- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

TurboID Labeling and Analysis of Proteins in the Primary Cilium
Published: Vol 15, Iss 9, May 5, 2025 DOI: 10.21769/BioProtoc.5303 Views: 3077
Reviewed by: Jibin SadasivanAlexandre SmirnovAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2024
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


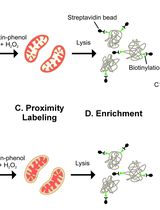
Abstract
Known as the cell’s antenna and signaling hub, the primary cilium is a hair-like organelle with a few micrometers in length and 200–300 nm in diameter. Due to the small size of the primary cilium, it is technically challenging to profile ciliary proteins from mammalian cells. Traditional methods, such as physical isolation of cilia, are susceptible to contamination from other cellular components. Other proximity-based labeling methods via APEX or BioID have been used to map ciliary proteins. However, these approaches have their inherent limitations, including the use of toxic reagents like H2O2 and prolonged labeling kinetics. Here, we show a new proximity-based labeling technique for primary cilia with TurboID. TurboID presents a distinct advantage over BioID and APEX2 due to its expedited labeling kinetics, taking minutes instead of hours, and its use of a non-toxic biotin substrate, which eliminates the need for H2O2. When targeted to the cilium, TurboID selectively labels ciliary proteins with biotin. The biotinylated proteins are then enriched with streptavidin beads and labeled with tandem mass tags (TMT), followed by mass spectrometry (MS) detection. This protocol eliminates the requirement of toxic labeling reagents and significantly reduces the labeling time, thus providing advantages in mapping signaling proteins with high temporal resolution in live cells.
Key features
• Compared to other proximity labeling enzymes, TurboID offers fast labeling kinetics and uses cell-permeable biotin as the labeling reagent [1].
• This protocol includes a straightforward subcellular fractionation step to remove the nuclei to reduce the non-specific background.
• This protocol has been successfully applied to the NIH 3T3 cell line and could also be applied in other cell lines and animal tissues.
Keywords: Proximity labelingGraphical overview
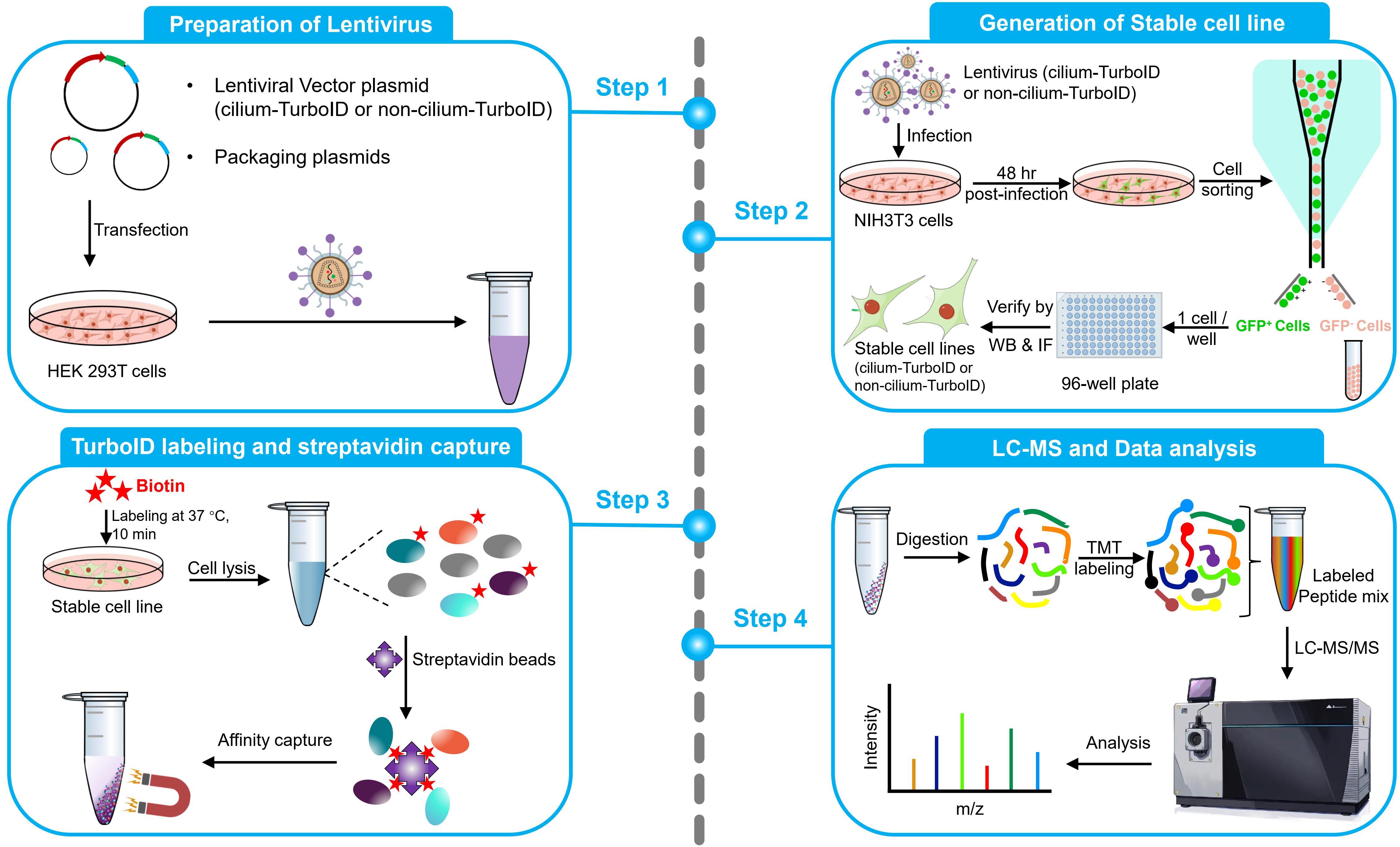
Background
The primary cilium is a hair-like organelle that protrudes from the surface of nearly all vertebrate cells. Most cilia are a few micrometers long and 200–300 nm in diameter. The ciliary membrane is approximately 1/1,000 to 1/5,000 of the total cellular surface, and the volume is around 1/30,000 of the total cellular volume [2,3]. Acting as cellular antennae, primary cilia sense a wide variety of extracellular cues and transduce them into intracellular signaling pathways [4,5]. Cilium dysfunction causes a wide range of human diseases, collectively known as ciliopathies. Ciliopathies affect the function of almost all human organs, including the brain, eyes, heart, kidneys, liver, skeleton, and reproductive system [6]. To understand the pathogenesis of ciliopathies, it is crucial to identify and characterize the signaling molecules within cilia.
However, progress along this line has been hindered by technical challenges associated with studying such a minuscule compartment of the cell. Conventional approaches of physically isolating cilia produce inconsistent results due to the significant amounts of contaminations from non-ciliary structures, such as microvilli [7–10]. In addition, some ciliary proteins are present in the cilium at very low abundance [11,12], and most signaling transducers transit through the cilia in a very dynamic manner during signal transduction. Physical isolation is not able to capture this dynamic protein transport into and out of the cilium. Proximity labeling–based approaches, circumventing these challenges, have been used to profile ciliary proteins in several studies [11,13–16]. Two categories of proximity labeling enzymes have been developed and used in proximity labeling. One category is peroxidase, such as APEX/APEX2, which adds biotin-phenol to its substrates in the presence of hydrogen peroxide (H2O2). The other category is biotin ligase, which catalyzes the covalent transfer of biotin to its neighboring proteins within 10–20 nm. Each proximity labeling enzyme has its advantages and inherent limitations. APEX/APEX2 has rapid labeling kinetics within a couple of minutes; however, its substrate biotin-phenol shows poor membrane permeability [17,18]. This limits its application to certain cell types and tissues. Using hydrogen peroxide in the labeling process is also a concern because it may affect the cellular oxidative status and induce stress responses [19]. The first generation of promiscuous biotin ligase, BioID, originates from bacteria. BioID requires a long labeling time (24 h) with biotin [15,16], which restricts its application in mapping the dynamic translocation of ciliary proteins during signal transduction.
The new generation of biotin ligase, TurboID, is an engineered biotin ligase derived from BioID. However, the labeling kinetics of TurboID are significantly improved in comparison with its predecessor. TurboID labels proteins within 5 min. Therefore, TurboID offers a fast labeling speed comparable to APEX/APEX2, while eliminating the need for the toxic labeling reagent hydrogen peroxide. Thus, TurboID is ideal for mapping dynamic protein composition with high temporal resolution in live cells. However, TurboID may exhibit elevated baseline biotinylation due to endogenous biotin, which may lead to increased background labeling concerns when mapping dynamic proteins.
This protocol aims to obtain quantitative cilium proteome with TurboID. It covers the full process of quantitative proteomics, including preparation of lentivirus, generation of a stable cell line, TurboID labeling and streptavidin capture, liquid chromatography–mass spectrometry (LC–MS), and quantitative analysis of mass spectrometry results. To probe the ciliary protein, we fused the TurboID to the ciliary-targeting domain (N+RVEP+PR) and non-ciliary-targeting domain (PR) of the small GTPase Arl13b (cilium-TurboID and non-cilium-TurboID). Our results confirmed the presence of over 100 proteins that were previously reported to localize to the cilium, underscoring its validation in mapping ciliary proteome. Meanwhile, we identified hundreds of new ciliary candidates that can be harvested for further studies [20]. Given that TurboID features a labeling radius of 10–20 nm, which enables precise labeling and identification of proteins close to the bait, leading to the effective identification of bait interactors, this protocol can also be adapted to map protein–protein interactions.
Materials and reagents
Biological materials
1. Bacterial strains: TOP10 (Thermo Fisher Scientific, catalog number: C404010), Stbl3 (Thermo Fisher Scientific, catalog number: C737303)
2. Cell lines: 293T cells (ATCC, catalog number: CRL-3216), NIH 3T3 cells (ATCC, catalog number: CRL-1658)
Reagents
1. psPAX2 plasmid (ampicillin resistance) (Addgene, catalog number: 12260); pMD2.G plasmid (ampicillin resistance) (Addgene, catalog number: 12259); Cilium-TurboID (pFUGW-NRVEPPR-GFP-TurboID, ampicillin resistance) and non-cilium-TurboID (pFUGW-PR-GFP-TurboID, ampicillin resistance) plasmids as described in [20]
2. Ampicillin, sodium salt (Research Products International, catalog number: A40040-5.0)
3. LB broth (Fisher Scientific, catalog number: BP1426-500)
4. Maxi Fast-Ion Plasmid kit (IBI Scientific, catalog number: Ib47122)
5. DMEM (Corning, catalog number: 10-013-CM)
6. Fetal bovine serum (FBS) (Corning, catalog number: 35-010-CV)
7. Penicillin/streptomycin (10,000 U/mL) (Thermo Fisher Scientific, catalog number: 15-140-122)
8. Calcium Phosphate Transfection kit (Takara Bio, catalog number: 631312); includes 2 M calcium solution and 2× HBS
9. Calf serum (CS) (Fisher Scientific, catalog number: 16-010-159)
10. 0.05% trypsin-EDTA (Fisher Scientific, catalog number: 25-300-120)
11. DPBS (Fisher Scientific, catalog number: 14-190-136)
12. Poly-D-lysine (Sigma, catalog number: A003E) (store at -20 °C)
13. Biotin (Sigma, catalog number: B4501-1G)
14. DMSO (Sigma, catalog number: D8418-50ML)
15. 20% PFA (Fisher Scientific, catalog number: 50-980-493)
16. Donkey serum (Sigma, catalog number: S30-100ML)
17. Triton X-100 (Sigma, catalog number: T8787-50ML)
18. HEPES (Fisher Scientific, catalog number: BP310-500)
19. KCI (Fisher Scientific, catalog number: P217-500)
20. MgCl2 (Fisher Scientific, catalog number: AC223211000)
21. 1 M sodium hydroxide (NaOH) (Sigma, catalog number: 1091371003)
22. Hydrochloric acid (HCl) 36.5% to 38.0% (Fisher Scientific, catalog number: A144-500)
23. 0.5 M EDTA (Fisher Scientific, catalog number: AAJ15694AE)
24. 0.5 M EGTA (Fisher Scientific, catalog number: 50-255-956)
25. Tris base (Fisher Scientific, catalog number: BP152-500)
26. NaCl (Fisher Scientific, catalog number: S271-500)
27. SDS (Fisher Scientific, catalog number: BP166-100)
28. Na2CO3 (Fisher Scientific, catalog number: S263-500)
29. Hoechst (Sigma, catalog number: 94403-1ML)
30. Chicken anti-GFP (Aves labs, catalog number: GFP-1020)
31. Mouse anti-Ac-tubulin (Sigma, catalog number: T6793)
32. Donkey anti-chicken AlexaFluor 488 (Jackson ImmunoResearch Labs, catalog number: 703-545-155)
33. Donkey anti-mouse rhodamine (Jackson ImmunoResearch Labs, catalog number: 715-025-151)
34. Alexa Fluor 647 Streptavidin (Jackson ImmunoResearch Labs, catalog number: 016-600-084)
35. Urea (Sigma, catalog number: U5128-500G)
36. Fluoromount-G mounting medium (SouthernBiotech, catalog number: 0100-01)
37. Protease inhibitor cocktail (Roche, catalog number: 11836170001)
38. 10% NP-40 (Thermo Fisher Scientific, catalog number: 28324)
39. BCA Protein Assay kit (Thermo Fisher Scientific, catalog number: 23225)
40. Streptavidin magnetic beads (Thermo Fisher Scientific, catalog number: 88817)
41. 6× Laemmli sample buffer (MP Biomedicals, catalog number: 04822261)
42. Rabbit anti-GFP (Thermo Fisher Scientific, catalog number: A-11122)
43. Mouse anti-GAPDH (Santa Cruz Biotechnology, catalog number: sc-32233)
44. HRP-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch Labs, catalog number: 711-035-152)
45. HRP-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch Labs, catalog number: 715-035-151)
46. HRP-conjugated streptavidin (Thermo Fisher Scientific, catalog number: N100)
47. 1 M Triethylammonium bicarbonate (TEAB) (Sigma, catalog number: 18597-100ML)
48. 16plex TMT label reagent set (Thermo Fisher Scientific, catalog number: A44521)
49. 0.4% trypan blue solution (Thermo Fisher Scientific, catalog number: 15250061)
Solutions
1. 50 mM biotin stock solution (see Recipes)
2. Biotin labeling medium (see Recipes)
3. Blocking buffer (see Recipes)
4. Subcellular fractionation buffer (see Recipes)
5. 10× TBS buffer (see Recipes)
6. 25× protease inhibitor cocktail buffer (see Recipes)
7. TEAB buffer (see Recipes)
8. 1 M KCl (see Recipes)
9. 0.1 M Na2CO3 (see Recipes)
10. 10 mM Tris-HCl buffer (see Recipes)
11. 2 M urea buffer (see Recipes)
Recipes
1. 50 mM biotin stock solution
Store at -20 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Biotin | 50 mM | 0.5 g |
| DMSO | n/a | 41 mL |
| Total | n/a | 41 mL |
2. Biotin labeling medium
Store at 4 °C.
| Reagent | Final concentration | Volume |
|---|---|---|
| Calf serum (CS) | 0.5% | 250 µL |
| 50 mM biotin stock (Recipe 1) | 50 µM | 50 µL |
| DMEM | n/a | 49.7 mL |
| Total | n/a | 50 mL |
3. Blocking buffer
Store at 4 °C.
| Reagent | Final concentration | Volume |
|---|---|---|
| Donkey serum | 2% | 2 mL |
| Triton X-100 | 0.2% | 200 µL |
| DPBS | n/a | 97.8 mL |
| Total | n/a | 100 mL |
4. Subcellular fractionation buffer
Store at 4°C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| HEPES | 20 mM | 4.77 g |
| KCI | 10 mM | 0.75 g |
| MgCl2 | 2 mM | 0.19 g |
| 0.5 M EDTA | 1 mM | 2 mL |
| 0.5 M EGTA | 1 mM | 2 mL |
| H2O (MilliQ water) | n/a | See note |
| Total | n/a | 1 L |
Note: Allow the reagents to mix completely and adjust the pH of the solution to 7.4 by adding 1 M NaOH while measuring with a pH meter. Then, bring the volume up to 1 L with MilliQ water. Just before use, add and dilute the 25× protease inhibitor cocktail buffer to 1×.
5. 10× TBS buffer
Store at 4 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tris | 200 mM | 24.23 g |
| NaCl | 1.5 M | 87.66 g |
| 0.5 M EDTA | 10 mM | 20 mL |
| H2O (MilliQ water) | n/a | See note |
| Total | n/a | 1 L |
Note: Allow the reagents to mix completely and adjust the pH of the solution to 7.4 by adding HCl (36.5% to 38.0%) while measuring with a pH meter. Then, bring the volume up to 1 L with MilliQ water.
6. 25 × protease inhibitor cocktail buffer
Store at -20 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Protease inhibitor cocktail | 25× | 1 tablet |
| H2O (MilliQ water) | n/a | 2 mL |
| Total | n/a | 2 mL |
7. TEAB buffer
Store at 4 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M TEAB | 50 mM | 50 mL |
| NaCl | 150 mM | 8.77 g |
| 0.5 M EDTA | 1 mM | 2 mL |
| H2O (MilliQ water) | n/a | See note |
| Total | n/a | 1 L |
Note: Allow the reagents to mix completely and adjust the pH of the solution to 7.4 by adding HCl (36.5% to 38.0%) while measuring with a pH meter. Then, bring the volume up to 1 L with MilliQ water.
8. 1 M KCl
Store at 4 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| KCl | 1 M | 7.46 g |
| H2O (MilliQ water) | n/a | 100 mL |
| Total | n/a | 100 mL |
9. 0.1 M Na2CO3
Store at 4 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Na2CO3 | 0.1 M | 1.06 g |
| H2O (MilliQ water) | n/a | 100 mL |
| Total | n/a | 100 mL |
10. 10 mM Tris-HCl buffer
Store at 4 °C.
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tris | 10 mM | 1.21 g |
| H2O (MilliQ water) | n/a | 1 L |
| Total | n/a | 1 L |
Note: Allow the Tris to mix completely and adjust the pH of the solution to 8.0 by adding HCl (36.5% to 38.0%) while measuring with a pH meter. Then, bring the volume up to 1 L with MilliQ water.
11. 2 M urea buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Urea | 2 M | 1.2 g |
| 10 mM Tris-HCl buffer (Recipe 10) | n/a | 10 mL |
| Total | n/a | 10 mL |
Note: Make this solution fresh on the day of use. No need to adjust the pH of the solution.
Laboratory supplies
1. 10 cm dish (Fisher Scientific, catalog number: FB012924)
2. 15 cm dish (Thermo Fisher Scientific, catalog number: 150468)
3. 0.45 µm syringe filter (Fisher Scientific, catalog number: 726-2545)
4. 15 mL conical tube (Corning, catalog number: 430791)
5. 70 µm cell strainer (Falcon, catalog number: 352350)
6. Polystyrene round-bottom tube (Falcon, catalog: 352058)
7. 96-well plate (Thermo Fisher Scientific, catalog number: 12-556-008)
8. 12 mm glass coverslips (Bellco Glass, catalog number: 194310012A)
9. 24-well plate (Fisher Scientific, catalog number: FB012929)
10. Parafilm (Fisher Scientific, catalog number: PM999)
11. Cell scraper (Falcon, catalog number: 353085)
12. 1.5 mL protein LoBind tube (Eppendorf, catalog number: 22431081)
13. 5 mL protein LoBind tube (Eppendorf, catalog number: 30108302)
14. 1 mL syringe (BD Biosciences, catalog number: 309628)
15. 27-gauge needle (BD Biosciences, catalog number: 305136)
16. Tweezers (Fine Science Tools, catalog number: 11252-00)
Equipment
1. Purifier Class II biological safety cabinet (Labconoco, model: 36212/36213 TYPE A2)
2. CO2 incubator (Eppendorf, model: CellXpert C170)
3. Vortex mixer (Fisher Scientific, catalog number: 02-215-414)
4. Centrifuge (Eppendorf, model: 5702R)
5. Cell sorter (BD Biosciences, model: FACSAria III)
6. Fluorescence microscope (Leica, model: DMi8 system)
7. Confocal microscope (Zeiss, model: LSM 880)
8. Sonicator equipped with a microtip, measuring 3/32 inches in diameter (Branson, model: SLPt)
9. Magnetic stand (Thermo Fisher Scientific, catalog number: CS15000)
10. Dry block heater (VWR, catalog number: 75838-282)
11. Savant SpeedVac vacuum concentrator (Thermo Fisher Scientific, model: SPD210-115)
12. UPLC system (Waters, model: ACQUITY UPLC M-Class)
13. Mass spectrometer (Thermo Fisher Scientific, model: Orbitrap Fusion Tribrid MS)
Software and datasets
1. FACSDiva (BD Biosciences, v7.0.x)
2. LAS X (Leica, v3.7.4.23463)
3. ImageJ (NIH, v1.53k)
4. ZEN (Zeiss, ZEN v2.3)
5. Preview (Protein Metrics, v3.5.0.0)
6. Proteome Discoverer (Thermo Fisher Scientific, v1.5)
7. Byonic (Protein Metrics, v3.5.0.0)
Procedure
A. Generation of stable cell lines with lentivirus-mediated DNA integration
A1. Lentivirus production
1. Perform heat-shock transformation of psPAX2 plasmid (ampicillin resistance) into Top10, pMD2.G plasmid (ampicillin resistance) into Stbl3, cilium-TurboID plasmid (ampicillin resistance) into Stbl3, and non-cilium-TurboID plasmid (ampicillin resistance) into Stbl3. Add antibiotics to LB broth medium, culture at 37 °C, and perform the plasmid maxiprep with the Maxi Fast-Ion Plasmid kit after 12 h.
Note: Use 0.5 µg of plasmid for transformation. Stbl3 is recommended for use when cloning unstable inserts such as lentiviral DNA containing direct repeats.
2. Maintain 293T cells in DMEM+10% FBS medium without penicillin/streptomycin at 37 °C with 5% CO2. Split cells the day before transfection.
a. Aspirate the culture medium, wash cells once with DPBS, and incubate with 0.05% trypsin-EDTA for 2 min at 37 °C.
b. Gently tap the dish to detach the cells, add 10 mL of DMEM+10% CS, resuspend, and collect all cells into a 15 mL conical tube.
c. Centrifuge at 100× g for 3 min at room temperature and aspirate supernatant.
d. Add 3 mL of DMEM+5% CS and resuspend cells. Seed a 10 cm dish with 6–7.5 × 106 cells per dish in 10 mL of DMEM+10% FBS without penicillin/streptomycin.
Note: Prepare 2 × 10 cm dishes at the same time, one for the cilium-TurboID and one for the non-cilium-TurboID. Perform the transfection when the cells are 90%–95% confluent.
3. Carefully replace the medium with 10 mL of DMEM+10% FBS.
Note: Prewarm the medium to 37 °C and do not include penicillin/streptomycin in the medium. The 293T cells have a tendency to contract after the medium change and then flatten and spread again after 0.5–1 h.
4. For each 10 cm dish, according to the manufacturer’s instructions, prepare the transfection mix in a 15 mL tube by adding the components in the order provided in Table 1.
Note: Take out the Calcium Phosphate Transfection kit from -20 °C and thaw the 2 M calcium solution and 2× HBS to room temperature.
Table 1. Components of the transfection mix
| Reagent | Quantity |
|---|---|
| Sterile H2O (Calcium Phosphate Transfection kit) | To 1,250 µL (total volume) |
| Cilium-TurboID or non-cilium-TurboID plasmid | 12 µg |
| psPAX2 plasmid | 12 µg |
| pMD2.G plasmid | 6 µg |
| 2 M calcium solution (Calcium Phosphate Transfection kit) | 140 µL |
Then, vortex the transfection mix using a vortex mixer, followed by adding 1,250 µL of 2× HBS (Calcium Phosphate Transfection kit) dropwise while continuing to vortex. Incubate the transfection mix at room temperature for 15 min before adding it to the 10 cm dish.
5. Add transfection solution dropwise to each 10 cm dish and incubate the plate overnight at 37 °C in a 5% CO2 incubator.
Note: Mix gently by rocking the dish back and forth before placing it into the incubator.
6. Twelve hours post-transfection, replace the medium with 10 mL of prewarmed DMEM+10% FBS.
Note: When working with lentiviruses, always wear appropriate PPE and perform all steps in a biosafety cabinet. Decontaminate all materials that come into contact with lentiviruses before disposal, using a 10% bleach solution.
7. Collect the lentivirus-containing supernatant at 48 h post-transfection and decontaminate the 293T cells using a 10% bleach solution.
8. Centrifuge the supernatant at 2,000× g for 10 min at 4 °C to remove cellular debris.
9. Filter the viral supernatant through a 0.45 µm syringe filter, aliquot 2–3 mL into each 15 mL conical tube, and store the viral supernatant at -80 °C.
Note: If the cells are prepared, it is preferable to use fresh virus-containing supernatant to infect NIH 3T3 cells. If this is not possible, aliquot 2–3 mL into each 15 mL conical tube and store them at -80 °C. Use 2–3 mL of the viral supernatant to infect one 10 cm cell dish.
A2. TurboID stable cell line generation
1. Maintain NIH 3T3 cells in DMEM+10% CS medium at 37 °C with 5% CO2. Split cells the day before infection, take out one-third (4 × 106 cells) of cells, and plate into a new 10 cm dish containing 10 mL of DMEM+10% CS.
Note: Include a dish of noninfected NIH 3T3 cells to optimize the purity and yield of the sorted cell populations when performing cell sorting.
2. After 12 h of culture, aspirate the culture medium and add one-fourth (2–3 mL) of the lentivirus mixture from one 10 cm dish of 293T cells with 10 mL of DMEM+10% CS to the NIH 3T3 cells.
3. Twelve hours post-infection, change the medium to 10 mL of prewarmed DMEM+10% CS.
4. Forty-eight hours post-transfection, prepare cells for sorting:
a. Aspirate the culture medium, wash cells once with DPBS, and incubate with 0.05% trypsin-EDTA for 2 min at 37 °C.
b. Gently tap the dish to detach the cells, add 10 mL of DMEM+10% CS, resuspend, and collect all cells into a 15 mL conical tube.
c. Centrifuge at 100× g for 3 min at room temperature and aspirate supernatant.
d. Add 3 mL of DMEM+5% CS and resuspend cells.
e. Run cells through a 70 µm cell strainer to get rid of potential cell clumps and transfer cells to a 5 mL polystyrene round-bottom tube.
Note: Include a dish of noninfected NIH 3T3 cells to set up the background for cell sorting. Also, treat these cells in parallel to the lentiviral-infected cells.
5. Perform cell sorting with FACSAria III cell sorter.
Note: Use the noninfected NIH 3T3 cells to adjust gate positions. Utilize FSC and SSC to identify and remove debris and dead cells. The fluorescent marker GFP can further refine the population of infected cells.
6. Prefill the wells of a 96-well plate with 100 µL of DMEM+10% CS containing penicillin/streptomycin.
7. Seed one cell each (GFP-positive cell) into one well of a 96-well plate.
8. Seed 3–4 96-well plates in total, incubate the plates at 37 °C in a 5% CO2 incubator, and wait until colonies emerge.
B. Validation of TurboID stable cell lines by immunofluorescence (IF)
1. Prepare 12 mm glass coverslips coated with Poly-D-lysine.
a. Transfer the autoclaved coverslips into a 24-well plate using tweezers.
b. Dilute the 1 mg/mL Poly-D-lysine (20×) with sterile water into 50 µg/mL (1×). Add 500 µL of diluted Poly-D-lysine into each well of a 24-well plate.
Note: Store 1 mg/mL Poly-D-lysine at -20 °C. Submerge all coverslips under the surface of the solution.
c. Incubate the 24-well plate at 37 °C for 2 h in a 5% CO2 incubator.
Note: Leave the coverslips in the Poly-D-lysine solution and seal the 24-well plate with parafilm. You can store it at 4 °C for 1 month.
d. Wash the coverslips by adding 500 µL of DPBS into each well and then aspirate. Repeat two more times.
2. Seed 1 × 105 cells onto 12 mm glass coverslips coated with Poly-D-lysine and culture cells in DMEM+10% CS medium to achieve 95% confluency.
3. Change media to 0.5 mL of serum-starving DMEM+0.5% CS and incubate for 24 h at 37 °C in a 5% CO2 incubator.
4. After 24 h starvation, change the medium to biotin labeling medium for 10 min.
Note: Include one coverslip that is not labeled with biotin to serve as a control. Prewarm the biotin labeling medium to 37 °C before labeling.
5. Rinse cells three times with DPBS, 5 min each time.
6. Fix cells for 10 min with cold 4% PFA at room temperature.
Note: Dilute 20% PFA to 4% buffered PFA with PBS.
7. Wash cells three times with DPBS, 5 min each time.
Note: You can store the fixed cells in DPBS at 4 °C for 1 week.
8. Take coverslips out of the plate using sharp tweezers and put them on parafilm in a humidified chamber.
Note: Make sure that the cells face up.
9. Permeabilize and block with blocking buffer for 1 h at room temperature.
10. Aspirate blocking buffer and add primary antibody (50 µL/coverslip, dilute antibody in blocking buffer).
Note: Dilute the primary antibodies using the specified ratios: chicken anti-GFP (1:500) and mouse anti-Ac-tubulin (1:1,000).
11. Incubate for 2 h at room temperature or overnight at 4 °C.
12. Rinse cells three times with DPBS, with 5 min between washes.
13. Add secondary antibody (50 µL/coverslip, dilute antibody in blocking buffer). Incubate for 1 h at room temperature.
Note: Dilute the secondary antibodies and fluorescent streptavidin using the specified ratios: donkey anti-chicken AlexaFluor 488 (1:200), donkey anti-mouse rhodamine (1:100), and Alexa Fluor 647 streptavidin (1:200). Do not overstain with secondary antibody!
14. Add 1 µg/mL Hoechst and incubate for 10 min at room temperature.
15. Wash three times with DPBS, with 5 min between washes.
16. Mount the cells by flipping coverslips on a drop of Fluoromount-G mounting medium (cells facing down) on the slide.
Note: Store slides in a cardboard slide tray.
17. Image the cells with a Leica DMi8 system with a 63× oil-immersion lens (Figure 1) and a confocal microscope with a 100× oil-immersion lens (Figure 2).
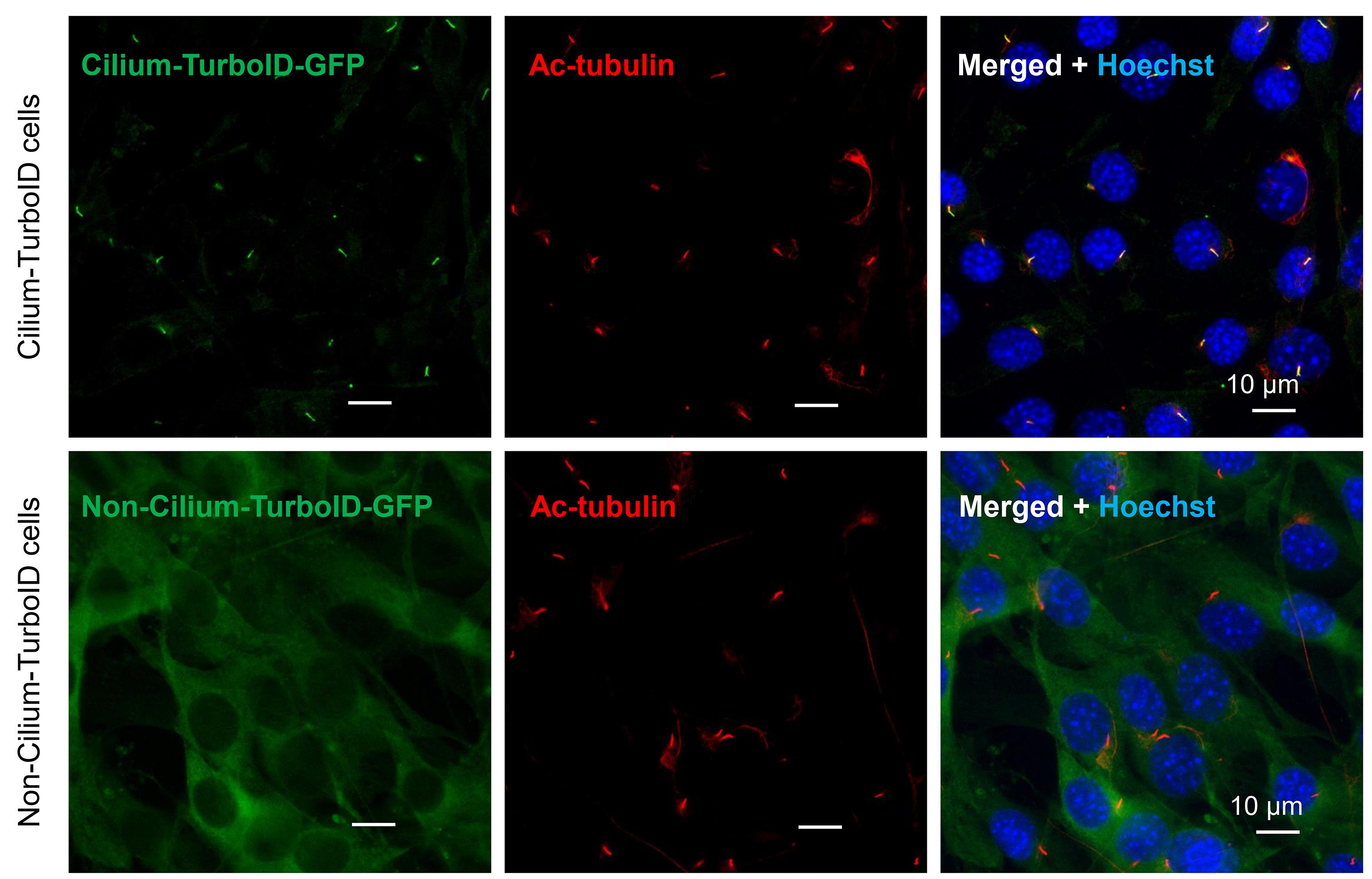
Figure 1. Immunostaining of cell colonies. Immunostaining of transgene-TurboID with anti-GFP antibody, cilia with anti-Ac-tubulin antibody, and nuclei with Hoechst in cilium-TurboID and non-cilium-TurboID cells.
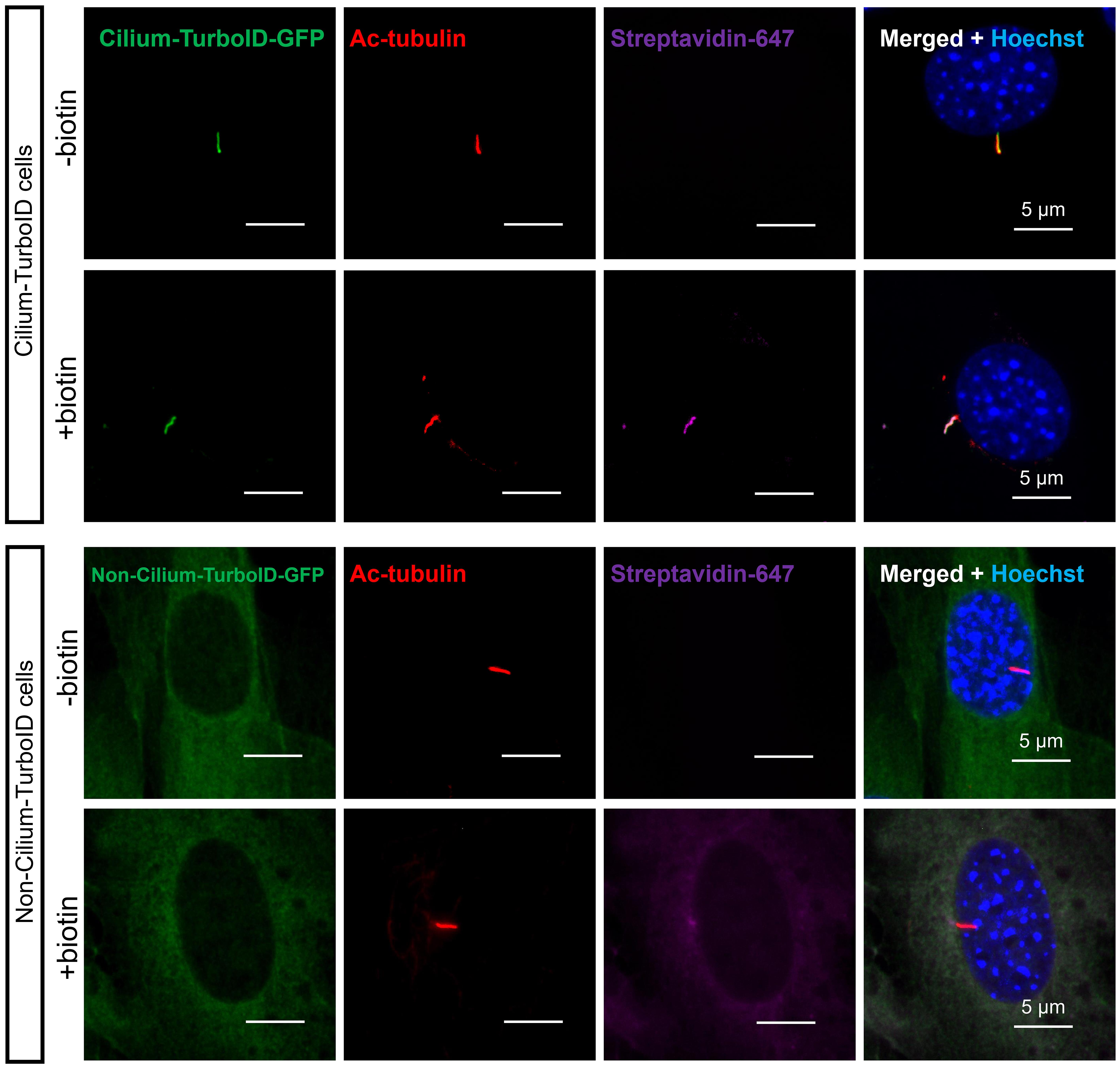
Figure 2. Validation of TurboID stable cell lines before and after biotin labeling by immunofluorescence (IF). Cells were serum-starved for 24 h, incubated with 50 µM biotin for 10 min at 37 °C, and fixed for immunostaining. The cilium was labeled by immunostaining for acetylated-tubulin (red, ac-tubulin), and biotinylated proteins were detected via streptavidin-Alexa Fluor 647 (magenta). Cilium-TurboID and non-cilium TurboID were detected by immunostaining for GFP (green).
C. Validation of TurboID stable cell lines by streptavidin pulldown and western blotting (WB)
1. Seed 4 × 106 cells in a 10 cm dish and culture cells in DMEM+10% CS medium to achieve 95% confluency.
2. Change media to 10 mL of serum-starving DMEM+0.5% CS and incubate for 24 h at 37 °C in a 5% CO2 incubator.
3. After 24 h starvation, change the medium to biotin labeling medium for 10 min.
Note: Prewarm the biotin labeling medium to 37 °C. It is recommended to include a dish that is not labeled with biotin to serve as a control.
4. Rinse cells three times with ice-cold DPBS, 5 min each time.
5. Add 10 mL of ice-cold DPBS to one 10 cm dish and scrape cells with a cell scraper into a microcentrifuge tube.
Note: Keep samples on ice throughout the procedure. Use the protein LoBind tubes.
6. Centrifuge at 150× g for 5 min at 4 °C and aspirate supernatant.
7. Add 400 µL of subcellular fractionation buffer (containing 1× protease inhibitor cocktail buffer) to the protein LoBind tube containing the cell pellet and incubate for 15 min on ice.
Note: Use 400 µL of subcellular fractionation buffer for one 10 cm cell dish.
8. Using a 1 mL syringe, pass the cell suspension through a 27-gauge needle 10 times (or until all cells are lysed). Leave it on ice for 20 min.
Note: To check if cells are lysed, stain the samples with trypan blue. Damaged cells appear blue under a microscope, while live cells with intact membranes remain unstained.
9. Centrifuge sample at 720× g for 5 min. Transfer the supernatant into a fresh tube and keep it on ice.
Note: The pellet contains nuclei, and the supernatant contains cytoplasm, membranes, and mitochondria. You can store samples at -80 °C.
10. Add ice-cold 10× TBS, 10% NP-40, and 1% SDS to the supernatant until the final concentration reaches 1× TBS, 0.5% NP-40, and 0.1% SDS. Pipette up and down several times to mix.
11. Sonicate the supernatant using a sonicator in a bath of ice water 5–10 times at a cycle of 3 s on and 10 s off.
Note: Use 50% amplitude.
12. Centrifuge at 10,000× g for 15 min at 4 °C and keep the supernatant on ice.
13. Test the protein concentration using a BCA Protein Assay kit.
Note: Dilute samples 2, 5, and 10 times to fit the protein concentration into the range of the standard curve. Typically, one 10 cm dish of NIH 3T3 cells produces 0.5 mg of protein.
14. Normalize all samples to the same protein amount and take out 50 µg as input.
15. Pre-washing streptavidin magnetic beads:
a. Add 15 µL (0.15 mg) of streptavidin magnetic bead slurry into a 1.5 mL microcentrifuge tube.
Note: To ensure homogeneity, mix the beads thoroughly before use by repeated inversion, gentle vortexing, or by using a rotating platform. Use the protein LoBind tubes.
b. Place the tube into a magnetic stand to collect the beads against the side of the tube. Remove and discard the supernatant.
Note: Do not allow beads to dry.
c. Add 1 mL of 1× TBS buffer to the tube. Invert the tube several times or vortex gently to mix. Collect the beads with a magnetic stand, then remove and discard the supernatant. Repeat this one more time.
Note: Do not allow beads to dry. Add TBS buffer immediately after removing the supernatant. Store beads in TBS buffer before proceeding with the samples.
16. Remove the TBS buffer, add the protein samples immediately to a microcentrifuge tube containing pre-washed magnetic beads, and incubate at room temperature for 2 h or at 4 °C overnight with rotation.
17. Collect the magnetic beads with a magnetic stand and remove the supernatant.
18. Wash two times with TEAB buffer, once with 1 M KCl, once with 0.1 M Na2CO3, once with 2 M urea buffer, twice with TEAB buffer, and three times with DPBS.
Note: For each washing, invert the tube several times or vortex gently to mix. Collect the beads with a magnetic stand, then remove and discard the supernatant. After the final wash, remove the remaining DPBS as much as possible.
19. Add 50 µL of 1× TBS and 13 µL of 6× Laemmli sample buffer to the microcentrifuge tube containing the washed magnetic beads and mix well. Boil for 5–10 min at 90–95 °C and then cool down immediately by placing on ice. Collect the supernatant by placing the tube into a magnetic stand and transfer the supernatant to a new 1.5 mL tube.
20. Run the 10% SDS-polyacrylamide gel electrophoresis and detect proteins with specific antibodies (Figure 3).
Notes:
1. Begin with a voltage of 100 V and increase it to 120 V when the samples are in the separating gel. Run for 1–2 h until the blue dye reaches the bottom of the gel.
2. Dilute the antibodies and streptavidin-HRP using the specified ratios: rabbit anti-GFP (1:2,000), mouse anti-GAPDH (1:2,000), HRP-conjugated donkey anti-rabbit IgG (1:10,000), HRP-conjugated donkey anti-mouse IgG (1:10,000), and HRP-conjugated streptavidin (1:10,000).
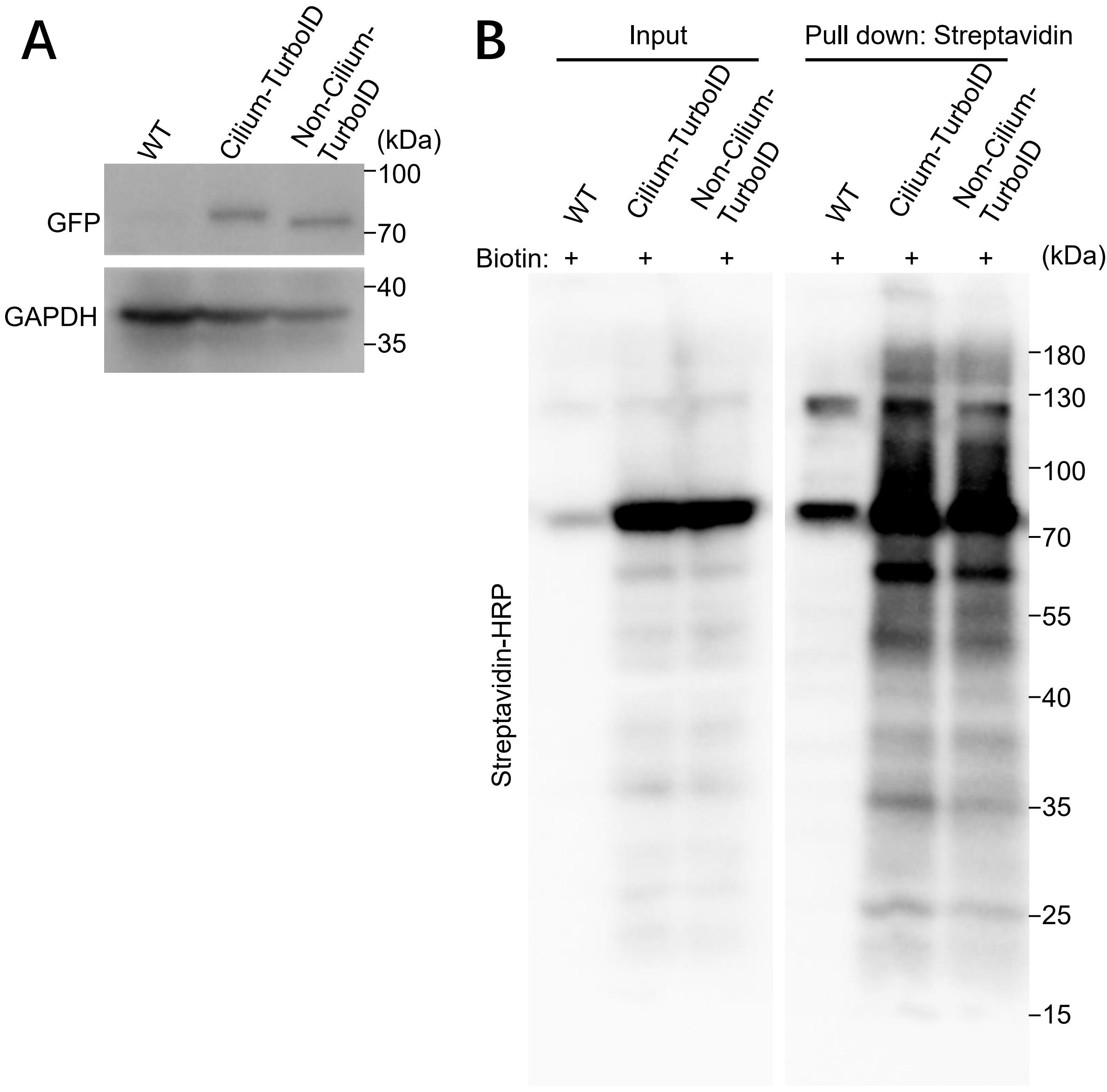
Figure 3. Validation of TurboID stable cell lines by western blotting (WB) and streptavidin pull down. (A) Detection of transgene-TurboID by western blotting. The bands at 75 kDa represent the transgene. (B) Biotinylated proteins from cell lysates of cilium-TurboID and non-cilium-TurboID cells were isolated by streptavidin beads and analyzed by WB. The overall biotinylated proteins were detected by streptavidin-HRP.
D. Cell lysis and affinity purification for mass spectrometry analysis
For one replicate of each condition (cilium-TurboID with biotin labeling, cilium-TurboID without biotin labeling, and non-cilium-TurboID with biotin labeling), we harvested fifteen 15 cm dishes of cells. The following steps are used for one 15 cm cell dish.
Note: To control for background biotinylation, we include a condition omitting biotin labeling. To control for non-ciliary protein biotinylation by TurboID, we collected samples from labeled non-cilium-TurboID cells.
1. Seed cells in 15 cm dishes and culture cells in DMEM+10% CS medium to achieve 95% confluency.
2. Change media to 30 mL of serum-starving DMEM+0.5% CS and incubate for 24 h at 37 °C in a 5% CO2 incubator.
3. After 24 h starvation, change the medium to biotin labeling medium for 10 min.
Note: Prewarm the biotin labeling medium to 37 °C.
4. Rinse cells three times with ice-cold DPBS, 5 min each time.
5. Add 15 mL of ice-cold DPBS to the 15 cm dish and scrape cells with the cell scraper into a microcentrifuge tube.
Note: Keep samples on ice throughout the procedure. Use the protein LoBind tubes.
6. Centrifuge at 150× g for 5 min at 4 °C and aspirate supernatant.
7. Add 700 µL of subcellular fractionation buffer (containing 1× protease inhibitor cocktail buffer) to the protein LoBind tube containing the cell pellet and incubate for 15 min on ice.
Note: Use 700 µL of subcellular fractionation buffer for one 15 cm cell dish.
8. Using a 1 mL syringe, pass the cell suspension through a 27-gauge needle 10 times (or until all cells are lysed). Leave it on ice for 20 min.
9. Centrifuge the sample at 720× g for 5 min. Transfer the supernatant into a fresh tube and keep it on ice.
Note: The pellet contains nuclei, and the supernatant contains cytoplasm, membranes, and mitochondria. You can store samples at -80 °C.
10. Add ice-cold 10× TBS, 10% NP-40, and 1% SDS to the supernatant until the final concentration reaches 1× TBS, 0.5% NP-40, and 0.1% SDS. Pipette up and down several times to mix.
11. Sonicate the supernatant using a sonicator in a bath of ice water 5–10 times at a cycle of 3 s on and 10 s off.
Note: Use 50% amplitude.
12. Centrifuge at 10,000× g for 15 min at 4 °C and keep the supernatant on ice.
13. Test the protein concentration using the BCA Protein Assay kit.
Note: Dilute samples 2, 5, and 10 times to fit the protein concentration into the range of the standard curve.
14. Normalize all samples to the same protein amount.
15. Pre-washing Streptavidin magnetic beads:
a. Add 35 µL (0.35 mg) of Streptavidin magnetic bead slurry into a 1.5 mL microcentrifuge tube.
Note: To ensure homogeneity, mix the beads thoroughly before use by repeated inversion, gentle vortexing, or by using a rotating platform. Use the protein LoBind tubes.
b. Place the tube into a magnetic stand to collect the beads against the side of the tube. Remove and discard the supernatant.
Note: Do not allow beads to dry.
c. Add 1 mL of 1× TBS buffer to the tube. Invert the tube several times or vortex gently to mix. Collect the beads with a magnetic stand, then remove and discard the supernatant. Repeat this one more time.
Note: Do not allow beads to dry. Add TBS buffer immediately after removing the supernatant. Store beads in TBS buffer before proceeding with the samples.
16. Remove the TBS buffer, immediately add the protein samples to a microcentrifuge tube containing pre-washed magnetic beads, and incubate at room temperature for 2 h with rotation.
17. Collect the magnetic beads with a magnetic stand and remove the supernatant.
18. Wash two times with TEAB buffer, once with 1 M KCl, once with 0.1 M Na2CO3, once with 2 M urea buffer, twice with TEAB buffer, and three times with DPBS.
Note: For each washing, invert the tube several times or vortex gently to mix. Collect the beads with a magnetic stand, then remove and discard the supernatant. After the final washing, remove the remaining DPBS as much as possible.
19. Store the microcentrifuge tube containing the washed magnetic beads at -80 °C.
E. On-beads trypsin digestion of biotinylated proteins and TMT labeling
We sent magnetic beads with bound proteins to Stanford University’s Mass Spectrometry Center (https://mass-spec.stanford.edu/) and performed mass spectrometry and MS data processing [20].
Briefly, beads with bound proteins were reduced, alkylated, and digested. The digested peptides were further quantified and labeled with 16plex TMT label reagent set. Tagged peptides were then pooled to generate a multiplexed sample and fractionated [20].
F. Mass spectrometry
Mass spectrometry was performed in Stanford University’s Mass Spectrometry Center. MS experiments were performed using liquid chromatography (LC) with an Acquity M-Class UPLC (Waters) followed by MS using an Orbitrap Fusion Tribrid MS (Thermo Scientific) [20].
G. MS data processing
MS data processing was performed in Stanford University’s Mass Spectrometry Center. Briefly, RAW data were checked using Preview (Protein Metrics) to verify calibration and quality prior to further analysis. Data were then processed using Proteome Discoverer v1.5 (Thermo) for quantitative analysis of reporter ion ratios and the Byonic (Protein Metrics) node to identify peptides and infer proteins against the Mus musculus database from Uniprot (including isoforms and concatenated with common contaminant proteins) [20].
H. MS data analysis
For data analysis, we defined the relative protein abundance as the ratio of abundance in each channel over that in the reference channel. Only proteins with more than seven unique peptides were used in data analysis. We then determined candidate proteins via statistical analysis of the relative enrichment between the cilium-TurboID and the non-cilium-TurboID datasets. To be scored as ciliary proteins, candidates had to fulfill four criteria: 1) greater than 2-fold enrichment (TMT ratio >2) in the labeled cilium-TurboID samples over the non-labeled cilium-TurboID samples and 2) over the labeled non-cilium-TurboID samples; 3) statistically significant (p-value < 0.05) enrichment in the labeled cilium-TurboID samples vs. the non-labeled cilium-TurboID samples and 4) vs. the labeled non-cilium-TurboID samples [20].
Validation of protocol
This protocol has been used and validated in the following research article:
• Liu et al. [20]. Numb positively regulates Hedgehog signaling at the ciliary pocket. Nature Communications. 15, 3365 (2024). (Figures 1 and 2 and Supplementary figures 1–4)
General notes and troubleshooting
Troubleshooting
Problem 1: Failure to package lentivirus.
Possible cause: The plasmid quality might be poor. 293T cell viability might be low or 293T cells are contaminated with mycoplasma.
Solution: Use endotoxin-free plasmid purification kits to extract plasmids. Minimize the plasmid damage caused by repeated freeze-thaw cycles. Use low-passaged 293T cells. Eliminate mycoplasma from 293T cells before packaging lentivirus.
Problem 2: The stable cell line grows slowly.
Possible cause: Cell viability might be low, or cells are contaminated with mycoplasma. Transgene might be integrated into the genome in a way that disrupts normal gene function.
Solution: Use low-passaged cells. Add antibiotics (such as plasmocin) and eliminate mycoplasma from stable cells. Select a few additional cell lines and retain the best one.
Problem 3: Cells detach from the coverslip when performing IF.
Possible cause: Cell density might be slightly too high. Coverslips might float up when coated with Poly-D-lysine.
Solution: Seed fewer cells on coverslips or starve cells before they contact each other. After the addition of Poly-D-lysine on the coverslips, gently poke the coverslips with a 1 mL tip and repeat this after 30 min incubation to improve the coating condition.
Problem 4: High background and faint streptavidin-HRP signal for WB.
Possible cause: Cells were labeled for too long with biotin. Too much biotin was used or left after washing with DPBS. Too much streptavidin-HRP was used.
Solution: Optimize the labeling time and biotin concentration. After labeling, wash cells thoroughly and remove free biotin as much as possible. Use less streptavidin-HRP.
Acknowledgments
We thank all authors from the corresponding original research paper in which this protocol was used: Nature Communications [20]. This work was supported by the National Institutes of Health (NIH)/National Cancer Institute (NCI) (CA235749), NIH/National Institute of General Medical Sciences (GM143276), NIH/NCI (CA274595), and National Science Foundation CAREER award (IOS-2143711).
Author contributions: X. Liu: writing–original draft, review, and editing. X. Ge: conceptualization and writing–original draft, review, and editing.
Competing interests
The authors declare no conflicts of interest.
References
- Branon, T. C., Bosch, J. A., Sanchez, A. D., Udeshi, N. D., Svinkina, T., Carr, S. A., Feldman, J. L., Perrimon, N. and Ting, A. Y. (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol. 36(9): 880–887. https://doi.org/10.1038/nbt.4201
- Delling, M., DeCaen, P. G., Doerner, J. F., Febvay, S. and Clapham, D. E. (2013). Primary cilia are specialized calcium signalling organelles. Nature. 504(7479): 311–314. https://doi.org/10.1038/nature12833
- Mukhopadhyay, S., Badgandi, H. B., Hwang, S. H., Somatilaka, B., Shimada, I. S. and Pal, K. (2017). Trafficking to the primary cilium membrane. Mol Biol Cell. 28(2): 233–239. https://doi.org/10.1091/mbc.E16-07-0505
- Singla, V. and Reiter, J. F. (2006). The primary cilium as the cell's antenna: signaling at a sensory organelle. Science. 313(5787): 629–633. https://doi.org/10.1126/science.1124534
- Eggenschwiler, J. T. and Anderson, K. V. (2007). Cilia and developmental signaling. Annu Rev Cell Dev Biol. 23: 345–373. https://doi.org/10.1146/annurev.cellbio.23.090506.123249
- Reiter, J. F. and Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 18(9): 533–547. https://doi.org/10.1038/nrm.2017.60
- Liu, Q., Tan, G., Levenkova, N., Li, T., Pugh, E. N., Jr., Rux, J. J., Speicher, D. W. and Pierce, E. A. (2007). The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics. 6(8): 1299–1317. https://doi.org/10.1074/mcp.M700054-MCP200
- Mayer, U., Ungerer, N., Klimmeck, D., Warnken, U., Schnolzer, M., Frings, S. and Mohrlen, F. (2008). Proteomic analysis of a membrane preparation from rat olfactory sensory cilia. Chem Senses. 33(2): 145–162. https://doi.org/10.1093/chemse/bjm073
- Mayer, U., Kuller, A., Daiber, P. C., Neudorf, I., Warnken, U., Schnolzer, M., Frings, S. and Mohrlen, F. (2009). The proteome of rat olfactory sensory cilia. Proteomics. 9(2): 322–334. https://doi.org/10.1002/pmic.200800149
- Ishikawa, H., Thompson, J., Yates, J. R., 3rd and Marshall, W. F. (2012). Proteomic analysis of mammalian primary cilia. Curr Biol. 22(5): 414–419. https://doi.org/10.1016/j.cub.2012.01.031
- Mick, D. U., Rodrigues, R. B., Leib, R. D., Adams, C. M., Chien, A. S., Gygi, S. P. and Nachury, M. V. (2015). Proteomics of Primary Cilia by Proximity Labeling. Dev Cell. 35(4): 497–512. https://doi.org/10.1016/j.devcel.2015.10.015
- Mill, P., Christensen, S. T. and Pedersen, L. B. (2023). Primary cilia as dynamic and diverse signalling hubs in development and disease. Nat Rev Genet. 24(7): 421–441. https://doi.org/10.1038/s41576-023-00587-9
- Kohli, P., Hohne, M., Jungst, C., Bertsch, S., Ebert, L. K., Schauss, A. C., Benzing, T., Rinschen, M. M. and Schermer, B. (2017). The ciliary membrane-associated proteome reveals actin-binding proteins as key components of cilia. EMBO Rep. 18(9): 1521–1535. https://doi.org/10.15252/embr.201643846
- May, E. A., Kalocsay, M., D'Auriac, I. G., Schuster, P. S., Gygi, S. P., Nachury, M. V. and Mick, D. U. (2021). Time-resolved proteomics profiling of the ciliary Hedgehog response. J Cell Biol. 220(5): e202007207. https://doi.org/10.1083/jcb.202007207
- Gupta, G. D., Coyaud, E., Goncalves, J., Mojarad, B. A., Liu, Y., Wu, Q., Gheiratmand, L., Comartin, D., Tkach, J. M., Cheung, S. W., et al. (2015). A Dynamic Protein Interaction Landscape of the Human Centrosome-Cilium Interface. Cell. 163(6): 1484–1499. https://doi.org/10.1016/j.cell.2015.10.065
- Bozal-Basterra, L., Gonzalez-Santamarta, M., Muratore, V., Bermejo-Arteagabeitia, A., Da Fonseca, C., Barroso-Gomila, O., Azkargorta, M., Iloro, I., Pampliega, O., Andrade, R., et al. (2020). LUZP1, a novel regulator of primary cilia and the actin cytoskeleton, is a contributing factor in Townes-Brocks Syndrome. eLife. 9: e55957. https://doi.org/10.7554/eLife.55957
- Hung, V., Udeshi, N. D., Lam, S. S., Loh, K. H., Cox, K. J., Pedram, K., Carr, S. A. and Ting, A. Y. (2016). Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc. 11(3): 456–475. https://doi.org/10.1038/nprot.2016.018
- Tan, B., Peng, S., Yatim, S., Gunaratne, J., Hunziker, W. and Ludwig, A. (2020). An Optimized Protocol for Proximity Biotinylation in Confluent Epithelial Cell Cultures Using the Peroxidase APEX2. STAR Protoc. 1(2): 100074. https://doi.org/10.1016/j.xpro.2020.100074
- Trinkle-Mulcahy, L. (2019). Recent advances in proximity-based labeling methods for interactome mapping. F1000Res. 8. https://doi.org/10.12688/f1000research.16903.1
- Liu, X., Yam, P. T., Schlienger, S., Cai, E., Zhang, J., Chen, W. J., Torres Gutierrez, O., Jimenez Amilburu, V., Ramamurthy, V., Ting, A. Y., et al. (2024). Numb positively regulates Hedgehog signaling at the ciliary pocket. Nat Commun. 15(1): 3365. https://doi.org/10.1038/s41467-024-47244-1
Article Information
Publication history
Received: Jan 29, 2025
Accepted: Apr 11, 2025
Available online: Apr 23, 2025
Published: May 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Liu, X. and Ge, X. (2025). TurboID Labeling and Analysis of Proteins in the Primary Cilium. Bio-protocol 15(9): e5303. DOI: 10.21769/BioProtoc.5303.
Category
Cell Biology > Cell structure > Primary cilium
Biochemistry > Protein > Labeling
Systems Biology > Proteomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.