulated CD8+ splenocytes (Figure 6).
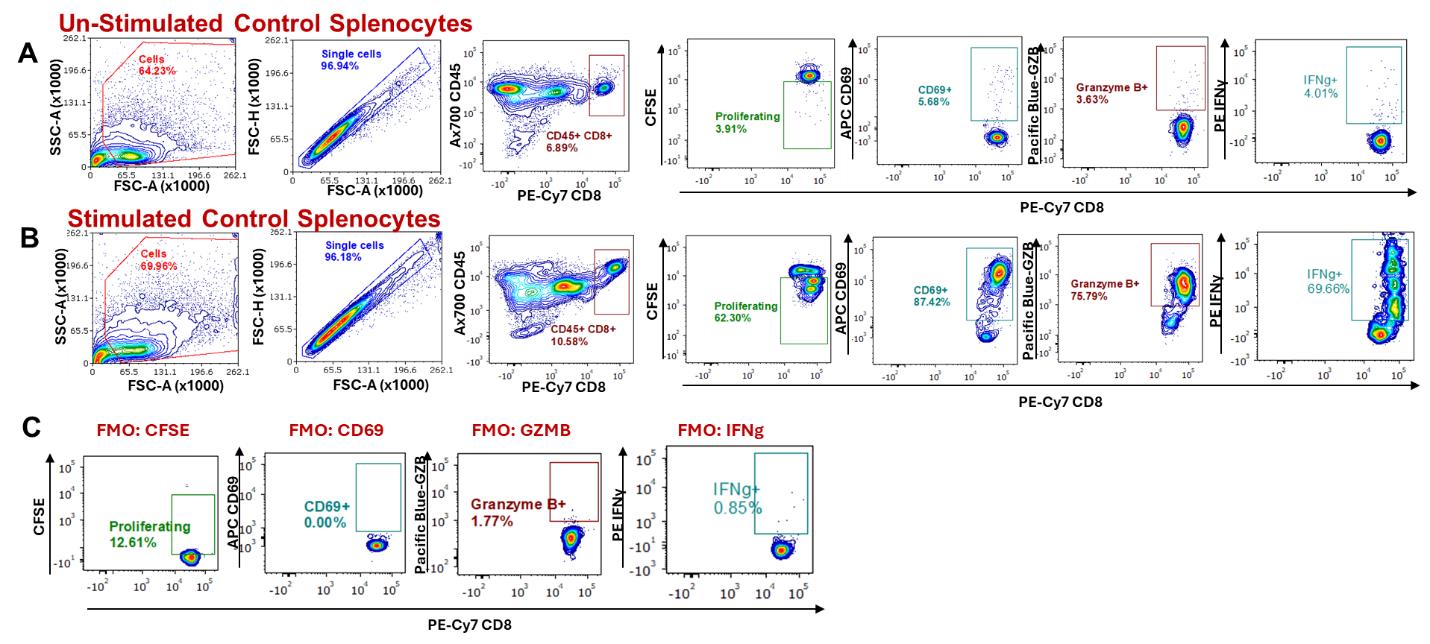
Figure 4. Stimulating splenocyte CD8+ T cells and analysis by flow cytometry. Gating strategy to analyze the activation status of CD8+ T cells from splenocytes stimulated with anti-CD3 and anti-CD28 antibodies after 3 days, compared to unstimulated splenocytes. Cells were gated based on forward and side scatter to eliminate debris, followed by singlet selection, and then gated on CD45+ and CD8+/hi cells. Proliferation, expression of CD69, Granzyme B (GZB), and IFNγ were determined by flow cytometry following a gating strategy based on comparing (A) unstimulated splenocytes, (B) stimulated splenocytes, and (C) fluorescence minus one (FMO) staining for key activation markers.
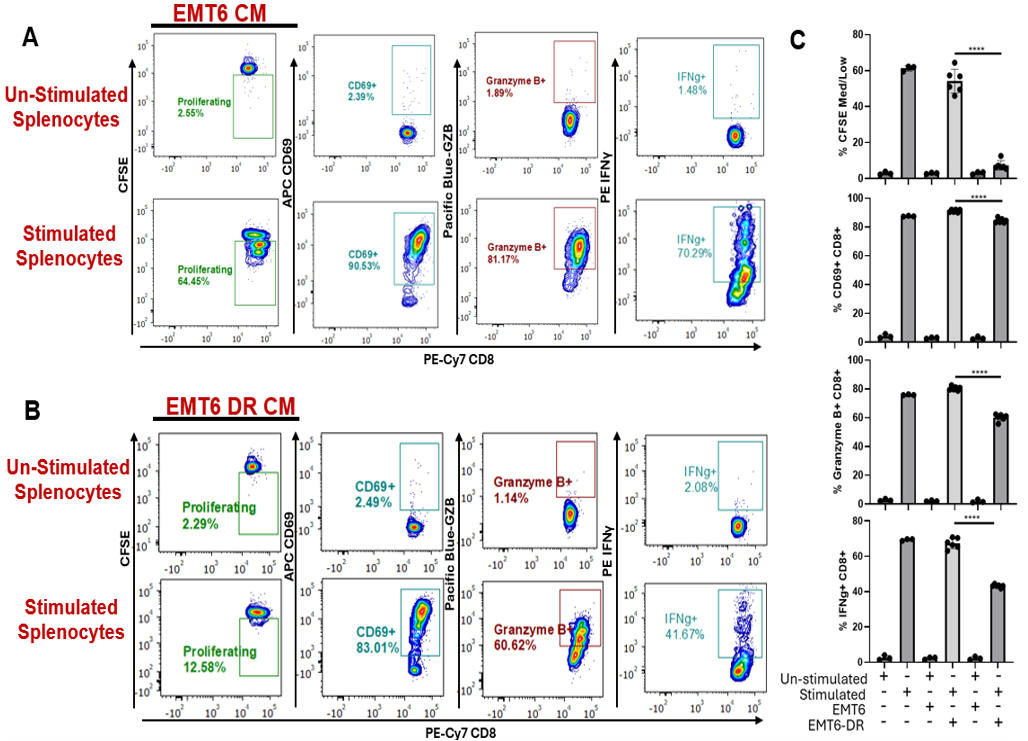
Figure 5. Immunophenotyping CD8+ T cells following incubation with cancer cell–derived conditioned media (CM) by flow cytometry. (A, B) Unstimulated and stimulated splenocytes containing CD8+ T cells were evaluated by flow cytometry for proliferation, surface expression of CD69, and intracellular expression of effector molecules GZB and IFNγ following incubation with conditioned media from EMT6 cancer cells or EMT6 drug-resistant (DR) cancer cells. (C) Quantification and comparisons of indicators of CD8 T-cell activation in tested conditions. Unstimulated splenocytes and stimulated splenocytes alone were used as controls (experimental replicates n = 3–6). Two-tailed T-test; mean ± SD is represented. All experiments were performed in an independent run.
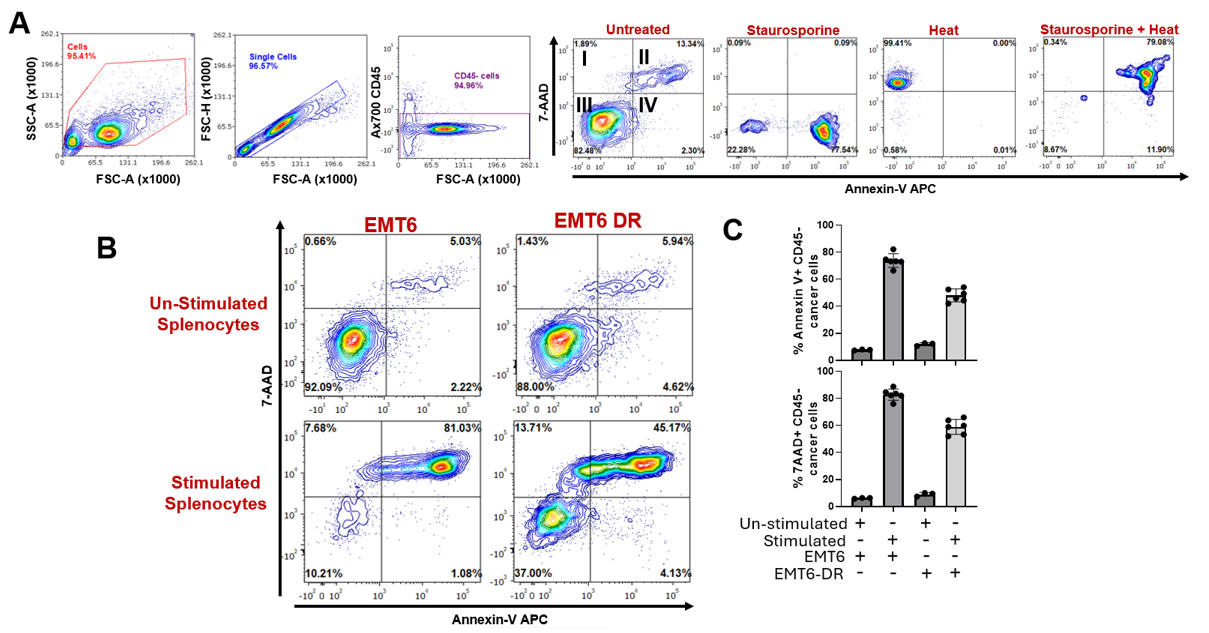
Figure 6. Cancer cell death evaluation following co-culture with stimulated splenocytes containing activated CD8+ T cells by flow cytometry. (A) Gating strategy to assess annexin V and 7-AAD staining in tumor cells following co-culture with unstimulated splenocytes or splenocytes stimulated with anti-CD3 and anti-CD28 antibodies. Cells were gated based on forward and side scatter to eliminate debris and selected for singlets, and then gated on CD45 expression to gate out splenocytes. (B) Gating representation of annexin-V and 7-AAD levels showing that the addition of activated splenocytes can induce cancer cell death. (C) Quantification and comparison of annexin-V and 7-AAD levels (experimental replicates n = 3–6). Two-tailed T-test; mean ± standard deviation is represented. All experiments were performed in an independent run.
In summary, this protocol describes the steps to evaluate the functional impact of cancer cells on CD8+ T-cell activation from splenocytes, as well as the impact of CD8+ T cells on cancer cell death. The expression of surface markers and intracellular effector molecules is used to assess activation status of CD8+ T cells, including CD69, GZB, and IFNγ, following stimulation via CD3/TCR and CD28/CD80 signaling.
Validation of protocol
All experiments herein were repeated independently at least once. All parts of this protocol were validated in Figure 6C, D and Figure 6F–K from Shi et al. EMBO Reports (2025) [4]. Contact-dependent and independent effects of cancer cells on CD8+ splenocytes were also validated in Figure 5D–J from Dolan et al. Molecular Cancer Therapeutics (2024) [5].
General notes and troubleshooting
| Problem | Possible reason | Solution |
|---|---|---|
| Cancer cells lifting off during splenocyte collection | Cancer cells are too confluent or weakly adherent. | Seed fewer cancer cells or use a separate plate for flow cytometry gating to separate cell populations. |
| Difficulty determining differences between experimental groups in apoptosis assays. | Use of positive apoptosis assay controls to establish range. | Certain cancer cell types may be more or less sensitive to Staurosporine and require studies to determine the proper treatment dose and duration. Other agents capable of inducing cell death can be substituted, e.g., methanol. |
| Lack of robust response or need for further confirmation | Bulk splenocyte activation may not be specific enough for some experiments. Alternative CD8+ T-cell-dependent assays may be needed. | Purify CD8+ T cells via magnetic or flow-based separation [13] or use transformed cell lines (e.g., MOHITO cells) as surrogates for T cells [14,15]. |
| Need for alternative readouts for cancer cell death | Flow cytometry might not fully capture cell death dynamics. Additional assays may improve rigor/reproducibility. | Supplement flow cytometry with microscopy assays (e.g., crystal violet colony assays). |
| Variability in results or lack of robust responses during co-culture duration at different timepoints | Immune activation kinetics vary with strain and context. | Optimize incubation time (e.g., 24–72 h). NOTE: BALB/c typically needs 72 h. Use age-matched mice between independent replicates, and if pooling spleens for larger experiments. |
| Plate density concerns | High density may impair cell–cell contact assessment. | Optimize seeding density and plate size (24-well vs. 48-well vs. 12-well formats). We recommend 24-well plates to fit all conditions in 2 plates. Processing time should be taken into consideration when setting up conditions. |
| Cancer cell line differences | Different cell lines vary in growth rates and immune interactions. | Adjust splenocyte-to-cancer cell ratio and incubation conditions for each cancer model. |
| Splenocyte viability after RBC lysis | Prolonged exposure to lysis buffer reduces viability. | Strictly limit RBC lysis to 5 min maximum. |
| CFSE staining issues | Over- or under-staining can compromise proliferation tracking. | Titrate CFSE concentration and optimize staining conditions for each experiment. |
| Antibody titration for flow cytometry | Using incorrect antibody concentrations can yield poor staining. | Perform titration curves for all antibodies before formal experiments. |
| Dead cell exclusion during flow cytometry | Dead cells can cause artifacts in flow cytometry data. | Use a viability dye (e.g., 7-AAD, Fixable zombie dye) during surface staining. |
| CM studies yield poor or inconsistent results. | Cytokine composition in CM can vary between batches. | Pool/condense CM to increase cytokine concentration or standardize to select cytokines to minimize batch-to-batch variability. Use cancer cells that are passage-matched as much as possible. |
| Nutrient depletion during CM exposure | Cancer CM alone may lack sufficient nutrients for 3-day splenocyte culture. | Supplement CM with splenocyte complete RPMI to maintain splenocyte health. We recommend a 1:1 ratio. |
Acknowledgments
This project used shared resources supported by the Roswell Park Comprehensive Cancer Center (RPCCC) Support Grant from the National Cancer Institute (P30CA016056). This work was supported by grants to JMLE from the American Cancer Society (ACS) via a Research Scholar Grant (RSG-18-064-01-TBG), Mission Boost Grant (MBGI-23-1038434-01-MBG), and Roswell Park Alliance Foundation (RPAF). This work was also supported by an NCI grant to YS (F30 CA243281). Parts of this protocol were adapted from the publication Shi et al. [4] and Dolan et al. [5]. All graphics were made using BioRender.com.
Competing interests
The authors declare no competing interests.
Ethical considerations
Animal cancer model studies and animal housing conditions were performed in strict accordance with the recommendations in the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and according to guidelines of the IACUC at RPCCC (Protocol: 1227M).
References
- Emens, L. A., Romero, P. J., Anderson, A. C., Bruno, T. C., Capitini, C. M., Collyar, D., Gulley, J. L., Hwu, P., Posey, A. D., Silk, A. W., et al. (2024). Challenges and opportunities in cancer immunotherapy: a Society for Immunotherapy of Cancer (SITC) strategic vision. J ImmunoTher Cancer. 12(6): e009063. https://doi.org/10.1136/jitc-2024-009063
- Wang, Y., Shelton, S. E., Kastrunes, G., Barbie, D. A., Freeman, G. J. and Marasco, W. A. (2022). Preclinical models for development of immune–oncology therapies. Immuno Oncology Insights. 3(8): 396–398. https://doi.org/10.18609/ioi.2022.41
- Franklin, M. R., Platero, S., Saini, K. S., Curigliano, G. and Anderson, S. (2022). Immuno-oncology trends: preclinical models, biomarkers, and clinical development. J ImmunoTher Cancer. 10(1): e003231. https://doi.org/10.1136/jitc-2021-003231
- Shi, Y., McKenery, A., Dolan, M., Mastri, M., Hill, J. W., Dommer, A., Benzekry, S., Long, M., Abrams, S. I., Puzanov, I., et al. (2024). Acquired resistance to PD-L1 inhibition enhances a type I IFN-regulated secretory program in tumors. EMBO Rep. 26(2): 521–559. https://doi.org/10.1038/s44319-024-00333-0
- Dolan, M., Shi, Y., Mastri, M., Long, M. D., McKenery, A., Hill, J. W., Vaghi, C., Benzekry, S., Barbi, J., Ebos, J. M., et al. (2024). A Senescence-Mimicking (Senomimetic) VEGFR TKI Side Effect Primes Tumor Immune Responses via IFN/STING Signaling. Mol Cancer Ther. 23(9): 1241–1260. https://doi.org/10.1158/1535-7163.mct-24-0139
- Lewis, M. D., de Leenheer, E., Fishman, S., Siew, L. K., Gross, G. and Wong, F. S. (2015). A reproducible method for the expansion of mouse CD8+ T lymphocytes. J Immunol Methods. 417: 134–138. https://doi.org/10.1016/j.jim.2015.01.004
- Look, T., Meister, H., Weller, M. and Weiss, T. (2023). Protocol for the expansion of mouse immune effector cells for in vitro and in vivo studies. STAR Protoc. 4(4): 102700. https://doi.org/10.1016/j.xpro.2023.102700
- Waldman, A. D., Fritz, J. M. and Lenardo, M. J. (2020). A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 20(11): 651–668. https://doi.org/10.1038/s41577-020-0306-5
- Montecino-Rodriguez, E., Berent-Maoz, B. and Dorshkind, K. (2013). Causes, consequences, and reversal of immune system aging. J Clin Invest. 123(3): 958–965. https://doi.org/10.1172/jci64096
- Xiao, T., Lee, J., Gauntner, T. D., Velegraki, M., Lathia, J. D. and Li, Z. (2024). Hallmarks of sex bias in immuno-oncology: mechanisms and therapeutic implications. Nat Rev Cancer. 24(5): 338–355. https://doi.org/10.1038/s41568-024-00680-z
- Pinchuk, L. M. and Filipov, N. M. (2008). Differential effects of age on circulating and splenic leukocyte populations in C57BL/6 and BALB/c male mice. Immun Ageing. 5(1): e1186/1742–4933–5–1. https://doi.org/10.1186/1742-4933-5-1
- Zhang, X. D., Gillespie, S. K. and Hersey, P. (2004). Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol Cancer Ther. 3(2): 187–197. https://doi.org/10.1158/1535-71
- Xie, L., Liu, G., Liu, Y. and Yu, Y. (2021). In vitro and In vivo CD8+ T Cell Suppression Assays. Bio Protoc. 11(10): e4020. https://doi.org/10.21769/bioprotoc.4020
- Liu, J., Wang, Y., Mu, C., Li, M., Li, K., Li, S., Wu, W., Du, L., Zhang, X., Li, C., et al. (2022). Pancreatic tumor eradication via selective Pin1 inhibition in cancer-associated fibroblasts and T lymphocytes engagement. Nat Commun. 13(1): 4308. https://doi.org/10.1038/s41467-022-31928-7
- Kleppe, M., Mentens, N., Tousseyn, T., Wlodarska, I. and Cools, J. (2010). MOHITO, a novel mouse cytokine-dependent T-cell line, enables studies of oncogenic signaling in the T-cell context. Haematologica. 96(5): 779–783. https://doi.org/10.3324/haematol.2010.035931
Article Information
Publication history
Received: May 2, 2025
Accepted: Jul 17, 2025
Available online: Aug 1, 2025
Published: Aug 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Dolan, M., Shi, Y., McKenery, A., Tzetzo, S., Chida, K., Takabe, K., Abrams, S. I. and Ebos, J. M. (2025). Isolation and Ex Vivo Testing of CD8+ T-Cell Division and Activation Using Mouse Splenocytes. Bio-protocol 15(16): e5423. DOI: 10.21769/BioProtoc.5423.
Category
Cancer Biology > Tumor immunology > Immunological assays
Cell Biology > Cell-based analysis > Flow cytometry
Immunology > Immune cell function > Antigen-specific response
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.