- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Optimized Midgut Tissue Dissociation of Mosquitoes and Sandflies for High-Quality Single-Cell RNA Sequencing
(§Technical contact) Published: Vol 15, Iss 12, Jun 20, 2025 DOI: 10.21769/BioProtoc.5352 Views: 2415
Reviewed by: Pilar Villacampa AlcubierreAnonymous reviewer(s)
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


Abstract
Single-cell RNA sequencing has revolutionized molecular cell biology by enabling the identification of unique transcription profiles and cell transcription states within the same tissue. However, tissue dissociation presents a challenge for non-model organisms, as commercial kits are often incompatible, and current protocols rely on tissue enzymatic digestion for extended periods. Tissue digestion can alter cell transcription in response to temperature and the stress caused by enzymatic treatment. Here, we propose a protocol to stabilize RNA using a deep eutectic solvent (Vivophix, Rapid Labs) prior to tissue dissociation, thereby avoiding transcription changes induced by the process and preventing RNase activity during incubation. We validated this methodology for three medically important insect vectors: Anopheles gambiae, Aedes aegypti, and Lutzomyia longipalpis. Single-cell RNA sequencing using our insect midgut dissociation protocol yielded high-quality sequencing results, with a high number of cells recovered, a low percentage of mitochondrial reads, and a low percentage of ambient RNA—two hallmark standards of cell quality.
Key features
• This protocol stabilizes tissue RNA before dissociation, avoiding RNase-mediated RNA degradation and transcription changes during the dissociation process.
• Current protocols for insect midgut dissociation use enzymatic digestion of live tissues at temperatures that are incompatible with insect physiology.
• We validated this new stabilization-dissociation methodology for insect midgut, yielding high-quality single-cell RNA sequencing with high gene counts, low mitochondrial RNA, and minimal ambient RNA contamination.
Keywords: Tissue dissociationGraphical overview
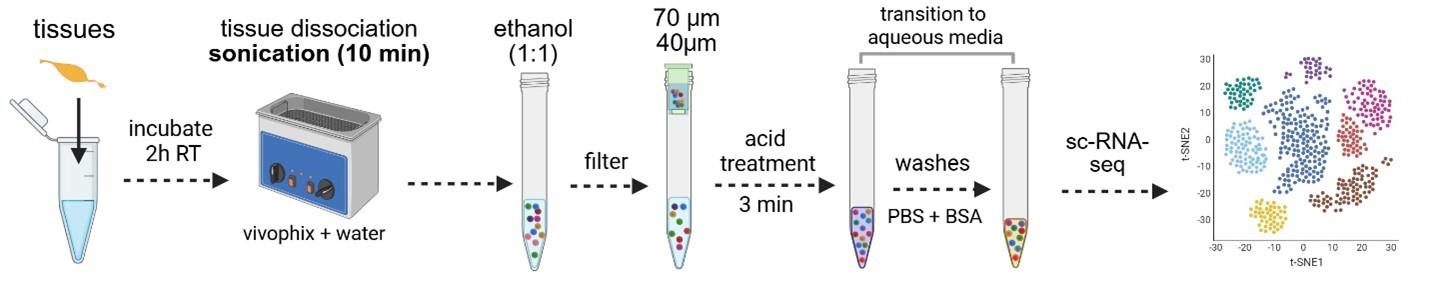
Mosquito and sand fly midgut RNA stabilization and dissociation protocol for single-cell sequencing applications
Background
Single-cell multi-omics approaches have become indispensable tools since they overcome the limitations of bulk sequencing to resolve cellular heterogeneity [1]. One advantage of these technologies is the ability to identify unique transcription profiles and different cell states within a single tissue; this requires cells to be dissociated and transformed into a single-cell suspension. However, dissociating cells while preserving nucleic acids, especially RNA, poses a significant challenge. RNA degradation can substantially affect downstream analysis, particularly in single-cell sequencing approaches that have less sequencing depth. While multiple dissociation methods that supposedly preserve RNA integrity before sequencing are available for mouse and human tissues, the same is not true for non-model organisms such as insects.
Insects are typically reared at 28 °C, a temperature within their physiological range. Still, the available methods for insect tissue dissociation, including those developed for Drosophila, involve enzymatic digestion of live tissues at room temperature [2–4] or high temperatures (37 °C) [5]. Therefore, in addition to the stress of enzymatic digestion, tissues are subjected to heat shock, which can lead to drastic transcriptional changes. Some methods describe the use of enzymes that work at low temperatures (4 °C) [6,7], but they require longer incubation periods, which can affect tissue oxygenation and viability, and consequently lead to transcriptional changes during sample processing. Furthermore, mosquito midguts express significant levels of RNases; therefore, longer incubation periods may also lead to RNA degradation. Of note, tissue dissociation can generate ambient RNA, which consists of cell-free RNA molecules present in the cell suspension, derived from ruptured, dead, or dying cells, and other sources [8,9]. Enzymatic digestion combined with mechanical disruption increases the amount of ambient RNA contamination in the sample, leading to a low fraction of quality reads in single cells [10].
Here, we propose an optimized protocol that dehydrates tissues and stabilizes RNA within cells before and during dissociation, thereby avoiding transcriptional changes caused by sample processing. Once the RNA is stabilized, dehydrated tissues can either be processed immediately or stored for up to several months. This is crucial for projects in which samples are harvested over time but need to be processed and sequenced simultaneously to avoid batch effects. It is also beneficial when the necessary infrastructure is not available or during field sample collection, because samples can be stored at room temperature, and the RNA remains stable.
The uniqueness and strength of this method rely on three key steps: 1) The dehydration of the tissue and stabilization of RNA within the cells using a deep eutectic solvent (presumably organic). This prevents any enzymatic reactions from occurring, keeping tissues stable and preserving their transcriptomes, although the tissues are no longer viable. After dissociation, the solvent is removed using ethanol. 2) Before transitioning to an aqueous medium, cells are incubated with acetic acid to inactivate any remaining RNase activity. 3) Dissociated cells are then washed in an aqueous medium and can be used for downstream single-cell RNA sequencing.
Using this procedure, we successfully dissociated midgut cells from three medically important insect vectors, preserving their RNA integrity and transcription patterns while achieving low ambient RNA contamination.
Materials and reagents
Biological materials
1. Anopheles gambiae (G3 – CDC strain) or Aedes aegypti (Liverpool strain) dissected midguts (30 midguts)
2. Lutzomya longipalpis (Jacobina strain) dissected midguts (50 midguts)
Reagents
1. Vivophix (RNA stabilization reagent for single cell analysis) (RapidLabs, UK, catalog number: RD-VIVO-50)
2. Nuclease-free water (NFW), no DEPC (Thermo Fisher Scientific, catalog number: AM2618)
3. RNASE inhibitor, protector RNASE inhibitor 10,000 units (Sigma-Aldrich, catalog number: 3335402001)
4. Phosphate buffer saline (PBS) 10× (90 g NaCl, 1.44 g KH2PO4, 7.95 g NaHPO4 in 1 L of water), pH 7.4. (KD-Medical, catalog number: RGF-3210)
5. Glacial acetic acid (Sigma-Aldrich, catalog number: A6283, 100 mL)
Caution: Flammable, skin burns/corrosion, eye damage, corrosive to metals. Handle inside of a safety cabinet.
6. Ethyl alcohol (Sigma-Aldrich, catalog number: E7023, 500 mL)
Caution: Flammable, irritant to skin and eyes.
7. Ultrapure BSA 50 mg/mL (Thermo Fisher Scientific, catalog number: AM2618)
8. RNase away (Molecular Bioproducts, catalog number: 7000)
9. 7-AAD (7-Aminoactinomycin D) (Thermo Fisher Scientific, catalog number: A1310) or Sytox (Thermo Fisher Scientific, catalog number: S7020)
10. Dimethyl sulfoxide (DMSO) (Sigma-Aldrich, catalog number: 472301)
11. WGA (wheat germ agglutinin) (Thermo Fisher Scientific, catalog number: W11261)
Solutions
1. Dissociation mixture (see Recipes)
2. Washing buffer (see Recipes)
3. Resuspension buffer (see Recipes)
4. Acid solution (see Recipes)
Recipes
Note: All recipes should be RNASE-free to keep RNA integrity during the procedure.
1. Dissociation mixture (per tube or sample)
| Reagent | Final proportion | Quantity or volume |
|---|---|---|
| Vivophix | 65% of final volume | 150 μL |
| Nuclease-free water | 35% of final volume | 350 μL |
Notes:
1. Add dissociation solution on top of 500 μL of Vivophix containing the sample to achieve the final proportion of 65/35).
2. Dissociation solution can be prepared proportionally to the number of samples.
3. Dissociation mixture cannot be stored and has to be prepared fresh before dissociation.
2. Washing buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| PBS | 1× (150 mM, 1 mM KH2PO4, 5.5 mM NaHPO4) | 5 mL |
| Ultrapure BSA 50 mg/mL | 1 mg/mL or 0.1% | 1 mL |
| RNASE inhibitor (40 U/μL) | 1 U/50 mL | 1 μL |
| Nuclease-free water | n/a | 44 mL |
| Total | n/a | 50 mL |
Notes:
1. Each sample will require the use of 20 mL of washing buffer.
2. Because of the BSA and RNASE inhibitor, the washing buffer must be freshly prepared before dissociation. Storage is not recommended.
3. Resuspension buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| PBS | 1× (150 mM, 1 mM KH2PO4, 5.5 mM NaHPO4) | 100 μL |
| Ultrapure BSA 50 mg/mL | 1 mg/mL or 0.1% | 20 μL |
| RNASE inhibitor (40 U/μL) | 0.2 U/μL | 5 μL |
| Nuclease-free water | n/a | 875 μL |
| Total | n/a | 1 mL |
Notes:
1. Each sample will require up to 200 μL.
2. Because of the BSA and RNASE inhibitor, the resuspension buffer must be freshly prepared before dissociation. Storage is not recommended.
4. Acid solution (per sample)
| Reagent | Final proportion | Quantity or volume |
|---|---|---|
| Vivophix | 60% | 600 μL |
| Glacial acetic acid | 40% | 400 μL |
| Total | n/a | 1 mL |
Notes:
1. The acid solution should preferably be freshly prepared and well mixed. However, it can be stored at room temperature for 72 h.
2. Acid solution can be prepared proportionally to the number of samples.
Caution: Prepare and handle this solution inside a safety cabinet.
Laboratory supplies
1. CellPro 15 mL conical centrifuge tubes (Alkali Scientific, catalog number: CW5600)
2. CellPro 50 mL conical centrifuge tubes (Alkali Scientific, catalog number: CW5603)
3. Open-top thick wall polycarbonate tubes, 8 × 51 mm, 25 Pk (1 mL) (Beckman Coulter, catalog number: 355657)
4. 1.5 mL microtubes (Axygen, catalog number: MCT-150-C-S)
5. 200 μL micropipette tips (TipOne 200 μL graduated filter tips) (USA Scientific, catalog number: 1180-8810)
6. 1250 μL micropipette tips (Purepoint barrier tips FT1250) (Alkali Scientific, catalog number: FT1250)
7. C-Chip disposable hemocytometer (iNCYTO, catalog number: DHC-N01-5 NI – Neubauer Improved)
9. 70 μm filter (Pluriselect, catalog number: 43-10040-70) or pre-separation filters (70 μm) (Miltenyl Biotec, catalog number: 130-095-823)
10. 40 μm filter (Pluriselect, catalog number: 43-10040-40)
11. MACS® SmartStrainers (30 μm) (Miltenyl Biotec, catalog number: 130-098-458)
12. Transfer pipette, sterile, polyethylene (Millipore Sigma, catalog number: Z350818-500EA)
13. Parafilm (to seal thick wall polycarbonate tubes) (Amcor, catalog number: PM-996)
14. Cellometer cell counting chamber (Nexcelom Bioscience, catalog number: SD100)
15. 15 μ-slide 18 well (Ibidi, catalog number: 81816) or any chamber that is compatible with a fluorescence microscope
16. Laboratory pipettes (10, 20, 200, and 1,000 μL)
Equipment
1. Professional benchtop ultrasonic cleaner, E-series, dual ultrasonic frequency 28/40 kHz
2. Thermo Sorvall Legend XTR refrigerated centrifuge (Thermo Fisher Scientific, catalog number: 75004520, discontinued)
3. TX-1000 4 × 1,000 mL swinging bucket rotor (Thermo Fisher Scientific, catalog number: 75003001)
Note: In this protocol, it is critical to use a swing bucket rotor type.
4. Cellometer Auto 2000 automated cell counter cell profiler (Nexcelom Bioscience, discontinued)
5. Transmitted light and epifluorescence microscopes
Procedure
A. Tissue dehydration and RNA stabilization with a deep eutectic solvent (Vivophix)
1. Dissect 30–50 midguts in PBS (1×) and place them directly into 500 μL of Vivophix in a 1.5 mL microcentrifuge tube (Figure 1).
Critical: Use a micropipette to measure the Vivophix volume for precision, as this will impact dissociation further in the procedure.
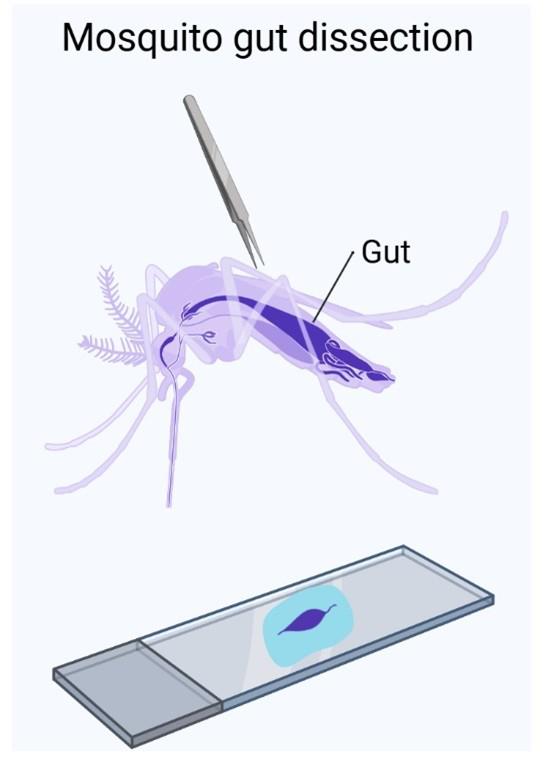
Figure 1. Graphical representation of mosquito midgut dissection
a. Transfer tissues into Vivophix using a disposable needle to carry as little liquid as possible, since dilution of Vivophix can trigger tissue dissociation.
b. Tap or vortex the microcentrifuge tube several times to ensure the tissues are in contact with Vivophix.
2. Leave for at least 2 h in a rotation shaker at room temperature.
Critical: Tissues must stay for a minimum of 2 h at room temperature after being transferred to Vivophix. If you are in a rush, it is acceptable to leave them at room temperature overnight. Early next day, samples can be placed at 4 °C (if they are going to be dissociated in the next 2–3 weeks) or -20 °C (if you are storing fixed tissues for 2–6 months before dissociation). In our experience, leaving samples at room temperature overnight has no impact on the dissociation yield and RNA quality, although there is no recommendation regarding this in the Vivophix datasheet.
B. Tissue dissociation
1. Before starting the dissociation:
a. Add ice cubes to the water sonicator bath to lower the temperature; it should be between 4 and 18 °C.
Notes:
1. Sonication increases the temperature over time, so if the temperature gets higher than 18 °C, add more ice.
2. Ideally, add ice cubes before preparing the reagents so they are partially melted at the time of dissociation. If the water is too cold (below 4 °C), it can cause precipitation of Vivophix (white cloud solution) during the procedure. If that happens, vortex your sample at room temperature until Vivophix goes back into solution (becomes clear) and proceed with the protocol. We found that precipitation of Vivophix does not alter the quality of the dissociation.
3. The temperature of the water bath should not be too cold (below 4 °C), which could impact the speed of dissociation, nor warmer than 18 °C, which could compromise tissue quality.
c. Place the following reagents and supplies on ice: washing buffer, resuspension buffer, 15 mL conical tubes, and 1 mL thick-walled polycarbonate tubes.
Note: Set the temperature of the centrifuge to 4 °C.
d. Equilibrate samples at room temperature.
e. Prepare the dissociation mixture. Dissociation is triggered by the dilution of Vivophix in water.
Critical: The amount of water in the mixture is crucial for a successful dissociation, so precision is important at this step. To dissociate midgut tissues, we used the following proportion: 65% Vivophix and 35% water. For each sample, prepare a solution with 150 μL of Vivophix and 350 μL of nuclease-free water, then vortex for 30 s.
Note: If you have multiple tubes (samples) to dissociate at one time, you can prepare a large mix respecting the proportions above.
2. Dissociation:
a. At room temperature, add the mixture prepared in step B1d to the tissues in Vivophix.
Note: Tissues are stored in 500 μL of Vivophix, so when you add the dissociation mixture to the sample, the final volume of Vivophix will be 650 μL (500 + 150), plus 350 μL of water, reaching the proportion of 65%/35% (65% Vivophix and 35% water).
b. Invert the tube several times to ensure that Vivophix, water, and tissues are well mixed. Vortex briefly. Alternatively, you can pipette up and down to mix. Vortexing is preferable to prevent tissues from sticking to the pipette.
c. Transfer the sample (1 mL) to a polycarbonate tube for sonication.
Critical: Use thick-walled polycarbonate tubes to ensure proper sonication frequency and to prevent the sample from overheating.
d. Seal polycarbonate tubes with parafilm.
Note: You can wipe the parafilm with RNASE away.
e. Place tubes in a floater or metal rack and put them inside the sonicator bath. Use a 28 kHz frequency.
Note: If you use a floater, make sure the liquid/air interface in the tube is visible (completely below the floater material) and immersed in water.
f. Sonicate the tubes for 7–10 min and mix every 2 min by vortexing.
Notes:
1. Monitor tissue dissociation by observing the turbidity of the media. Usually, midguts start to dissociate around minute 6–7. If the tissues have been longitudinally opened, it may take even less time.
2. Avoid removing the tubes from the floater because it increases the chances of rupturing the parafilm. Invert the tubes every 2 min or vortex while the tubes are still on the floater.
3. Preferably use a rack so you can easily remove tubes every 2 min to invert or vortex.
Optional: Monitor dissociation by taking 10 μL of the sample during the dissociation process and observing it in a disposable Neubauer chamber.
g. After sonication, place samples on ice and move to the next steps.
Notes:
1. The temperature of the water bath may increase due to the sonication. Keep the temperature between 4 and 18 °C by adding ice cubes along the process.
2. The dissociation efficiency will depend on the correct proportion of Vivophix and water. Be precise with the pipetting.
C. Sample filtering

Figure 2. Anopheles gambiae midgut dissociation progression. (A) Turbidity of the tubes every 2 min during dissociation. Pictures go from time 0 until 10 min, when we end dissociation. (B) Dissociation progresses every 2 min. As dissociation progresses, we can see more individual cells. This panel matches the turbidity with the actual dissociation observed in the Neubauer chamber.
1. Attach the 70 μm filter to a prechilled 15 mL conical tube and filter the 1 mL containing the dissociated tissue. The filter itself does not need to be chilled on ice, just the conical tube attached to it.
2. Then, filter the flowthrough through a 40 μm filter attached to a new prechilled 15 mL conical tube.
Note: From this step on, we begin to remove the Vivophix, transitioning the sample into the aqueous phase.
3. Pass the same sample volume of 100% ethyl alcohol (in this case, 1 mL) through the filters. Pass through the 70 μm filter first, then collect the flowthrough and pass it through the 40 μm filter. At the end, the sample will be topped with 1 mL of ethyl alcohol, and the total volume after this step will be 2 mL.
Note: We pass the ethyl alcohol through the filters after the sample to remove any potential single cells that get caught during filtration.
4. Mix the sample with the ethyl alcohol by inverting the 15 mL conical tube at least 10 times until the liquids are well mixed.
5. Centrifuge the cells at 150× g for 10 min at 4 °C.
Critical: Use a swing bucket rotor to pellet the cells. This has less impact on the cells.
Notes:
1. This step creates a soft pellet located at the bottom of the tube.
2. Always keep samples on ice.
6. Remove the supernatant using a transfer pipette, as much as you can, leaving less than half an inch of liquid above the pellet (around 200 μL). If you want, as you get closer to the pellet, it is okay to use a micropipette.
Note: At this point, cells are slightly dehydrated because of the mixture of the eutectic solvent and the ethyl alcohol.
D. Acid treatment
Note: This step is critical to maintain the integrity of the mRNA and prevent the reactivation of RNases once we change the cells to aqueous media. We use a mixture of Vivophix and acetic acid (acid solution) to inactivate RNases before the washes.
1. Slowly add 1 mL of the acid solution to the pellet. Use a micropipette and pipette on the side so you do not disrupt the pellet.
Notes:
1. Sometimes, the pellet can detach from the bottom. That is fine because we are going to centrifuge the cells again in 2 steps.
2. Vortex the acid solution right before applying it to the cells to ensure it is well mixed. Sometimes, the acid can separate from Vivophix.
2. Incubate cells with the acid solution for 3 min at room temperature.
Note: This is the only step in which samples are kept at room temperature after the start of the dissociation process. All the other steps should be performed on ice or using prechilled centrifuges, buffers, and tubes.
3. Add 2 mL of 100% ethyl alcohol on top of the cells with the acid solution. The final volume will be 3 mL. This step is to form ethyl acetate, which is easier to wash and remove from cells in the washing steps.
Critical: We must remove all the acid from the cells before proceeding with single-cell applications. Any acid residue could result in reagent precipitation and failure during GEM formation (10× technologies).
4. Mix by inversion (10–15 times) until all the ethyl alcohol is well mixed with the acid solution. The pellet will get resuspended in this step.
5. Centrifuge the sample at 150× g for 10 min at 4 °C to re-form a soft pellet.
Critical: Use a swing bucket rotor to pellet the cells.
6. Remove most of the supernatant using a transfer pipette (3 mL) and leave less than half an inch of liquid above the pellet.
E. Cell washes and transfer to aqueous media
1. Add 10 mL of prechilled washing buffer to the pellet. Add the first 1 mL of washing buffer using a micropipette on the side of the tube. Then, for the remaining 9 mL, use the transfer pipette gently on the side.
Note: Do not resuspend the pellet in this step. The idea is to keep the pellet intact in this step, so we do not stress the cells by pelleting them at all the steps.
2. Centrifuge at 150× g for 5 min at 4 °C.
3. Remove most of the supernatant using a transfer pipette (3 mL) and leave less than half an inch of liquid above the pellet. As you get closer to the pellet, switch to a micropipette (p1000) to have more control while removing the liquid and avoiding pellet disruptions.
4. Repeat the wash with 10 mL of washing buffer. This time, add the entire 10 mL gently to the side of the tube with a transfer pipette (3 mL). The pellet must be resuspended in washing buffer by inverting the tube at least 15 times.
5. Pass the 10 mL through a 30 μm SmartStrainer to filter any remaining clumps that may still exist and collect the flowthrough in a 15 mL conical tube.
Notes:
1. We found that, for midguts, this filtration step is crucial to decrease the frequency of sequencing doublets.
2. Previous filtration steps were performed when cells were dehydrated and in aqueous media.
6. Centrifuge at 300× g for 10 min at 4 °C to pellet the cells for the last time.
7. Remove most of the supernatant with a transfer pipette (3 mL).
8. Then, using a micropipette, gently remove the remaining liquid, leaving around 20–30 μL volume above the pellet.
9. Resuspend cells in a final volume of 200 μL by adding 170–180 μL of resuspension buffer.
10. After adding the resuspension buffer, gently pipette cells up and down until the pellet is resuspended.
F. Counting and visualizing cells
1. Counting cells:
We have tested two different methods to estimate cell number after dissociation: automated cell counter (Cellometer) and manual counting using a Neubauer improved chamber under the microscope. Counts from the Cellometer were usually 2–3 times higher than the manual counts on a Neubauer chamber. For single-cell applications, automated cell counting was more reliable to estimate cell number.
2. Visualizing cells
Note: To check the morphology of the cells after dissociation, we tested a couple of nuclear and membrane dyes. For nuclei staining, the best option was 7-AAD (1:1,000), but Sytox 504/523 (1:1,000) also works. For membrane staining, we used WGA 488 nm (1:1,000), but any other color will also work.
a. Prepare a solution by adding 1 μL of the DMSO stock of 7-AAD and 1 μL of the 1 mg/mL of WGA (488 nm) into 498 μL of resuspension buffer. The dilutions of the dyes will be 1:500.
b. Mix 100 μL of cell suspension with 100 μL of the staining solution prepared in step F2a. The final dilution of the dyes together with the cells is 1:1,000.
c. Transfer the 200 μL to a microscope chamber and incubate at room temperature for 20 min, protected from light.
d. Visualize the cells under an epifluorescent inverted microscope.
Validation of protocol
The protocol described here was tested on three different insect vectors and yielded robust and reproducible results. The number of single cells recovered for sequencing was high, but more importantly, we obtained high-quality cells (Table 1). The dissociation preserved morphology (Figure 3A, D, and G) and generated high-quality mRNA and cDNA libraries that were synthesized using 10× technologies. Libraries were sequenced, and a summary is shown in Table 1. Recovered cells had a low mitochondrial percentage, and a high proportion of reads were from intracellular RNA, which demonstrates that the amount of ambient RNA generated with this procedure is low. Indeed, this protocol does not require any software to remove ambient RNA in downstream analysis. The stabilization and dehydration of the cells prior to the dissociation process, together with the sustained inactivation of RNases before the transition to the aqueous media, results in high-quality cells, prevents transcriptional responses to cellular stress during dissociation, and preserves the quality of the RNA obtained from single cells.
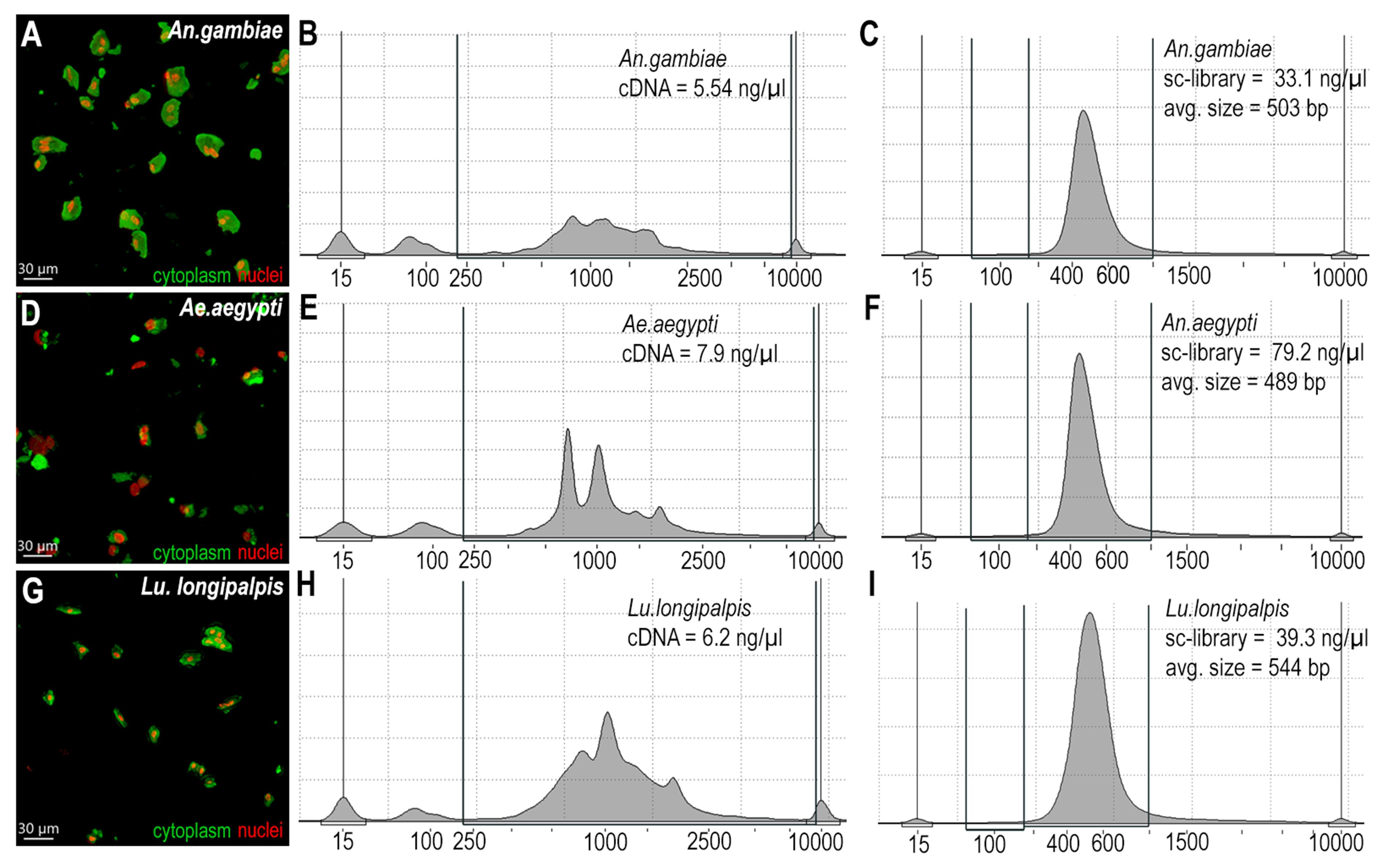
Figure 3. Midgut tissue dissociation, cDNA, and library quality. (A) Anopheles gambiae dissociated cells stained with WGA (green, cytoplasm) and 7-AAD (red, nuclei). (B) An. gambiae cDNA sample intensity normalized and acquired with TapeStation. (C) An. gambiae library sample intensity normalized and acquired with TapeStation. (D) Ae. aegypti dissociated cells stained with WGA (green, cytoplasm) and 7-AAD (red, nuclei). (E) Ae. aegypti cDNA sample intensity normalized and acquired with TapeStation. (F) Ae. Aegypti library sample intensity normalized and acquired with TapeStation. (G) Lu. longipalpis dissociated cells stained with WGA (green, cytoplasm) and 7-AAD (red, nuclei). (H) Lu. longipalpis cDNA sample intensity normalized and acquired with TapeStation. (I) Lu. longipalpis library sample intensity normalized and acquired with TapeStation.
Table 1. Summary of parameters after sc-RNA seq with dissociated tissues
| Parameters | Anopheles gambiae | Aedes aegypti | Lutzomyia longipalpis |
| Loaded cells | 5,000 cells | 10,000 cells | 7,000 cells |
| Recovered cells | 7,820 cells | 7,756 cells | 3,015 cells |
| Mean reads/cell | 31,021 | 35,471 | 323,985* |
| Median genes/cell | 422 | 412 | 1197* |
| Average % mitochondria | 7%–8% | 1.36%–1.38% | 1.11%–2.02% |
| Fraction reads in cells | 70% | 70.8% | 51.3%** |
*A high number of reads and genes were obtained because we used a high-capacity lane for sequencing.
**Low coverage genome annotation could impact the number of mapped reads inside cells.
***The number of recovered cells in Anopheles gambiae is higher than the number of loaded cells. That is a common observation when cell concentration is lower than the optimal range, which is between 700 and 1,200 cells per microliter.
Acknowledgments
A.B.B.F. conceptualized and planned the study. A.B.B.F, O.A.T.C., and P.A.C. performed test trials and troubleshooting. A.B.B.F. performed immunofluorescence and image analysis. A.B.B.F. and C.B.-M. wrote the manuscript. A.B.B.F., C.B.-M., O.A.T.C., and P.A.C. edited the manuscript. We thank Kevin Lee, Yonas Gebremicale, Andre Laughinghouse, and Claudio Meneses for insectary support. This work was supported by the Intramural Research Program of the Division of Intramural Research Z01AI000947, NIAID. The graphical overview created in BioRender. Barletta Ferreira, A. (2025) https://BioRender.com/hdyntdv
Competing interests
The authors declare no conflicts of interest.
References
- Jovic, D., Liang, X., Zeng, H., Lin, L., Xu, F. and Luo, Y. (2022). Single‐cell RNA sequencing technologies and applications: A brief overview. Clin Transl Med. 12(3): e694. https://doi.org/10.1002/ctm2.694
- Hung, R. J., Hu, Y., Kirchner, R., Liu, Y., Xu, C., Comjean, A., Tattikota, S. G., Li, F., Song, W., Ho Sui, S., et al. (2020). A cell atlas of the adultDrosophilamidgut. Proc Natl Acad Sci USA. 117(3): 1514–1523. https://doi.org/10.1073/pnas.1916820117
- Dutta, D., Buchon, N., Xiang, J. and Edgar, B. A. (2015). Regional Cell Specific RNA Expression Profiling of FACS Isolated Drosophila Intestinal Cell Populations. Curr Protoc Stem Cell Biol. 34(1): esc02f02s34. https://doi.org/10.1002/9780470151808.sc02f02s34
- Guo, X., Yin, C., Yang, F., Zhang, Y., Huang, H., Wang, J., Deng, B., Cai, T., Rao, Y., Xi, R., et al. (2019). The Cellular Diversity and Transcription Factor Code of Drosophila Enteroendocrine Cells. Cell Rep. 29(12): 4172–4185.e5. https://doi.org/10.1016/j.celrep.2019.11.048
- Pederzolli, M., Barion, E., Valerio, A., Cuda, A. V., Manara, V. and Bellosta, P. (2025). Optimized protocol for single-cell isolation and alkaline comet assay to detect DNA damage in cells of Drosophila wing imaginal discs. STAR Protoc. 6(1): 103590. https://doi.org/10.1016/j.xpro.2024.103590
- Vial, T., Lopez-Maestre, H., Couderc, E., Pinaud, S., Howick, V., Akorli, J., Lawniczak, M., Marti, G. and Merkling, S. H. (2024). Single-cell transcriptional landscapes ofAedes aegyptimidgut and fat body after a bloodmeal. bioRxiv : e622039. https://doi.org/10.1101/2024.11.08.622039
- Fitzmeyer, E. A., Dutt, T. S., Pinaud, S., Graham, B., Gallichotte, E. N., Hill, J. L., Campbell, C. L., Ogg, H., Howick, V., Lawniczak, M. K. N., et al. (2025). A single-cell atlas of the Culex tarsalis midgut during West Nile virus infection. PLoS Pathog. 21(1): e1012855. https://doi.org/10.1371/journal.ppat.1012855
- Caglayan, E., Liu, Y. and Konopka, G. (2022). Neuronal ambient RNA contamination causes misinterpreted and masked cell types in brain single-nuclei datasets. Neuron. 110(24): 4043–4056.e5. https://doi.org/10.1016/j.neuron.2022.09.010
- Madissoon, E., Wilbrey-Clark, A., Miragaia, R. J., Saeb-Parsy, K., Mahbubani, K. T., Georgakopoulos, N., Harding, P., Polanski, K., Huang, N., Nowicki-Osuch, K., et al. (2019). scRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 21(1): e1186/s13059–019–1906–x. https://doi.org/10.1186/s13059-019-1906-x
- Ilicic, T., Kim, J. K., Kolodziejczyk, A. A., Bagger, F. O., McCarthy, D. J., Marioni, J. C. and Teichmann, S. A. (2016). Classification of low quality cells from single-cell RNA-seq data. Genome Biol. 17(1): e1186/s13059–016–0888–1. https://doi.org/10.1186/s13059-016-0888-1
Article Information
Publication history
Received: Mar 10, 2025
Accepted: May 9, 2025
Available online: Jun 3, 2025
Published: Jun 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Barletta, A. B. F., Talyuli, O. A., Cecilio, P. and Barillas-Mury, C. (2025). Optimized Midgut Tissue Dissociation of Mosquitoes and Sandflies for High-Quality Single-Cell RNA Sequencing. Bio-protocol 15(12): e5352. DOI: 10.21769/BioProtoc.5352.
Category
Cell Biology > Tissue analysis > Tissue isolation
Cell Biology > Single cell analysis
Systems Biology > Transcriptomics > RNA-seq
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.