- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

RACE-Nano-Seq: Profiling Transcriptome Diversity of a Genomic Locus
Published: Vol 15, Iss 13, Jul 5, 2025 DOI: 10.21769/BioProtoc.5374 Views: 2804
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2024

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
The complexity of the human transcriptome poses significant challenges for complete annotation. Traditional RNA-seq, often limited by sensitivity and short read lengths, is frequently inadequate for identifying low-abundant transcripts and resolving complex populations of transcript isoforms. Direct long-read sequencing, while offering full-length information, suffers from throughput limitations, hindering the capture of low-abundance transcripts. To address these challenges, we introduce a targeted RNA enrichment strategy, rapid amplification of cDNA ends coupled with Nanopore sequencing (RACE-Nano-Seq). This method unravels the deep complexity of transcripts containing anchor sequences—specific regions of interest that might be exons of annotated genes, in silico predicted exons, or other sequences. RACE-Nano-Seq is based on inverse PCR with primers targeting these anchor regions to enrich the corresponding transcripts in both 5' and 3' directions. This method can be scaled for high-throughput transcriptome profiling by using multiplexing strategies. Through targeted RNA enrichment and full-length sequencing, RACE-Nano-Seq enables accurate and comprehensive profiling of low-abundance transcripts, often revealing complex transcript profiles at the targeted loci, both annotated and unannotated.
Key features
• This protocol is highly sensitive and can detect low-abundance transcripts.
• This protocol can be performed in a typical molecular biology laboratory.
• This protocol allows RACE reactions with single or multiple primers, supporting various research scales.
• This protocol enables characterization of complex genomic loci and discovery of novel transcripts, exons, and alternative splicing events.
Graphical overview
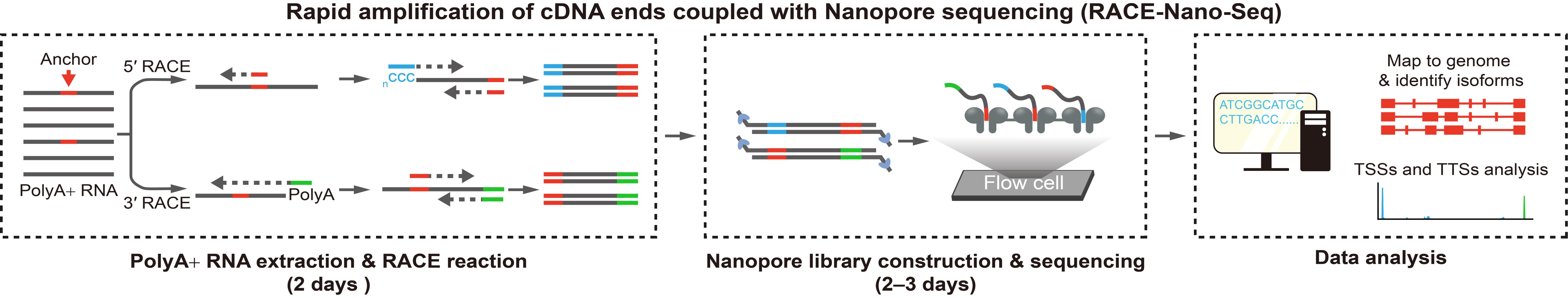
Background
The well-documented transcriptome complexity can be viewed as a combination of several hallmark features, including the multitude of alternative splicing events [1,2], multiple alternative transcription start and termination sites (TSSs and TTSs) [3,4], overlapping or chimeric transcripts [5–9], and pervasive transcription [10,11]. Collectively, the molecular processes that give rise to these features orchestrate the sophisticated landscape of mammalian gene expression.
Comprehensive characterization of complex transcriptome landscapes presents several challenges, particularly in detecting low-abundance transcripts, accurately defining transcript structures, and efficiently obtaining full-length sequences. While conventional transcriptome studies effectively identify and annotate highly expressed, ubiquitously present transcripts, they frequently miss elusive transcripts exhibiting tightly regulated expression patterns, produced only in specific tissues, cell types, or under specific biological conditions [12–16]. The restricted expression patterns of these transcripts result in extremely low abundance that often falls below the sensitivity limits of standard RNA-seq assays, hindering their effective detection.
Given these limitations, targeted RNA enrichment approaches have become essential for accessing low-abundance transcripts, especially in bulk samples. Currently, two primary targeted RNA enrichment strategies are employed: CaptureSeq and rapid amplification of cDNA ends (RACE). CaptureSeq, a hybridization-based method, utilizes specifically designed DNA oligonucleotide probes to capture transcripts of interest, thereby increasing sequencing coverage for target regions [7]. This method enhances the sensitivity and accuracy of low-abundance transcript detection and has been applied in the discovery of novel genes and transcripts [7,17]. Moreover, combining CaptureSeq with long-read sequencing platforms like Nanopore [18] or PacBio [19] enables high-precision sequencing without the need for transcript assembly. However, its implementation is costly and highly dependent on probe design accuracy.
RACE, an inverse PCR-based technique, amplifies the 5' and 3' terminal sequences of RNA molecules by targeting specific anchor sequences [20,21]. 3' RACE utilizes a poly(dT) primer to target the poly(A) tail, facilitating the retrieval of 3' terminal sequences [22]. 5' RACE employs various strategies to amplify the 5' end, including terminal transferase tailing, adapter ligation, and the switching mechanism at the 5' end of the RNA template (SMART) approach [22,23]. RACE is a cost-effective and readily accessible technique for most molecular biology laboratories. In a direct comparison study, this method demonstrated higher sensitivity than CaptureSeq in detecting splice junctions [24]. RACE has been combined with tiling arrays or next-generation sequencing (NGS) to resolve complex transcriptional patterns. Kapranov et al. integrated RACE with high-density tiling arrays, revealing the extensive complexity of the human transcriptome, including transcript fusion and interlacing structures [5]. Lagarde et al. developed RACE-Seq, performing 5' and 3' RACE on 398 known long noncoding RNA (lncRNA) exons, followed by high-throughput sequencing using the Roche 454 FLX+ NGS platform, yielding reads with an average length of approximately 600 base pairs (bp) [24]. In our recent studies, RACE was integrated with Nanopore long-read sequencing to determine the complete structure of novel intragenic or intergenic transcripts, using GENSCAN-predicted exons as anchors [25,26]
The inherent complexity of the transcriptome, characterized by dynamic splicing patterns and structural diversity, poses significant analytical challenges. Conventional RACE coupled with Sanger sequencing, while capable of generating relatively long reads, suffers from low throughput and labor-intensive workflows. Conversely, short-read NGS technologies have intrinsic limitations in adequately resolving complex splicing patterns. Although long-read sequencing overcomes read length limitations and enables full-length transcript detection [27], its modest throughput remains a bottleneck for capturing the full diversity of low-abundance transcripts. Therefore, combining targeted RNA enrichment techniques with long-read sequencing offers an effective strategy, providing a practical and streamlined solution for targeted analysis of complex loci. Among long-read sequencing techniques, Nanopore sequencing achieves significantly longer read lengths than PacBio and is more cost-effective, providing unprecedented capability for de novo gene annotation and structural variant detection [19,28,29].
Here, we introduce RACE coupled with Nanopore sequencing (RACE-Nano-Seq), a method designed to efficiently capture full-length transcripts. This approach leverages target locus sequences (annotated exons, predicted exons, or other sequences) as anchors for full-length transcript enrichment via 5'/3' RACE. The enriched cDNA products are then analyzed with Nanopore sequencing and aligned to the corresponding reference genome to enable sensitive transcriptome characterization. This approach is particularly well-suited for detecting low-abundance transcripts, identifying novel exons, and characterizing splicing patterns at specific gene loci. In this protocol, we detail the experimental procedures and analytical pipelines for implementing RACE-Nano-Seq. Additionally, we provide an example demonstrating its application in profiling the transcriptome diversity at a specific gene locus.
Materials and reagents
Biological materials
1. K562 (Cell Bank of Chinese Academy of Sciences, catalog number: TCHu191)
Reagents
1. TRNzol universal reagent (Tiangen, catalog number: DP424)
2. Chloroform (Guoyao, catalog number: 10006818)
3. E.Z.N.A.® Total RNA kit (OMEGA, catalog number: R6834-02). Kit components used in this protocol: HiBind® RNA Mini column, collection tube, RNA wash buffer I, RNA wash buffer II
4. Library preparation VAHTSTM mRNA capture beads (Vazyme, catalog number: N401-02)
5. VAHTS DNA clean beads (Vazyme, catalog number: N411)
6. UltraPureTM DNase/RNase-free distilled water (Invitrogen, catalog number: 10977035)
7. Ethanol (Guoyao, catalog number: 10009218)
8. PrimeScriptTM II 1st Strand cDNA Synthesis kit (Takara, catalog number: 6210A). Kit components used in this protocol: 10 mM dNTP mix, 5× PrimeScript II buffer, 200 U/μL PrimeScript II reverse transcriptase, 40 U/μL RNase inhibitor
9. Terminal transferase (NEB, catalog number: M0315): 20 U/μL terminal transferase, 10× terminal transferase buffer, 10× CoCl2
10. PrimeSTAR® GXL DNA polymerase (Takara, catalog number: R050A). Kit components used in this protocol: 5× PrimeSTAR GXL buffer, PrimeSTAR GXL DNA polymerase (1.25 U/μL), dNTP mix (2.5 mM each)
11. Agarose (Invitrogen, catalog number: 75510-019)
12. 50× TAE buffer (Solarbio, catalog number: T1060)
13. 10,000× SuperRed (Biosharp, catalog number: BS354A)
14. Ligation Sequencing kit (Oxford Nanopore Technologies, catalog number: SQK-LSK114). Kit components used in this protocol: AMPure XP beads, ligation adapters, ligation buffer, flow cell tether, flow cell flush, sequencing buffer, library beads, short fragment buffer, elution buffer
15. NEBNext FFPE Repair Mix (NEB, catalog number: M6630), includes: NEBNext FFPE DNA repair mix, NEBNext FFPE DNA repair buffer
16. NEBNext Ultra II End Repair/dA-tailing module (NEB, catalog number: E7546), includes: ultra II end-prep reaction mix, ultra II end-prep enzyme buffer
17. NEBNext Quick Ligation module (NEB, catalog number: E6056), includes NEBNext Quick T4 DNA ligase
18. Qubit dsDNA HS Assay kit (ThermoFisher, catalog number: Q32851)
19. InvitrogenTM UltraPureTM BSA (Invitrogen, catalog number: AM2616)
20. Equalbit 1× dsDNA HS Assay kit (Vazyme, catalog number: EQ121-01)
Solutions
1. 70% ethanol (see Recipes)
2. 80% ethanol (see Recipes)
3. 1× TAE buffer (see Recipes)
4. 1% agarose gel (see Recipes)
Recipes
1. 70% ethanol
| Reagent | Final concentration | Volume |
|---|---|---|
| Ethanol | 70% (v/v) | 700 μL |
| UltraPureTM DNase/RNase-free distilled water | 30% (v/v) | 300 μL |
Note: Prepare fresh 70% and 80% ethanol solutions immediately before use and adjust the volume based on reaction needs.
2. 80% ethanol
| Reagent | Final concentration | Volume |
|---|---|---|
| Ethanol | 80% (v/v) | 800 μL |
| UltraPureTM DNase/RNase-free distilled water | 20% (v/v) | 200 μL |
3. 1× TAE buffer
| Reagent | Final concentration | Volume |
|---|---|---|
| 50× TAE buffer | 1× | 1 mL |
| Ultrapure water (lab-purified) | n/a | 49 mL |
4. 1% agarose gel
| Reagent | Final concentration | Quantity/volume |
|---|---|---|
| Agarose | 1% (w/v) | 0.3 g |
| 10,000× SuperRed | 1× | 3 μL |
| 1× TAE buffer | n/a | 30 mL |
Laboratory supplies
1. 1.5 mL EP tubes (Axygen, catalog number: MCT-150-C)
2. 200 μL PCR tubes (Axygen, catalog number: PCR-02-C)
3. 10 μL pipette tips (Axygen, catalog number: TF-300)
4. 20 μL pipette tips (Axygen, catalog number: TF-20)
5. 200 μL pipette tips (KIRGEN, catalog number: KG5213-L)
6. 1,000 μL pipette tips (KIRGEN, catalog number: KG5313-L)
Equipment
1. Spectrophotometer (Merinton, model: SMA6000)
2. Fluorescence imaging system (Tanon, model: Tanon 3500R)
3. Qubit 4 fluorometer (Thermo Fisher, catalog number: Q33238)
4. C1000 TouchTM thermal cycler (Bio-Rad, model: C1000)
5. Oxford Nanopore PromethION (Oxford Nanopore Technologies)
6. FLO-PRO002 R10.4 flow cell (Oxford Nanopore Technologies)
7. Rotator mixer (Qilinbeier, model: BE-1100)
8. Magnetic rack (Promega, catalog number: Z5332)
9. Direct-Pure UP Ultrapure & RO Lab Water System (Rephile)
Software and datasets
1. Guppy (v4.3.6)
2. NanoFilt (2.8.0, https://github.com/wdecoster/nanofilt/)
3. Minimap2 (v2.17-r941, https://github.com/lh3/minimap2/)
4. Samtools (1.10, https://github.com/samtools/samtools/)
5. BEDTools (v2.30.0, https://bedtools.readthedocs.io/en/latest/)
6. GRCh38/hg38 (https://hgdownload.cse.ucsc.edu/goldenpath/hg38/bigZips/)
7. Bedparse (v0.2.3, https://github.com/tleonardi/bedparse/tree/b2833706a006504b267b9a0692334a7d18e44e5c/)
8. Encyclopedia of DNA Elements (ENCODE) Candidate Cis-Regulatory Elements (https://genome.ucsc.edu/)
9. ENCODE H3K4Me3 Mark in K562 cell line (https://genome.ucsc.edu/)
10. Functional Annotation of the Mammalian Genome 5 (FANTOM5) CAGE (https://fantom.gsc.riken.jp/5/datafiles/reprocessed/hg38_latest/basic/)
Procedure
Note: The workflow of the protocol is illustrated in Figure 1, with each substep indicated.
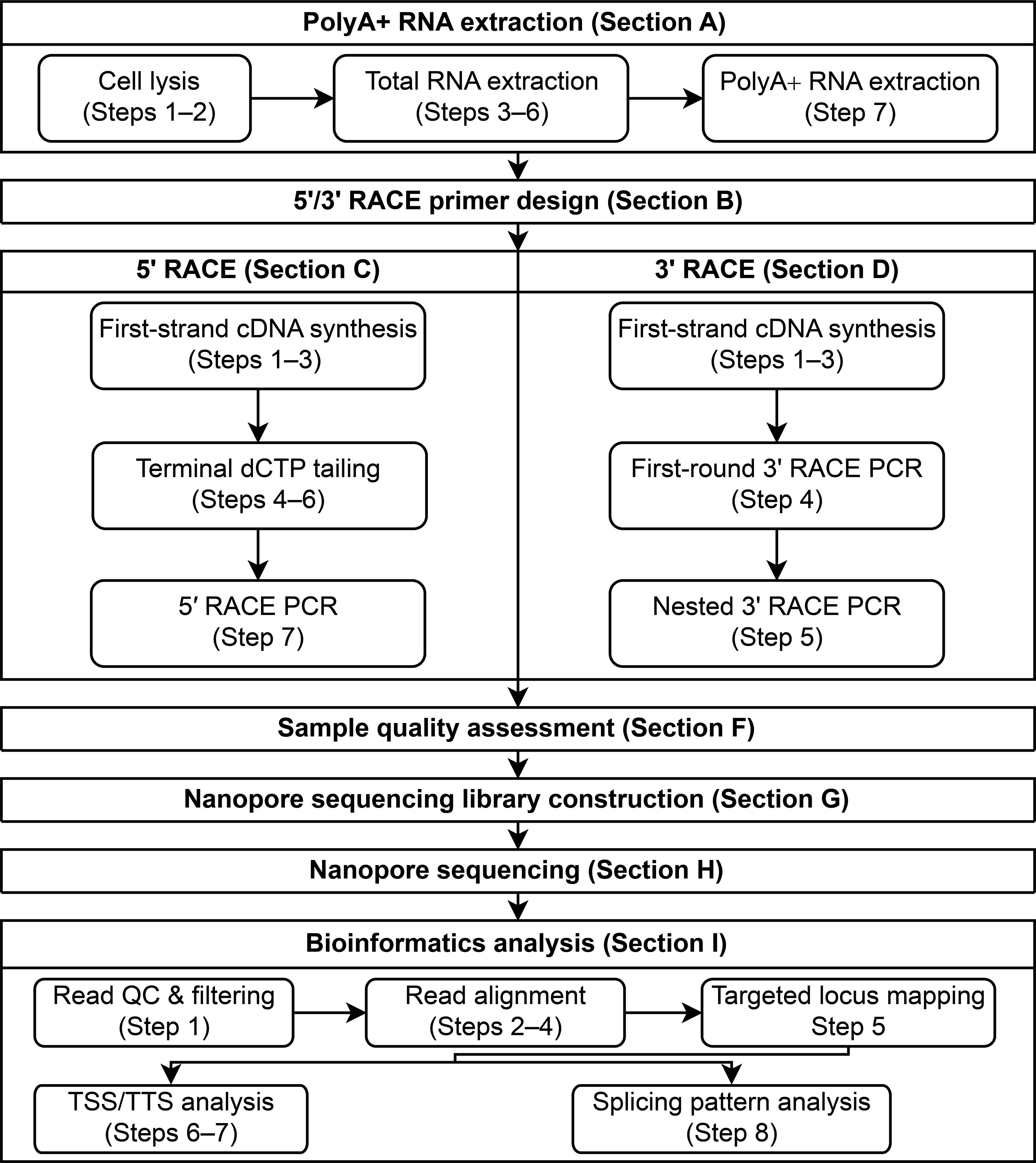
Figure 1. Workflow of the RACE-Nano-Seq protocol
A. PolyA+ RNA extraction
1. Collect 1 × 106 cells in a 2 mL EP tube and centrifuge at 200× g for 5 min at 4 °C. Discard the supernatant.
2. Add 1 mL TRNzol universal reagent and vortex for 20 s to ensure complete cell lysis.
3. Add 200 μL of chloroform, vortex for 30 s, and incubate at room temperature for 5 min.
4. Centrifuge at 13,500× g for 15 min at 4 °C to separate the mixture into three layers: the lower pink organic phase, the middle interphase, and the upper colorless aqueous phase.
5. Transfer 500 μL of the aqueous phase to a new tube and add an equal volume of 70% ethanol. Mix by vortexing.
Note: Avoid centrifugation after vortexing to prevent sedimentation. Centrifuge only after transferring the mixture to the HiBind® RNA mini column (see steps A6a–c).
6. Perform centrifugation and washing steps at room temperature according to the E.Z.N.A.® Total RNA Kit manual.
a. Insert a HiBind® RNA mini column into a 2 mL collection tube.
b. Load up to 700 μL of the mixture from step A5 (including any precipitate that may have formed) to the HiBind® RNA mini column.
c. Centrifuge at 10,000× g for 1 min.
d. Discard the filtrate and reuse the collection tube.
e. Repeat steps A6b–d until the entire sample has been transferred to the column.
f. Add 500 μL of RNA wash buffer I to the HiBind® RNA mini column.
g. Centrifuge at 10,000× g for 30 s.
h. Discard the filtrate and reuse the collection tube.
i. Add 500 μL of RNA wash buffer II to the HiBind® RNA mini column.
Note: RNA wash buffer II must be diluted with ethanol before use. The ethanol volume required is noted on the bottle.
j. Centrifuge at 10,000× g for 1 min.
k. Discard the filtrate and reuse the collection tube.
l. Repeat steps A6i–k for a second RNA wash buffer II wash step.
m. Centrifuge at the maximum speed for 2 min to completely dry the HiBind® RNA mini column.
Note: It is important to dry the HiBind® RNA mini column matrix before elution. Residual ethanol may interfere with downstream cDNA synthesis.
n. Transfer the HiBind® RNA mini column to a clean 1.5 mL EP tube.
o. Add 55 μL of DNase/RNase-free distilled water.
Note: For 1 × 10 cells, 55 μL of DNase/RNase-free distilled water is recommended. The volume can be adjusted between 30 and 70 μL depending on the cell number. Ensure that the water is added directly onto the HiBind® RNA mini column matrix.
p. Centrifuge at top speed for 2 min and measure the RNA concentration using a spectrophotometer. Store RNA at -80 °C.
Note: Typical RNA yields are in the range of 40–100 µg.
7. Take 12.5 μg of total RNA from step A6 to extract the polyA+ RNA. Perform the washing and eluting step according to the Library Preparation VAHTSTM mRNA capture beads manual.
a. Remove the mRNA capture beads from 2–8 °C storage and let them equilibrate to room temperature.
b. Dilute 12.5 μg of total RNA to 50 μL using DNase/RNase-free distilled water in a PCR tube. Keep the sample on ice until use.
c. Mix the mRNA capture beads thoroughly by inverting them at least 10 times. Pipette 50 μL of the beads into the total RNA sample and mix thoroughly by pipetting up and down six times.
d. Incubate the sample in a thermal cycler under the following conditions: 65 °C for 5 min, 25 °C for 5 min, followed by a 4 °C hold. This step allows polyA+ RNA to bind to the beads.
Note: It is recommended to proceed to the next step promptly once the temperature reaches 4 °C.
e. Place the sample on a magnetic rack for 5 min to separate polyA+ RNA from total RNA. Carefully remove the supernatant.
f. Remove the sample from the magnetic rack. Resuspend the beads thoroughly in 200 μL of bead wash buffer by pipetting up and down six times. Incubate the tube on a magnetic stand for 5 min, then carefully remove the supernatant.
g. Remove the sample from the magnetic rack, add 10.5 μL of DNase/RNase-free distilled water, and mix thoroughly by pipetting six times. Incubate at 80 °C for 2 min, then immediately place the sample on the magnetic rack for 5 min. Once the solution clarifies, carefully transfer 8–10 μL of the supernatant to a new nuclease-free PCR tube.
Note: Aspirate as much supernatant as possible without disturbing the beads.
h. Measure the concentration using a spectrophotometer. The expected yield of polyA+ RNA is approximately 1%–2% of the starting total RNA.
Pause point: The sample can be kept on ice for a short time (less than 1 h) or put at -80 °C for long-term storage. We recommend using it immediately for the next steps of the procedure below.
B. 5'/3' RACE primer design
Note: For 5' RACE, first-strand cDNA synthesis employs the 5' RACE gene-specific primer (5RACE_GS), followed by nested-PCR amplification using the 5' RACE gene-specific nested primer (5RACE_GSN). For 3' RACE, first-strand cDNA is synthesized using an oligo(dT)-based primer that anneals to the polyA+ tail, followed by first-round PCR amplification using the 3' RACE gene-specific primer (3RACE_GS) and a subsequent nested PCR using the 3' RACE gene-specific nested primer (3RACE_GS).
1. Primer selection guidelines:
Note: Guidelines are prioritized in the order below, with guidelines B1d and B1e being critical for primer specificity.
a. Length: 18–25 nucleotides (nt), optimally 22–24 nt.
b. GC content: 40%–60%.
c. No more than 3 G/C bases in the last 5 nt of the 3' end; G or C at the 3' terminus preferred.
d. Average uniqueness score = 1 (per USCS ENCODE/OpenChrom 20 bp window track).
e. No overlap with repetitive elements.
f. No homopolymer stretches >3 bases.
g. No dinucleotide repeats >2 consecutive pairs.
h. Balanced A/T/C/G distribution preferred.
2. Guidelines for selecting anchor exons within a gene locus and primer targeting sequences within anchor exons:
Note: While anchor sequences may include annotated exons, predicted exons, or other sequences within a target locus, these guidelines specifically address targeting of known or predicted exons.
a. Anchor exon selection within a gene locus
i. Avoid selecting the first or last exon of a gene as the anchor, as this may yield short RACE products unsuitable for Nanopore sequencing.
ii. For genes with ≤10 exons, select at least one anchor exon. For genes with >10 exons, increase the number of anchor exons proportionally.
b. Primer targeting site selection within an anchor exon
i. Primers should preferentially target the central region of the exon sequence.
ii. If no suitable central sequence is available, shift toward the exon boundaries while maintaining a minimum 5-base distance from either boundary.
C. 5' RACE
1. Prepare a 10 μL reaction containing 85 ng of polyA+ RNA, 1 μL of 10 mM dNTP mix, and 1 μL of 10 μM 5RACE_GS primer described in our previous publication [26]. Adjust the volume to 10 μL with DNase/RNase-free distilled water. Incubate at 65 °C for 2 min, then immediately place on ice.
Notes:
1. Total RNA, in addition to polyA+ RNA, can serve as the template for RACE.
2. This step is scalable and can utilize either a single gene-specific primer or a primer pool. When using a primer pool, add 1 μL of primer mix with each primer at 10 μM concentration.
2. Add 4 μL of 5× PrimeScript II buffer, 0.5 μL of 40 U/μL RNase inhibitor, and 1 μL of 200 U/μL PrimeScript II reverse transcriptase. Adjust the volume to 20 μL with DNase/RNase-free distilled water. Incubate the mixture in a thermal cycler (with heated lid on) at 50 °C for 60 min, followed by enzyme inactivation at 70 °C for 15 min.
3. Purify cDNA using VAHTS DNA clean beads at 1.5× the sample volume (e.g., add 30 μL of beads to 20 μL of reaction mixture from step C2). Elute the purified cDNA in 15 μL of DNase/RNase-free distilled water. For detailed purification procedures, refer to section E.
4. Add 2 μL of 10× terminal transferase buffer and 2 μL of 2.5 mM CoCl2 to the first-strand cDNA product. Incubate at 90 °C for 2 min, then immediately place on ice.
5. Add 5 U of terminal transferase and 2 μL of 10 mM dCTP. Incubate at 37 °C for 30 min, followed by enzyme inactivation at 70 °C for 10 min.
6. Purify using VAHTS DNA clean beads at 1.5× the sample volume and elute in 15 μL of DNase/RNase-free distilled water (See section E for detailed procedures).
7. Perform 5' RACE PCR in a 25 μL reaction (Table 1) using 15 μL of polyC-tailed cDNA as template. Use the 5RACE_GSN primer in combination with N10G10HN (5'-AGTTGCGGATGGGGGGGGGGHN-3'). PCR conditions: 30 cycles of 98 °C for 10 s, 55 °C for 30 s, 68 °C for 5 min; then, 68 °C for 7 min.
Note: 5RACE_GSN nested primer sequences were described in our previous publication [26]. See Note 2 in step C1.
Pause point: Post-amplification, samples may be maintained at 4 °C in the thermal cycler or transferred to refrigeration for storage if not processed immediately.
Table 1. 5' RACE PCR reaction
| Reagent | Volume | Final concentration |
|---|---|---|
| Products from last step | 15 μL | n/a |
| 5× PrimeSTAR GXL buffer | 5 μL | 1× |
| PrimeSTAR GXL DNA polymerase (1.25 U/μL) | 1 μL | 0.05 U/μL |
| N10G10HN (10 μM) | 1 μL | 0.4 μM |
| 5RACE_GSN (10 μM) | 1 μL | 0.4 μM |
| dNTP mix (2.5 mM each) | 2 μL | 0.2 mM |
| Total | 25 μL | n/a |
D. 3' RACE
1. Use 350 ng of polyA+ RNA as template in a 10 μL reaction containing 1 μL of 2.5 mM dNTP mix and 1 μL of 10 μM N20T12(VN) primer (5'-GCAATCATCGAGTTGCGGATTTTTTTTTTTTTVN-3').
Note: Total RNA, in addition to polyA+ RNA, can serve as the template for RACE.
2. Add 4 μL of 5× PrimeScript II buffer, 0.5 μL of 40 U/μL RNase inhibitor, and 1 μL of 200 U/μL PrimeScript II reverse transcriptase. Perform the reaction in a thermal cycler using the following program: ramp temperature from 37 °C to 50 °C at 2 °C/min, incubate at 50 °C for 50 min, then inactivate the enzyme at 70 °C for 15 min.
3. Purify cDNA using VAHTS DNA clean beads at 1.5× the sample volume and elute the purified cDNA in 15 μL of DNase/RNase-free distilled water (see section E for detailed procedures).
4. Perform PCR using first-strand cDNA as template in a 25 μL reaction (Table 2) with the 3RACE_GS primer described in our previous publication [26] and N20T12(VN) (5'-GCAATCATCGAG TTGCGGATTTTTTTTTTTTTVN-3'). PCR conditions: 15 cycles of 98 °C for 10 s, 55 °C for 30 s, and 68 °C for 5 min; then, 68 °C for 10 min.
Note: See Note 2 in step C1.
Table 2. 3' RACE PCR reaction
| Reagent | Volume | Final concentration |
|---|---|---|
| Products from last step | 2.8 μL | n/a |
| 5× PrimeSTAR GXL buffer | 5 μL | 1× |
| PrimeSTAR GXL DNA polymerase (1.25 U/μL) | 1 μL | 0.05 U/μL |
| N20T12(VN) (10 μM) | 1 μL | 0.4 μM |
| 3RACE_GS (10 μM) | 1 μL | 0.4 μM |
| dNTP mix (2.5 mM each) | 2 μL | 0.2 mM |
| UltraPureTM DNase/RNase-free distilled water | 12.2 μL | n/a |
| Total | 25 μL | n/a |
5. Use 1 μL of first-round 3' RACE PCR products as template in a 25 μL (Table 3) reaction with the nested primers 3RACE_GSN and N20T12(VN). Maintain the same thermal cycling conditions as the first-round PCR but extend to 30 amplification cycles.
Note: See Note 2 in step C1.
Table 3. 3' RACE nested PCR reaction
| Reagent | Volume | Final concentration |
|---|---|---|
| Products from last step | 1 μL | n/a |
| 5× PrimeSTAR GXL buffer | 5 μL | 1× |
| PrimeSTAR GXL DNA polymerase (1.25 U/μL) | 1.25 μL | 0.0625 U/μL |
| N20T12(VN) (10 μM) | 1 μL | 0.4 μM |
| 3RACE_GSN (10 μM) | 1 μL | 0.4 μM |
| dNTP mix (2.5 mM each) | 2 μL | 0.2 mM |
| UltraPureTM DNase/RNase-free distilled water | 13.75 μL | n/a |
| Total | 25 μL | n/a |
Pause point: Post-amplification, samples may be maintained at 4 °C in the thermal cycler or transferred to refrigeration for storage if not processed immediately.
E. Purification using VAHTS DNA clean beads
1. Remove the VAHTS DNA clean beads from 2–8 °C storage 30 min in advance and allow them to equilibrate to room temperature.
2. Vortex or invert the tube to thoroughly mix the bead suspension. Transfer the beads into the reaction. Mix thoroughly by gently pipetting up and down 10 times.
Note: The bead-to-sample volume ratio is specified in each purification step of the protocol.
3. Incubate the mixture at room temperature for 10 min to allow DNA to bind to the beads.
4. Place the sample on a magnetic rack and wait approximately 5 min until the solution clears. Carefully remove the supernatant.
5. Keeping the sample on the magnetic rack, add 200 μL of freshly prepared 80% ethanol to wash the beads. Incubate at room temperature for 30 s, then carefully remove the supernatant.
6. Repeat step E5 for a total of two washes.
7. Keep the sample on the magnetic rack with the lid open and allow the beads to air-dry at room temperature for 5–10 min.
8. Remove the sample from the magnetic rack and add an appropriate volume of DNase/RNase-free distilled water. Vortex or gently pipette up and down to mix thoroughly, then incubate at room temperature for 2 min.
9. Place the sample back on the magnetic rack and let it sit for 5 min until the solution clears. Carefully transfer the supernatant to a new 1.5 mL EP tube for further use.
F. Sample quality assessment
1. Mix equal volumes of 5' RACE and 3' RACE products. Purify using VAHTS DNA clean beads at 2× the sample volume and elute in 55 μL of DNase/RNase-free distilled water (see section E for detailed procedures).
2. Measure the concentration using a spectrophotometer.
3. Verify the RACE product lengths with 1% agarose gel electrophoresis. The quality standards for RACE products used in Nanopore sequencing library preparation are shown in Table 4. An expected gel result is shown in Figure 2.
Table 4. Quality standards for Nanopore sequencing library preparation
| Measurement category | Quality criteria | Measured values |
|---|---|---|
| Sample characteristics | Clear and transparent, with no insoluble material or viscosity | Pass |
| Spectrophotometry | A260/A280 = 1.8–2.0 A260/A230 ≥ 1.5 Total amount ≥ 10 μg | A250/A280 = 1.84 A260/A230 = 1.91 Total amount = 19.66 μg |
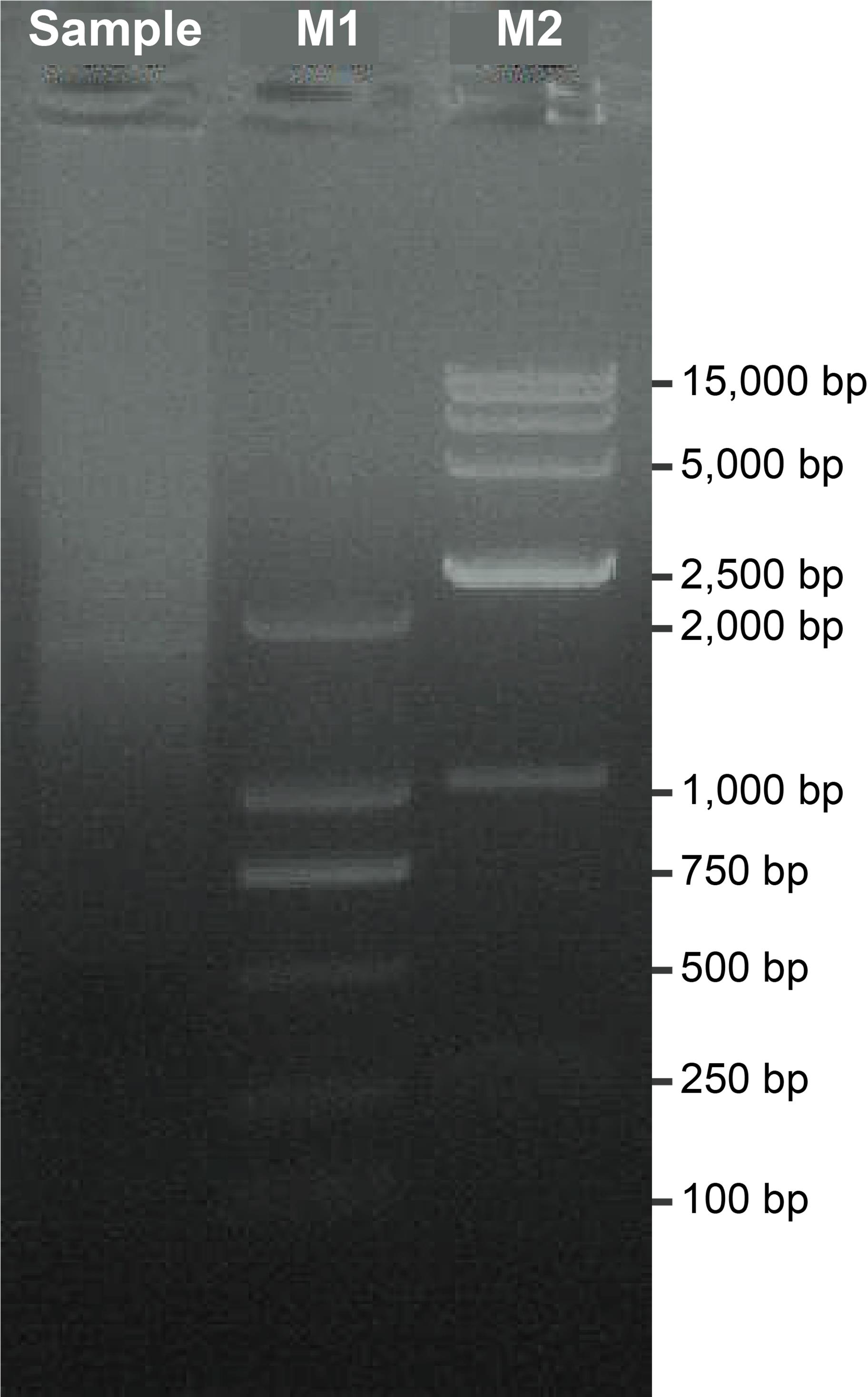
Figure 2. Gel electrophoresis of 5'/3' RACE products. The sample represents the purified 5'/3' RACE mixture. M1 indicates a 2 kb DNA marker (2,000, 1,000, 750, 500, 250, and 100 bp). M2 indicates a 15 kb DNA marker (15,000, 10,000, 7,500, 5,000, 2,500, 1,000, and 250 bp).
G. Nanopore sequencing library construction
Note: Procedures follow the instructions from the Ligation Sequencing kit.
1. Adjust 2 μg of the RACE product to 48 μL, then add the reagents to the PCR tube in the order specified in Table 5. After each addition, pipette up and down around 15 times to ensure thorough mixing. Gently pipette to avoid bubbles and briefly spin down before proceeding.
Table 5. DNA repair and end-prep reaction
| Reagent | Volume |
|---|---|
| RACE product | 48 μL |
| NEBNext FFPE DNA repair buffer | 3.5 μL |
| NEBNext FFPE DNA repair mix | 2 μL |
| Ultra II End-prep reaction buffer | 3.5 μL |
| Ultra II End-prep enzyme mix | 3 μL |
| Total | 60 μL |
2. Incubate at 20 °C for 5 min, then at 65 °C for 5 min.
3. Resuspend the AMPure XP beads by vortexing. Incubate AMPure XP beads at room temperature for at least 30 min before use.
4. Transfer the RACE product to a clean 1.5 mL EP tube.
5. Add 60 μL of resuspended AMPure XP beads to the end-prep reaction and mix by flicking the tube.
6. Incubate on the rotator mixer for 5 min at room temperature.
7. Prepare 500 μL of fresh 70% ethanol in DNase/RNase-free distilled water.
8. Spin down the sample and pellet on a magnetic rack until the supernatant is clear and colorless. Keep the tube on the magnetic rack and pipette off the supernatant.
9. Keep the tube on the magnetic rack and wash the beads with 200 μL of freshly prepared 70% ethanol without disturbing the pellet. Remove the ethanol using a pipette and discard.
10. Repeat step G9 for a total of two washes.
11. Spin down and place the tube back on the magnetic rack. Pipette off any residual ethanol. Allow to dry for ~30 s, but do not dry the pellet to the point of cracking.
12. Remove the tube from the magnetic rack and resuspend the pellet in 61 μL of DNase/RNase-free distilled water. Incubate for 2 min at room temperature.
13. Place the tube on a magnetic rack for at least 1 min until the eluate is clear and colorless.
14. Transfer 60 μL of eluate into a clean 1.5 mL EP tube. Add 25 μL of ligation buffer, 10 μL of NEBNext Quick T4 DNA ligase, and 5 μL of ligation adapters to the 60 μL eluate.
Note: Thaw ligation buffer at room temperature, spin down, and mix by pipetting. Due to viscosity, vortexing this buffer is ineffective. Place on ice immediately after thawing and mixing. Spin down the ligation adapter and Quick T4 ligase, and place on ice. Pipette mix 10–20 times between each addition. Ensure thorough mixing by gently pipetting to avoid creating bubbles and spin down briefly.
15. Incubate the reaction for 20 min at 25 °C.
16. Resuspend the AMPure XP beads by vortexing.
17. Add 40 μL of resuspended AMPure XP beads to the reaction and mix by flicking the tube.
18. Incubate on the rotator mixer for 5 min at room temperature.
19. Spin down the sample and pellet on a magnetic rack. Keep the tube on the magnetic rack and remove the supernatant.
20. Wash the beads by adding 250 μL of short fragment buffer. Flick the beads to resuspend, spin down, then return the tube to the magnetic rack and allow the beads to pellet. Remove the supernatant using a pipette and discard.
Note: Before use, thaw short fragment buffer at room temperature, vortex to mix, spin down, and place on ice before use.
21. Repeat step G20 for a total of two washes.
22. Spin down and place the tube back on the magnetic rack. Pipette off any residual supernatant. Allow to dry for ~30 s, but do not dry the pellet to the point of cracking.
23. Remove the tube from the magnetic rack and resuspend the pellet in 25 μL of elution buffer. Spin down and incubate for 10 min at 37 °C.
Note: Before use, thaw the elution buffer at room temperature, vortex to mix, spin down, and place on ice before use.
24. Pellet the beads on a magnetic rack for at least 1 min until the eluate is clear and colorless.
25. Move 25 μL of eluate containing the DNA library into a clean 1.5 mL EP tube.
26. Quantify 1 μL of eluted sample using a Qubit fluorometer.
Note: For optimal Nanopore sequencing, the eluate should contain 20–40 fmol of DNA in a total volume of 24 μL.
H. Nanopore sequencing
1. Add 5 μL of 50 mg/mL BSA and 30 μL of flow cell tether to flow cell flush, then vortex to prepare the priming mix.
2. Prime the flow cell twice with 200 μL of the priming mix.
3. Load 150 μL of prepared sequencing mix (Table 6).
Table 6. Sequencing mix
| Reagent | Volume |
|---|---|
| Sequencing buffer | 75 μL |
| Library beads | 51 μL |
| RACE Nanopore library | 24 μL |
| Total | 150 μL |
4. Initiate sequencing on the Oxford Nanopore PromethION platform, running for 48–72 h under default parameters.
5. Perform real-time base-calling using MinKNOW (v20.10.6) integrated with Guppy (v4.2.3) in high-accuracy mode to generate base-called FASTQ files.
6. Monitor sequencing quality and flow cell performance throughout the run to ensure optimal yield and pore activity.
I. Bioinformatics analysis
Note: A personal computer with a Linux- or Unix-based operating system is required to run this analysis pipeline. All software used in the pipeline are listed in the Software and datasets section. Installation instructions and functional descriptions for each tool are available in their respective official documentation.
1. Filter raw Nanopore sequencing data to obtain clean data with an average quality score ≥7 using NanoFilt [30]:
NanoFilt -q 10 $Raw.Data.fq > $Data.fq
Note: $Raw.Data.fq indicates the raw data from Nanopore sequencing, and $Data.fq indicates clean data after filtering.
2. Run the following command to generate an index from the reference genome using Minimap2 [31]:
minimap2 -k14 -d hg38.14k.mmi hg38.fa
Note: hg38.14k.mmi indicates the minimap2 index file created for the reference genome hg38.fa. The -k14 option sets the k-mer size to 14.
3. Align reads to the human GRCh38/hg38 reference genome using Minimap2 in spliced alignment mode with the command:
minimap2 -ax splice -ub -G400000 --end-seed-pen 30 hg38.14k.mmi $Data.fq -o $Data.sam
Note: hg38.14k.mmi is the reference genome index generated in step I2. $Data.fq indicates input fastq data containing clean reads generated in step I1. $Data.sam indicates alignment results in SAM format.
4. Filter low-quality alignments using Samtools [32] and generate BED12 file using BEDtools [33] suite's bamtobed function with the command:
samtools view -bh -F0×900 -q 60 $Data.sam | samtools sort -n - | bedtools bamtobed -bed12 -i stdin | bedtools sort -i stdin > $Data.filter.bed
Note: $Data.sam indicates the alignment file from step I3. $Data.filter.bed indicates filtered data in BED12 format.
5. Map the alignments to anchors. The 5'/3' RACE read count derived from two anchor exons of a gene locus is shown in Table 7.
Note: This step is essential for identifying RACE products that successfully mapped to primer-targeted regions.
a. Determine the overlap between alignments and the RACE nested primer using the BEDTools suite's intersect function:
bedtools intersect -a $ Data.filter.bed -b $primer.bed -sorted -wo -split > $bed.sum.txt
Note: This command identifies and reports all overlaps between $Data.filter.bed (from step I4) and $primer.bed (RACE nested primers).
b. Select alignments specifically mapped to the anchors for downstream analysis.
i. Positive-strand anchor filtering.
5' RACE alignments:
awk '$25>0&&$25>=($15-$14)' $bed.sum.txt | awk '$3<=$15+5&&$3>=$15-5' | cut -f 1-12 > $target.alignment.bed
3' RACE alignments:
awk '$25>0&&$25>=($15-$14)' $bed.sum.txt | awk '$2<=$14+5&&$2>=$14-5' | cut -f 1-12 > $target.alignment.bed
ii. Negative-strand anchor filtering.
5' RACE alignments:
awk '$25>0&&$25>=($15-$14)' $bed.sum.txt | awk '$2<=$15+5&&$2>=$15-5' | cut -f 1-12 > $target.alignment.bed
3' RACE alignments:
awk '$25>0&&$25>=($15-$14)' $bed.sum.txt | awk '$3<=$14+5&&$3>=$14-5' | cut -f 1-12 > $target.alignment.bed
Notes:
1. $bed.sum.txt indicates the overlapping analysis results from the previous step. $target.alignment.bed indicates the output file containing alignments mapped to the anchors.
2. It is crucial to acknowledge that not all reads originate from the target locus; the proportion of on-target reads fluctuates, often influenced by target expression levels. This step is essential to ensure that only reads specifically mapped to primer-targeted regions are utilized for downstream analysis.
Table 7. Read count of 5'/3' RACE products derived from two anchor exons of a gene locus
| Anchor exon | Read count | ||
| 5′ RACE | 3′ RACE | Total | |
| Anchor 1 | 5,651 | 11,677 | 17,328 |
| Anchor 2 | 19073 | 7,155 | 26,228 |
| Total | 24,724 | 18,832 | 43,556 |
Note: Adapted from our previous publication [26].
6. Determine the TSS and TTS positions. First, adjust all selected alignments from step I4 to match the anchor’s strand orientation. For positive-strand alignments, the TSS corresponds to the first base of the 5' RACE product, while the TTS is defined by the last base of the 3' RACE product. For negative-strand alignments, the TSS is defined as the last base of the 5' RACE product, and the TTS corresponds to the first base of the 3' RACE product.
Note: As this protocol employs a non-strand-specific sequencing library construction method, all strand alignments must be inferred from the anchor’s orientation.
7. Calculate the relative abundance of TSSs and TTSs using the following formula:
where C represents the count of each TSS or TTS from the 5' or 3' RACE-Nano-Seq assays, and T denotes the total number of reads in the corresponding assay.
8. Analyze splicing patterns from 5' RACE and 3' RACE data for each anchor exon using the intron function of bedparse [34] to extract BED12 intron coordinates, with unique intron combinations classified as distinct splicing patterns. This approach revealed extensive transcriptome diversity in a novel gene locus represented by at least 48 transcript isoforms, as summarized in Table 8.
Note: This method has consistently identified numerous novel exons across all loci tested by us, including both known and previously unannotated genomic regions.
Table 8. Count of sequences with unique splicing patterns derived from the two anchor exons
| Anchor exon | RACE | Count | |
| All | ≥5 Reads | ||
| Anchor 1 | 5′ RACE | 19 | 4 |
| 3′ RACE | 244 | 48 | |
| Anchor 2 | 5′ RACE | 62 | 17 |
| 3′ RACE | 94 | 22 | |
9. Representative results in a novel gene locus are shown in Figure 3.
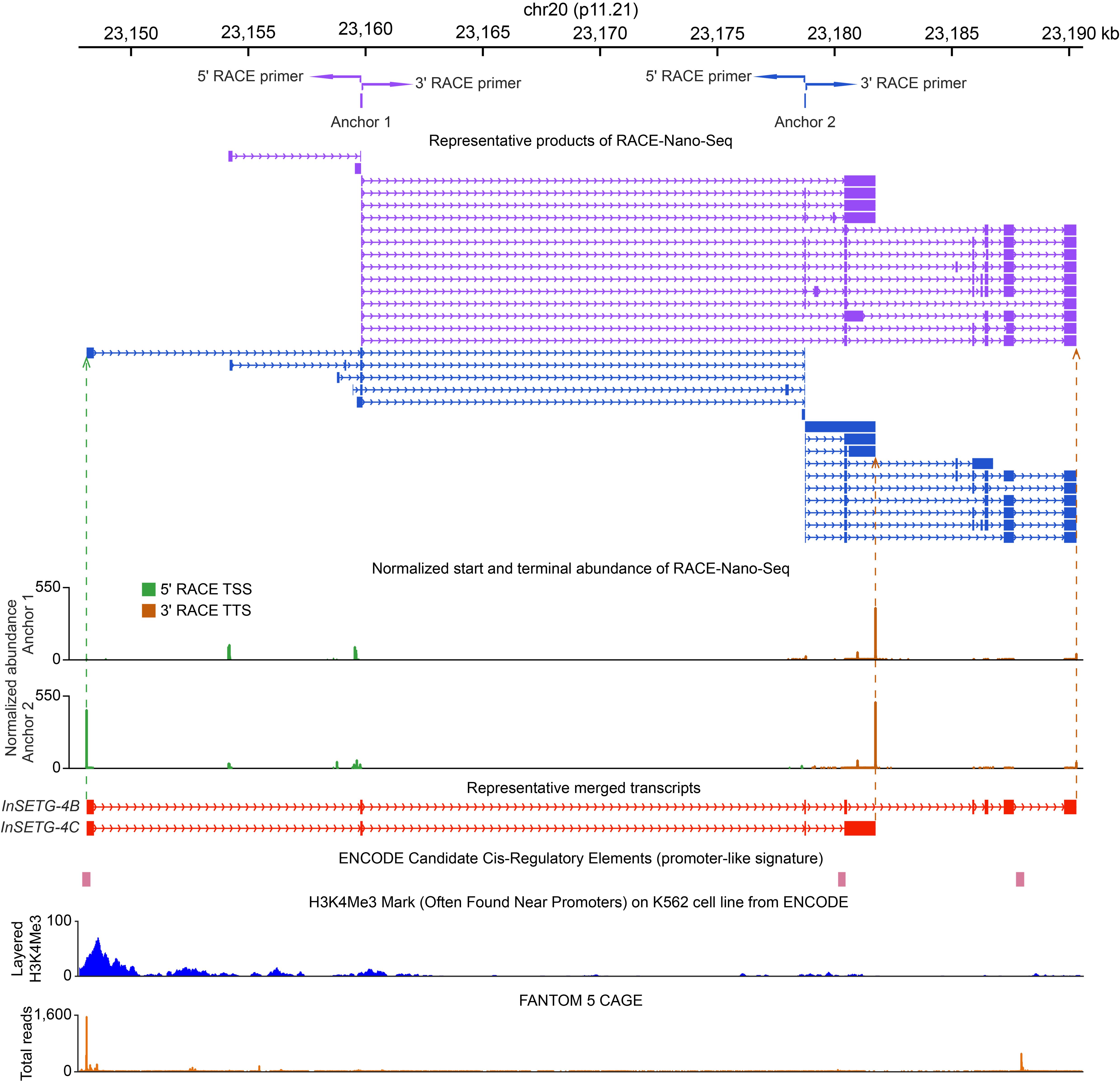
Figure 3. Representative results of RACE-Nano-Seq. A novel human gene locus located over 50 kb away from any annotated protein-coding genes was identified in the K562 cell line. The two anchors shown in the figure were derived from our initial RACE-Nano-Seq analysis [25]. RACE primers (paired arrows corresponding to different anchor exons), representative transcripts (colored to match the anchors), transcription start sites (TSSs), transcription termination sites (TTSs), and their normalized abundances are presented. Representative merged full-length transcripts (shown in red) represent the most abundant transcript (InSETG-4C) and the transcript with the largest number of exons (InSETG-4B). The authentic nature of the TSSs identified by RACE is supported by the overlap with the ENCODE candidate cis-regulatory elements [35–37], the ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) data for the H3K4me3 promoter-associated chromatin mark [36,38,39], and the FANTOM 5 CAGE track, which represents the total count of CAGE reads from all CAGE samples. Adapted from our previous publication [26].
Validation of protocol
RACE-Nano-Seq combines 5' RACE or 3' RACE with the long-read Nanopore sequencing to selectively enrich specific transcripts, including low-abundance RNAs emanating from the targeted loci, and to analyze the sequences of these RNA species. These low-abundance transcripts are often missed by conventional techniques. This protocol has been previously validated in the following research articles:
• Xu et al. [25]. Evidence for widespread existence of functional novel and non-canonical human transcripts. BMC Biology (Figure 4G, Figure 5, and Figure 6).
• Tang et al. [26]. A novel human protein-coding locus identified using a targeted RNA enrichment technique. BMC Biology (Figure 2A and Figure 3A, B).
Xu et al. (2023) implemented the multiplex primer strategy, while Tang et al. (2024) validated the single-primer approach, demonstrating the protocol’s adaptability to different experimental needs.
General notes and troubleshooting
General notes
1. RACE-Nano-Seq enables multiplexed amplification by allowing (1) simultaneous use of multiple primers per reaction (up to 10 primers tested), and (2) pooling of products from multiple RACE reactions (up to 14 reactions validated) for Nanopore sequencing. While the protocol is scalable and can be adapted to other primer/reaction quantities, users should note that read depth per primer decreases proportionally with increasing multiplexing scale.
2. The current implementation of the RACE-Nano-Seq protocol uses a single RNA sample as input. However, the approach can conceivably be adapted for multiplexed analysis of multiple samples by incorporating index sequences into the primers during the library preparation.
3. RACE-Nano-Seq is particularly suited for in-depth isoform characterization at specific gene loci, using a known or in silico predicted exon as the anchor. Notably, this method allows for the identification of transcript boundaries, alternative splicing events, and novel exons in both established and previously uncharacterized gene loci.
Troubleshooting
Problem 1: Failure to obtain Nanopore sequencing results when the selected anchor region is too close to the transcript boundary.
Possible cause: The choice of anchor region is critical for successful transcript discovery. If the selected region is too close to the 5' or 3' end of the transcript, the adjacent terminal sequences may not be captured effectively, leading to sequencing failure.
Solution: To maximize the likelihood of obtaining full-length isoforms, it is advisable to select an anchor region located closer to the central region of the transcript. This increases the chances of capturing both upstream and downstream sequences, facilitating the discovery of novel isoforms.
Problem 2: Difficulty in assembling full-length transcripts when using a single anchor region.
Possible cause: Using only one anchor region may limit the ability to reconstruct complete transcript structures, especially for highly complex isoforms.
Solution: To enhance transcript recovery and improve isoform detection, select at least 2–3 anchor regions instead of a single one to achieve a broader coverage of transcript variants. This approach enables deeper exploration of gene locus complexity and facilitates more accurate transcript assembly.
Problem 3: No reads map to the target anchor region(s).
Possible causes: Nonspecific amplification during RACE PCR or extremely low abundance of the target transcript(s).
Solution: Optimize primer specificity by strictly following the design criteria in step B3, with particular attention to the criteria d and e, as these are critical for reducing off-target amplification. If the issue persists due to low target transcript abundance, perform an additional nested PCR round using 1μL of the initial PCR product as template, while being aware that excessive amplification cycles may also increase nonspecific products. To confirm that the protocol is functioning properly and to distinguish between technical failure and true transcript absence, it is recommended to include a positive control sample known to express the target locus/loci.
Acknowledgments
1) Specific contributions of each author: Conceptualization, P.K. and D.X.; Writing—Original Draft, T.L.; Writing—Review & Editing, D.X. and P.K.; Funding acquisition, P.K. and D.X.; Supervision, P.K. and D.X.
2) Funding sources that supported the work: National Natural Science Foundation of China (32170619) and Research Fund for International Senior Scientists from the National Natural Science Foundation of China (32150710525) to P.K.; National Natural Science Foundation of China (32000441), Natural Science Foundation of Fujian Province, China (2023J01130), Fundamental Research Funds for the Central Universities of Huaqiao University (ZQN-924), and Scientific Research Funds of Huaqiao University (18BS205) to D.X.
This protocol was derived from the original research papers [25,26].
Competing interests
The authors declare no competing interests.
References
- Mazin, P. V., Khaitovich, P., Cardoso-Moreira, M. and Kaessmann, H. (2021). Alternative splicing during mammalian organ development. Nat Genet. 53(6): 925–934. https://doi.org/10.1038/s41588-021-00851-w
- Karlebach, G., Steinhaus, R., Danis, D., Devoucoux, M., Anczuków, O., Sheynkman, G., Seelow, D. and Robinson, P. N. (2024). Alternative splicing is coupled to gene expression in a subset of variably expressed genes. NPJ Genom Med. 9(1): 1–10. https://doi.org/10.1038/s41525-024-00432-w
- Carninci, P., Kasukawa, T., Katayama, S., Gough, J., Frith, M. C., Maeda, N., Oyama, R., Ravasi, T., Lenhard, B., Wells, C., et al. (2005). The Transcriptional Landscape of the Mammalian Genome. Science. 309(5740): 1559–1563. https://doi.org/10.1126/science.1112014
- Alfonso-Gonzalez, C. and Hilgers, V. (2024). (alternative) transcription start sites as regulators of RNA processing. Trends Cell Biol. 34(12): 1018–1028. https://doi.org/10.1016/j.tcb.2024.02.010
- Kapranov, P., Drenkow, J., Cheng, J., Long, J., Helt, G., Dike, S. and Gingeras, T. R. (2005). Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 15(7): 987–997. https://doi.org/10.1101/gr.3455305
- Denoeud, F., Kapranov, P., Ucla, C., Frankish, A., Castelo, R., Drenkow, J., Lagarde, J., Alioto, T., Manzano, C., Chrast, J., et al. (2007). Prominent use of distal 5′ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res. 17(6): 746–759. https://doi.org/10.1101/gr.5660607
- Mercer, T. R., Gerhardt, D. J., Dinger, M. E., Crawford, J., Trapnell, C., Jeddeloh, J. A., Mattick, J. S. and Rinn, J. L. (2012). Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol. 30(1): 99–104. https://doi.org/10.1038/nbt.2024
- Wang, Y., Zou, Q., Li, F., Zhao, W., Xu, H., Zhang, W., Deng, H. and Yang, X. (2021). Identification of the cross-strand chimeric RNAs generated by fusions of bi-directional transcripts. Nat Commun. 12(1): Article 1. https://doi.org/10.1038/s41467-021-24910-2
- Djebali, S., Lagarde, J., Kapranov, P., Lacroix, V., Borel, C., Mudge, J. M., Howald, C., Foissac, S., Ucla, C., Chrast, J., et al. (2012). Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One. 7(1): e28213. https://doi.org/10.1371/journal.pone.0028213
- Kapranov, P., Cawley, S. E., Drenkow, J., Bekiranov, S., Strausberg, R. L., Fodor, S. P. A. and Gingeras, T. R. (2002). Large-Scale Transcriptional Activity in Chromosomes 21 and 22. Science. 296(5569): 916–919. https://doi.org/10.1126/science.1068597
- Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., Stadler, P. F., Hertel, J., Hackermüller, J., Hofacker, I. L., et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 316(5830): 1484–1488. https://doi.org/10.1126/science.1138341
- Han, X., Luo, S., Peng, G., Lu, J. Y., Cui, G., Liu, L., Yan, P., Yin, Y., Liu, W., Wang, R., et al. (2018). Mouse knockout models reveal largely dispensable but context-dependent functions of lncRNAs during development. J Mol Cell Biol. 10(2): 175–178. https://doi.org/10.1093/jmcb/mjy003
- Chen, L., Zhang, Y.-H., Pan, X., Liu, M., Wang, S., Huang, T. and Cai, Y.-D. (2018). Tissue Expression Difference between mRNAs and lncRNAs. Int J Mol Sci. 19(11): 3416. https://doi.org/10.3390/ijms19113416
- Plasek, L. M. and Valadkhan, S. (2021). lncRNAs in T lymphocytes: RNA regulation at the heart of the immune response. Am J PhysiolCell Physiol. 320(3): C415–C427. https://doi.org/10.1152/ajpcell.00069.2020
- Ducoli, L., Agrawal, S., Sibler, E., Kouno, T., Tacconi, C., Hon, C.-C., Berger, S. D., Müllhaupt, D., He, Y., Kim, J., et al. (2021). LETR1 is a lymphatic endothelial-specific lncRNA governing cell proliferation and migration through KLF4 and SEMA3C. Nat Commun. 12(1): 925. https://doi.org/10.1038/s41467-021-21217-0
- Goñi, E., Mas, A. M., Gonzalez, J., Abad, A., Santisteban, M., Fortes, P., Huarte, M. and Hernaez, M. (2024). Uncovering functional lncRNAs by scRNA-seq with ELATUS. Nat Commun. 15(1): 9709. https://doi.org/10.1038/s41467-024-54005-7
- Deveson, I. W., Brunck, M. E., Blackburn, J., Tseng, E., Hon, T., Clark, T. A., Clark, M. B., Crawford, J., Dinger, M. E., Nielsen, L. K., et al. (2018). Universal alternative splicing of noncoding exons. Cell Syst. 6(2): 245–255.e5. https://doi.org/10.1016/j.cels.2017.12.005
- Hardwick, S. A., Bassett, S. D., Kaczorowski, D., Blackburn, J., Barton, K., Bartonicek, N., Carswell, S. L., Tilgner, H. U., Loy, C., Halliday, G., et al. (2019). Targeted, High-Resolution RNA Sequencing of Non-coding Genomic Regions Associated With Neuropsychiatric Functions. Front Genet. 10: 309. https://doi.org/10.3389/fgene.2019.00309
- Lagarde, J., Uszczynska-Ratajczak, B., Carbonell, S., Pérez-Lluch, S., Abad, A., Davis, C., Gingeras, T. R., Frankish, A., Harrow, J., Guigo, R., et al. (2017). High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat Genet. 49(12): 1731–1740. https://doi.org/10.1038/ng.3988
- Frohman, M. A., Dush, M. K. and Martin, G. R. (1988). Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci USA. 85(23): 8998–9002. https://doi.org/10.1073/pnas.85.23.8998
- Loh, E. Y., Elliott, J. F., Cwirla, S., Lanier, L. L. and Davis, M. M. (1989). Polymerase Chain Reaction with Single-Sided Specificity: Analysis of T Cell Receptor δ Chain. Science. 243(4888): 217–220. https://doi.org/10.1126/science.2463672
- Schaefer, B. C. (1995). Revolutions in Rapid Amplification of cDNA Ends: New Strategies for Polymerase Chain Reaction Cloning of Full-Length cDNA Ends. Anal Biochem. 227(2): 255–273. https://doi.org/10.1006/abio.1995.1279
- Zhu, Y. Y., Machleder, E. M., Chenchik, A., Li, R. and Siebert, P. D. (2001). Reverse Transcriptase Template Switching: A SMARTTM Approach for Full-Length cDNA Library Construction. BioTechniques. 30(4): 892–897. https://doi.org/10.2144/01304pf02
- Lagarde, J., Uszczynska-Ratajczak, B., Santoyo-Lopez, J., Gonzalez, J. M., Tapanari, E., Mudge, J. M., Steward, C. A., Wilming, L., Tanzer, A., Howald, C., et al. (2016). Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat Commun. 7(1): 12339. https://doi.org/10.1038/ncomms12339
- Xu, D., Tang, L., Zhou, J., Wang, F., Cao, H., Huang, Y. and Kapranov, P. (2023). Evidence for widespread existence of functional novel and non-canonical human transcripts. BMC Biol. 21(1): 271. https://doi.org/10.1186/s12915-023-01753-5
- Tang, L., Xu, D., Luo, L., Ma, W., He, X., Diao, Y., Ke, R. and Kapranov, P. (2024). A novel human protein-coding locus identified using a targeted RNA enrichment technique. BMC Biol. 22(1): 273. https://doi.org/10.1186/s12915-024-02069-8
- Deamer, D., Akeson, M. and Branton, D. (2016). Three decades of nanopore sequencing. Nat Biotechnol. 34(5): 518–524. https://doi.org/10.1038/nbt.3423
- Weirather, J. L., de Cesare, M., Wang, Y., Piazza, P., Sebastiano, V., Wang, X.-J., Buck, D. and Au, K. F. (2017). Comprehensive comparison of pacific biosciences and oxford nanopore technologies and their applications to transcriptome analysis. F1000Res. 6: 100. https://doi.org/10.12688/f1000research.10571.2
- Bolisetty, M. T., Rajadinakaran, G. and Graveley, B. R. (2015). Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol. 16: 204. https://doi.org/10.1186/s13059-015-0777-z
- De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M. and Van Broeckhoven, C. (2018). NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics. 34(15): 2666–2669. https://doi.org/10.1093/bioinformatics/bty149
- Li, H. (2018). Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34(18): 3094–3100. https://doi.org/10.1093/bioinformatics/bty191
- Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., Whitwham, A., Keane, T., McCarthy, S. A., Davies, R. M., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience. 10(2). https://doi.org/10.1093/gigascience/giab008
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26(6): 841–842. https://doi.org/10.1093/bioinformatics/btq033
- Leonardi, T. (2019). Bedparse: feature extraction from BED files. J Open Source Softw. 4(34): 1228. https://doi.org/10.21105/joss.01228
- Moore, J. E., Purcaro, M. J., Pratt, H. E., Epstein, C. B., Shoresh, N., Adrian, J., Kawli, T., Davis, C. A., Dobin, A., Kaul, R., et al. (2020). Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 583(7818): 699–710. https://doi.org/10.1038/s41586-020-2493-4
- ENCODE Project Consortium. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature. 489(7414): 57–74. https://doi.org/10.1038/nature11247
- ENCODE Project Consortium. (2011). A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9(4): e1001046. https://doi.org/10.1371/journal.pbio.1001046
- Luo, Y., Hitz, B. C., Gabdank, I., Hilton, J. A., Kagda, M. S., Lam, B., Myers, Z., Sud, P., Jou, J., Lin, K., et al. (2020). New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48(D1): D882–D889. https://doi.org/10.1093/nar/gkz1062
- Hitz, B. C., Lee, J.-W., Jolanki, O., Kagda, M. S., Graham, K., Sud, P., Gabdank, I., Strattan, J. S., Sloan, C. A., Dreszer, T., et al. (2023). The ENCODE uniform analysis pipelines. bioRxiv. e535623 https://doi.org/10.1101/2023.04.04.535623
Article Information
Publication history
Received: Mar 31, 2025
Accepted: Jun 3, 2025
Available online: Jun 19, 2025
Published: Jul 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Tang, L., Xu, D. and Kapranov, P. (2025). RACE-Nano-Seq: Profiling Transcriptome Diversity of a Genomic Locus. Bio-protocol 15(13): e5374. DOI: 10.21769/BioProtoc.5374.
Category
Bioinformatics and Computational Biology
Systems Biology > Transcriptomics > RNA-seq
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.