- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Cardiac-Specific Gene Editing via an AAV9-Tnnt2-SaCas9-miR122TS Vector
(*contributed equally to this work) Published: Vol 15, Iss 5, Mar 5, 2025 DOI: 10.21769/BioProtoc.5235 Views: 2725
Reviewed by: Xiaokang WuPooja MukherjeeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2024
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The adeno-associated virus serotype 9 (AAV9)-delivered gene expression driven by the cardiac troponin T (Tnnt2) promoter is broadly considered to be cardiac-specific. However, in cases where low AAV expression is sufficient to trigger a profound biological effect in CRISPR/Cas9 gene editing, the ectopic AAV9-Tnnt2 expression and gene editing in the liver becomes non-negligible. MicroRNA122 is a microRNA that is specifically expressed in the liver. The incorporation of the microRNA122 target sequence (miR122TS) into the 3' untranslated region (UTR) of the AAV transgene could reduce ectopic gene expression in the liver. Here, we provide a protocol for sgRNA design, plasmid construction, AAV packaging, and in vivo validation of a new AAV9-Tnnt2-SaCas9-miR122TS vector using publicly available materials and tools. The application of this new vector enables cardiac-specific gene editing while circumventing leakages in the liver.
Key features
• This protocol describes a detailed procedure to construct and validate AAV-based cardiac-specific gene editing in mice.
• MicroRNA-122 target sequences (miR122TS) in combination with a Tnnt2 promoter are used to enhance the cardiac specificity in genome editing.
• Amplicon sequencing analysis is applied to precisely and sensitively quantify the genome editing efficiency and tissue specificity in mice.
Keywords: Adeno-associated virusGraphical overview
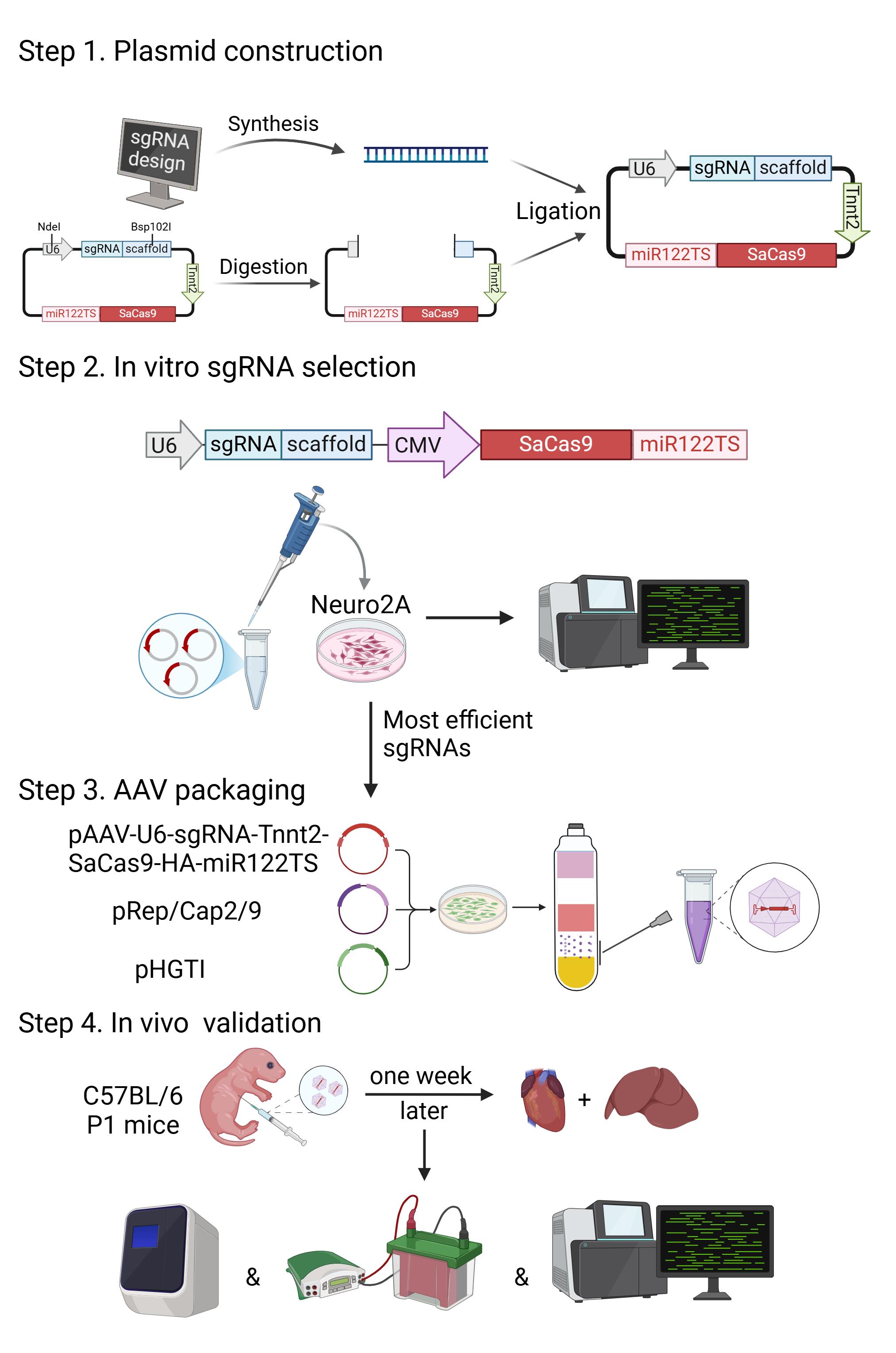
Construction and validation of AAV9-Tnnt2-SaCas9-miR122TS-based cardiac-specific gene editing. Step 1: In silico sgRNA design and plasmid construction. Step 2: sgRNA selection via gene editing in Neuro2A cells. Step 3: AAV packaging. Step 4: Validation of adeno-associated virus (AAV) expression, gene editing, and gene expression in murine tissues. P1, postnatal day 1. Graphics were generated with biorender.com.
Background
Adeno-associated virus (AAV) is a major gene delivery vector in basic and translational cardiology [1]. When a cardiac-specific promoter, such as the Tnnt2 promoter, is incorporated to drive gene expression via a cardiac tropic AAV capsid like AAV9, this technique is broadly believed to be cardiac-specific [2]. However, with increasingly favored AAV applications that could work under low expression levels, such as the Cre-LoxP and CRISPR/Cas9 genetic manipulation, the leaky expression of AAV9-Tnnt2 vector in the liver becomes non-negligible [3]. This caveat could potentially lead to data misinterpretation due to assumed cardiac specificity by AAV9-Tnnt2 vectors.
MicroRNAs are small non-coding RNAs that regulate gene expression at the post-transcriptional level. One major mechanism underlying the function of microRNA involves recognition and binding to complementary target sequences in mRNA [4]. This action leads to targeted mRNA cleavage followed by degradation and thereby gene silencing. Many microRNAs are expressed in a tissue-specific or cell type–specific manner and regulate key pathophysiological functions, which have been utilized to modulate exogenous transgene expression in a gene delivery system [5].
MicroRNA-122 is the most abundant microRNA that is specifically expressed in the liver [6]. Incorporation of microRNA-122 target sequence (miR122TS) in mRNA was known to exhibit a robust liver de-targeting effect on AAV-based gene transfer [7,8]. However, with the wide application of cardiac-specific promoters, the necessity of adding miR122TS into cardiac-targeted AAV vectors was underestimated. Recently, we showed that miR122TS was both necessary and sufficient to avoid hepatic leaky expression by the AAV9-Tnnt2 vector, which was of particular importance in Cre-LoxP and CRISPR/Cas9 applications that could work at a very low expression level [3]. Here, we describe how to design, construct, and validate an AAV9-Tnnt2-SaCas9-miR122TS vector to achieve cardiac-specific genome editing in mice with publicly available materials. Similar strategies could be applied to other efforts in cardiac-specific AAV gene transfer.
Materials and reagents
Animals
Studies described here were in conformity with the protocol authorized by the IACUC of Peking University with the approval number DLASBD0203. C57BL/6 mice were obtained from the Department of Laboratory Animal Science of Peking University Health Science Center.
For AAV injection, animals were anesthetized by isoflurane (RWD Life Science, catalog number: R510-22). Then, a 29G-needle insulin syringe (BD Ultra-FineTM, catalog number: 320312) was used to inject AAV via either subcutaneous or venous routes.
Plasmids
1. pAAV-U6-sgRNA-Tnnt2-SaCas9-HA-miR122TS (AAV vector plasmid) (Addgene, #209783)
2. pHGTI (AAV packaging helper plasmid) (Addgene, #112867)
3. pRepCap2/9 (AAV replication and capsid plasmid) (Addgene, #112865)
4. pAAV-CMV-GFP-LA (Addgene, #206197)
Cell lines
1. HEK293T cell (Wuhan Pricella Biotechnology, CL-0005)
2. Neuro2A cell (Wuhan Pricella Biotechnology, CL-0168)
Reagents
For plasmid construction
1. LB broth (Sangon Biotech, catalog number: A507002)
2. Ampicillin (Sangon Biotech, catalog number: B541011)
3. TIANprep Mini Plasmid kit (Tiangen, catalog number: DP103)
4. Stbl4 competent cells (Acmec, catalog number: AC10839-20×100 μL)
5. EndoFree Maxi Plasmid kit (Tiangen, catalog number: GDP117)
6. Genomic DNA extraction kit (Tiangen, catalog number: DP304)
7. E.Z.N.ATM Endo-Free Plasmid Giga kit (Omega, catalog number: D6234-01)
8. NdeI (Transgene, catalog number: JN201-01)
9. Bsp120I (Thermo, catalog number: ER0131)
10. Seamless Cloning kit (Sangon, catalog number: B632219)
11. 2× Taq DNA polymerase (Tiangen, catalog number: ET101)
12. 10× rCutsmart buffer (New England Biolabs, catalog number: B6004V)
For cell culture
1. DMEM (Gibco, catalog number: C11995500BT)
2. Fetal bovine serum (FBS) (Vistech, catalog number: SE100-B)
3. Penicillin-streptomycin (P/S) (TransGen, catalog number: FG101-01)
4. Phosphate-buffered saline (PBS) (FreeMoreBio, catalog number: FM58031)
5. 0.25% Trypsin-EDTA (Macgene, catalog number: CC017)
6. Opti-MEM (Cellworld, catalog number: c2656-862)
7. Polyetherimide (PEI), 1 mg/mL (Yeasen, catalog number: 40816ES03)
8. Lipo8000 transfection reagent (Beyotime, catalog number: C0533)
For AAV purification and analysis
1. Optiseal ultracentrifuge tube (Beckman, catalog number: 361625)
2. Amicon ultra centrifugal filter, 100 kDa MWCO (Millipore, catalog number: UFC9100)
3. Luer lock syringe (DengMiBio, catalog number: 302149)
4. Polyethylene glycol 8000 (PEG 8000) (Yeasen, catalog number: 60304ES76)
5. NaCl (HarveyBio, catalog number: SR5029)
6. 1 M Tris-HCl pH 8.0 (Yuanye, catalog number: R21105-500mL)
7. MgCl2 (HarveyBio, catalog number: SR4155)
8. KCl (HarveyBio, catalog number: SR1743)
9. 10× PBS (HarveyBio, catalog number: CT0035042)
10. Optiprep (Gibco, catalog number: 31985062)
11. Phenol red (Yuanye, catalog number: R22048)
12. Benzonase (Yeasen, catalog number: 20156ES60)
13. PluronicTM F-68 (Gibco, catalog number: 24040032)
14. DNase I (NEB, catalog number: M0303S)
15. Proteinase K (Beyotime, catalog number: ST532)
16. 10× reaction buffer (NEB, catalog number: B0303S)
For amplicon sequencing
1. TIANamp Genomic DNA kit (Tiangen, catalog number: GDP304-03)
2. TansNGC Dual Index Primers kit for Illumina (TransGen Biotech, catalog number: KI231)
3. Agarose (GenStar, catalog number: VA10252)
4. 50× TAE DNA electrophoresis buffer (GenStar, catalog number: E102-50)
For RT-qPCR
1. TransZol Up Plus RNA kit (TransGen Biotech, catalog number: ER501-01)
2. Taq Pro Universal SYBR qPCR master mix (Vazyme, catalog number: Q712-02)
3. HiScript III All-in-one RT SuperMix perfect for qPCR (Vazyme, catalog number: R333-01)
For western blot
1. SurePAGETM, Bis-Tris, 4%–12% (GenScript, catalog number: M00654)
2. Tris-MOPS-SDS (Shanghai yuanye Bio-Technology, catalog number: R23170)
3. 4× SDS-PAGE loading buffer (Solarbio, catalog number: P1016)
4. InStabTM protease cocktail (Yeasen, catalog number: 20124ES03)
5. Tween 20 (Sigma-Aldrich, catalog number: STS0200)
6. 0.45 μm PVDF membrane (Merck, catalog number: IPVH00010)
7. CaMKII delta antibody (GeneTex, catalog number: GTX111401)
8. HA-Tag antibody (Cell Signaling Technology, catalog number: 3724)
9. GAPDH antibody (TransGen Biotech, catalog number: HC301-01)
10. HRP-conjugated goat anti-rabbit IgG (TransGen Biotech, catalog number: HC101-01)
11. HRP-conjugated goat anti-mouse IgG (TransGen Biotech, catalog number: HC201-01)
12. Super ECL detection reagent (Yeasen, catalog number: 36208ES76)
13. Methyl alcohol (TGREAG, catalog number: 105099)
Solutions
1. Escherichia coli culture medium (see Recipes)
2. Complete mammalian cell culture medium (see Recipes)
3. Serum-free cell culture medium (see Recipes)
4. AAV precipitation solution (see Recipes)
5. Cell lysis buffer (see Recipes)
6. Iodixanol gradient (see Recipes)
7. AAV washing buffer (see Recipes)
8. RIPA buffer (see Recipes)
9. 5× transfer buffer (see Recipes)
10. 10×TBS (see Recipes)
Recipes
1. Escherichia coli culture medium (1 L)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| LB broth | 25 g/L | 25 g |
| Ampicillin | 100 mg/mL | 1 mL* |
| ddH2O | n/a | up to 1 L |
*Note: LB broth dissolved in water should be sterilized via a pressure steam sterilizer. Ampicillin should be added after the solution has cooled down to 50–60 °C.
2. Complete mammalian cell culture medium (500 mL)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| DMEM | n/a | 445 mL |
| FBS | 10% (v/v) | 50 mL |
| P/S | 1% (v/v) | 5 mL |
3. Serum-free cell culture medium (500 mL)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| DMEM | n/a | 495 mL |
| P/S | 1% (v/v) | 5 mL |
4. AAV precipitation solution (1 L)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| PEG 8000 | 40% (w/v) | 400 g* |
| NaCl | 2.5 M | 146.25 g |
| ddH2O | n/a | up to 1 L |
*Note: PEG 8000 makes the large quantity of NaCl difficult to dissolve, thus put the mixture in a 37 °C water bath overnight or heat the mixture above 50 °C for 2 h.
5. Cell lysis buffer (50 mL)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl pH 8.0 | 20 mM | 1 mL |
| 5 M NaCl | 150 mM | 1.5 mL |
| 1 M MgCl2 | 1 mM | 50 μL |
| ddH2O | n/a | up to 50 mL |
Note: Filter the solution and store it at 4 °C.
6. Iodixanol gradient (50 mL)
| % of iodixanol | 10× PBS (mL) | 1 M MgCl2 (mL) | 5 M NaCl (mL) | 1 M KCl (mL) | Optiprep (mL) | Phenol red (mL) | ddH2O (mL) |
|---|---|---|---|---|---|---|---|
| 17 | 5 | 0.05 | 10 | 0.125 | 12.5 | 0 | 22.3 |
| 25 | 5 | 0.05 | 0 | 0.125 | 20 | 0.5 | 24.325 |
| 40 | 5 | 0.05 | 0 | 0.125 | 33.3 | 0 | 11.525 |
| 60 | 0 | 0.05 | 0 | 0.125 | 50 | 0.5 | 0 |
Note: Filter and store at 4 °C.
7. AAV washing buffer (500 mL)
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| PBS | n/a | 500 mL |
| F-68 | 0.001% | 50 μL |
Note: Filter and store at 4 °C.
8. RIPA buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 1 M Tris-HCl pH 8.0 | 25 mM | 1.25 mL |
| 5 M NaCl | 150 mM | 1.5 mL |
| 10% SDS | 0.1% | 500 μL |
| 10% sodium deoxycholate | 0.5% | 2.5 mL |
| 10% Triton X-100 | 1% | 5 mL |
| ddH2O | n/a | Up to 50 mL |
Note: Add Protease Cocktail at 1:100 before use. Sterilize with 0.22 μm filter and store at -20 °C.
9. 5× transfer buffer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Tris | 0.125 M | 15.14 g |
| Glycine | 1 M | 75.07 g |
| ddH2O | n/a | Up to 1 L |
Note: Dilute to 1× transfer buffer with ddH2O. Add methyl alcohol at 1:4 before use.
10. 10×TBS
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| NaCl | 1.37 M | 80 g |
| KCl | 27 mM | 2 g |
| Tris | 25 mM | 30 g |
| ddH2O | n/a | Up to 1 L |
Note: Dilute to 1× TBS with dd H2O. Add Tween 20 at 1:1,000 before use.
Equipment
1. Water bath (Bluepard, model: HWS-12)
2. Dry thermostat (Kylin-Bell, model: GL-1900)
3. Constant temperature incubator (Bluepard, model: DHP-9032B)
4. Shaking incubator (ZhiCheng, model: ZWYR-211C)
5. Vortex mixer (Qlinbeier, model: VORTEX-6)
6. Heating plate with magnetic stirrer (Weibosci, model: 3483949)
7. Benchtop centrifuge (SCILOGEX, model: SCI-24)
8. High-speed centrifuge (Thermo Fisher, model: Sorvall RC 6 plus, 46910)
9. Ultracentrifuge (Beckman, model: Optima XPN Series, A99846)
10. CO2 incubator (ESCO, model: CLM-170B-8-NF)
11. All-in-one fluorescence microscope (Keyence, model: BZ-X800)
12. Autoclave sterilizer (ZEALWAY, model: GR85SA)
13. Vertical electrophoresis tank (Junyi Electrophoresis, model: JY-SCZ2+)
14. Horizontal electrophoresis tank (Junyi Electrophoresis, model: JY-SPCT)
15. AriaMx Real-Time PCR system (Agilent Technologies, model: AriaMx)
16. PCR thermal cycler (LongGene, model: T30D)
Software and datasets
1. CRISPick (https://portals.broadinstitute.org/gppx/crispick/public) (for sgRNA design)
2. Benchling (https://www.benchling.com/) (for vector cloning and primer design)
3. CRISPresso2 (https://github.com/pinellolab/CRISPResso2) (for amplicon sequencing analysis)
Procedure
A. Candidate sgRNA design
1. Navigate to the online sgRNA design tool CRISPick.
2. To design sgRNA for mice, select Mouse GRCm38 in the Reference Genome section.
3. To knock out a given gene, select CRISPRko in the Mechanism section.
4. To design sgRNA for SaCas9, select SaurCas9 (NNGRR) in the Enzyme section.
5. To design sgRNA for Camk2d, choose the Quick Gene Search option in the Target section and enter the gene name “Camk2d.”
6. In the CRISPick Quota section, we typically choose to output the top 5 sgRNA. See Figure 1 for details about the parameter selection.
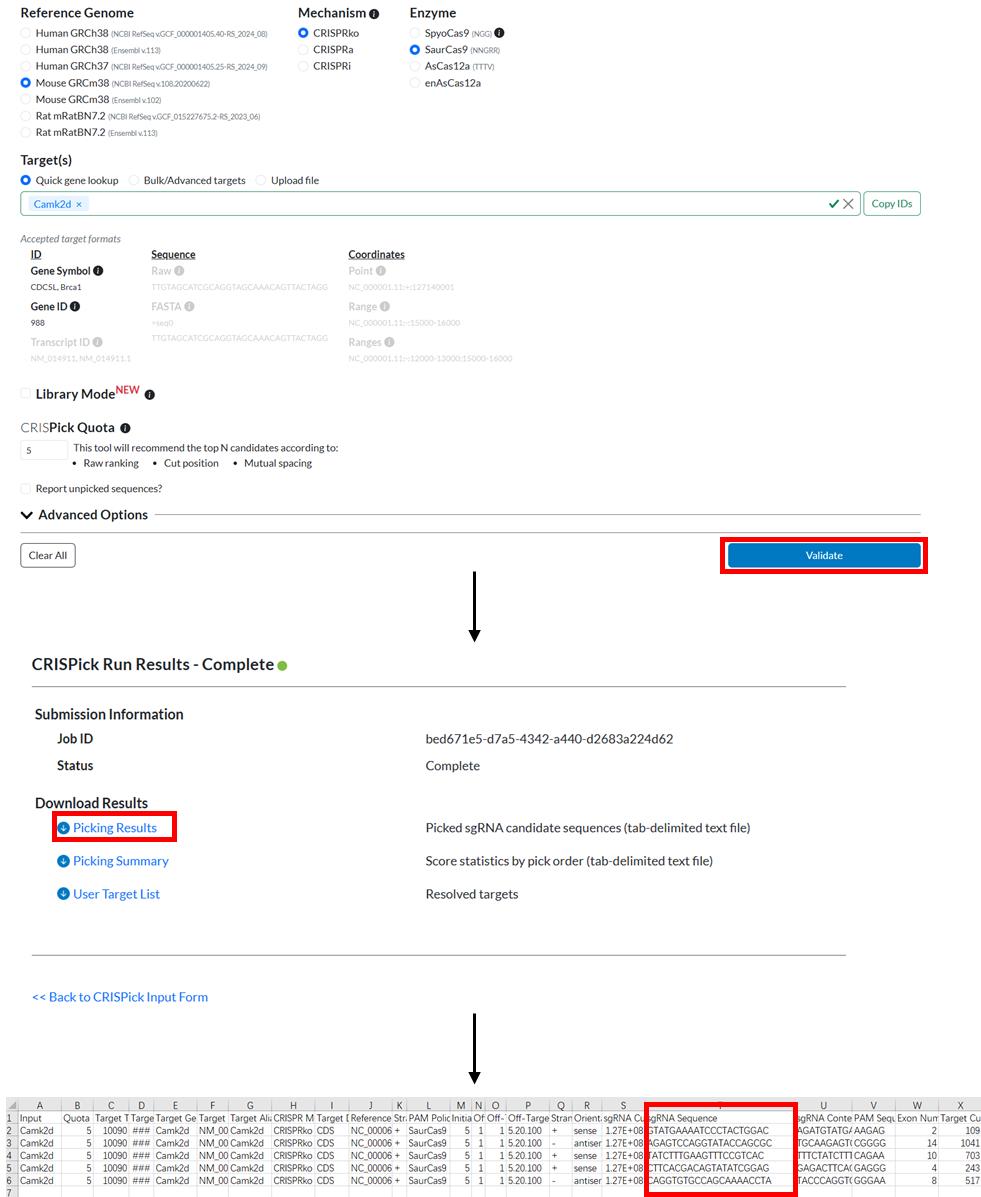
Figure 1. Workflow of an example of sgRNA design using CRISPick. SgRNA sequences are shown in the red box in the CSV file.
7. Select the four top sgRNAs with high on-target scores, low off-target scores, and targeting exons located at the N-terminal part of the protein domains.
Note: If the gene expresses alternative splicing variants, such as in the case of Camk2d, please ensure that the candidate sgRNAs target the shared exons by all splicing variants.
8. If the first base of the sgRNA is A, C, or T, add a G at the front as the transcription start site for the U6 promoter.
B. Vector construction
1. Digest the AAV-U6-sgRNA-Tnnt2-SaCas9-miR122TS plasmid with NdeI and Bsp120I to produce the vector ready for cloning with the designed insert DNA (Figure 2). Table 1 shows the digestion reaction mixtures and Table 2 shows the digestion reaction program.
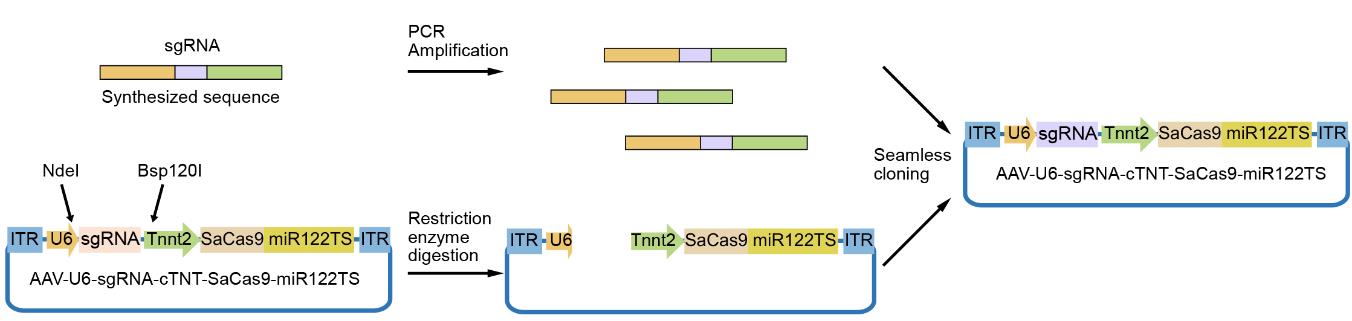
Figure 2. Workflow of sgRNA cloning into adeno-associated virus (AAV)-U6-sgRNA-Tnnt2-SaCas9-miR122TS backbone. The inserted DNA fragment is synthesized and amplified by PCR and then cloned into the vector between the NdeI and Bsp120I restriction digestion sites.
Table 1. Digestion reaction mixtures
| Reagent | Volume |
|---|---|
| DNA vector (AAV-U6-sgRNA-Tnnt2-SaCas9-miR122TS) | 3 μg |
| 10× rCutsmart buffer | 3 μL |
| NdeI | 0.5 μL |
| Bsp120I | 0.5 μL |
| Nuclease-free H2O | To 30 μL |
Table 2. Digestion reaction program.
| Step | Condition | Duration |
|---|---|---|
| Digestion | 37 °C | 2 h |
| Heat inactivation | 85 °C | 15 min |
| Hold | 4 °C | ∞ |
2. Synthesize the insert DNA with the specific sequence (Figure 3) as follows:
5'-TGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTT GGCTTTATATATCTTGTGGAAAGGACGAAACACC-[sgRNA sequence]-TTTTAGTAC TCTGGAAACAGAATCTACTAAAACAAGGCAAAATGCCGTGTTTATCTCGTCAACTTGTTGGCGAGATTTTTTTCTAGACGGGCCCCCCCTCGAGGTCGGGATAA-3'
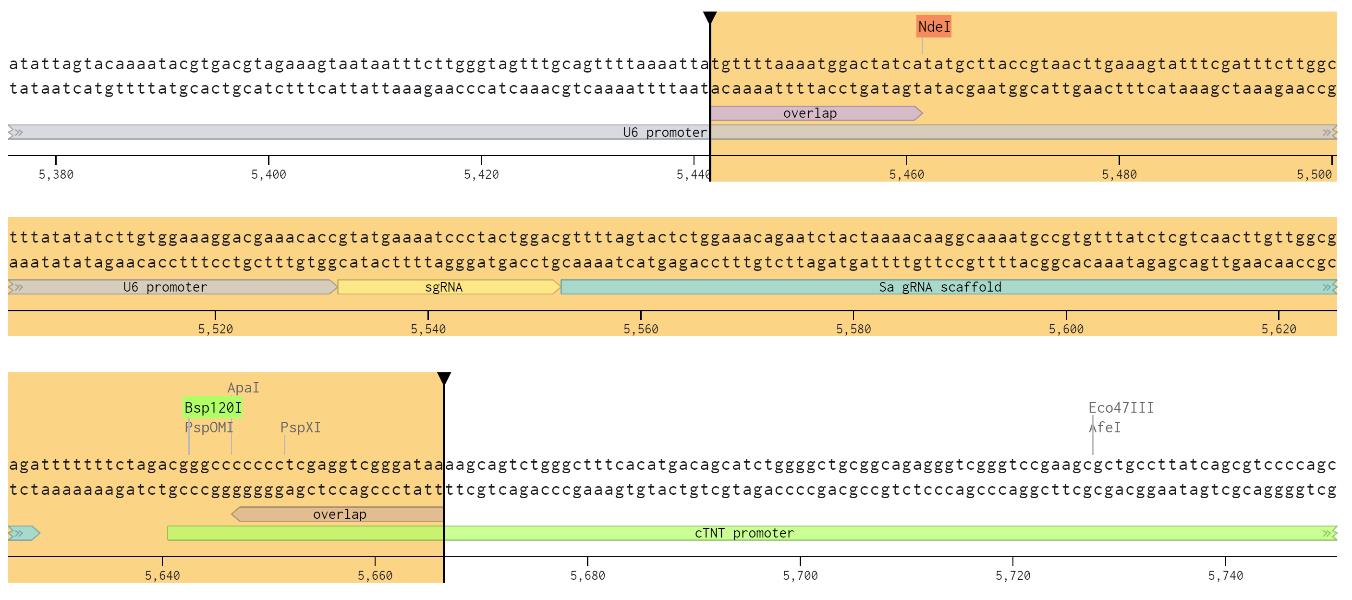
Figure 3. Benchling screenshot showing the inserted DNA and the restriction enzyme sites. A sgRNA for Camk2d is shown in the figure.
3. Use the Seamless Cloning kit to ligate the digested vector with the synthesized DNA sequence and transform the resultant plasmids into Stbl4 competent cells. Table 3 shows the seamless cloning reaction mixtures.
Table 3. Seamless cloning reaction mixtures
| Reagent | Volume |
|---|---|
| Digested vector | 50 ng |
| Synthesized DNA sequence | 1.6 ng |
| 2× Seamless cloning enzyme | 5 μL |
| Nuclease-free H2O | To 10 μL |
5. The reaction condition for seamless cloning is 50 °C for 15 min. After the reaction, take 5 μL of the recombinant plasmid from the system and add it to 50 μL of Stbl4 competent cells. Then, proceed with transformation (Table 4):
Table 4. Transformation program
| Step | Condition | Duration |
|---|---|---|
| Ice bath | 4 °C | 30 min |
| Heat shock | 42 °C | 90 s |
| Resuscitation | 37 °C, 200 rpm | 60 min |
6. Perform Sanger sequencing to validate the cloned plasmids.
7. The plasmids expressing the designed sgRNAs are usually tested in Neuro2A cells to select the sgRNA with the highest editing activity. Thus, the Tnnt2 promoter needs to be replaced with the CMV promoter using the molecular cloning strategy shown in Figure 4.
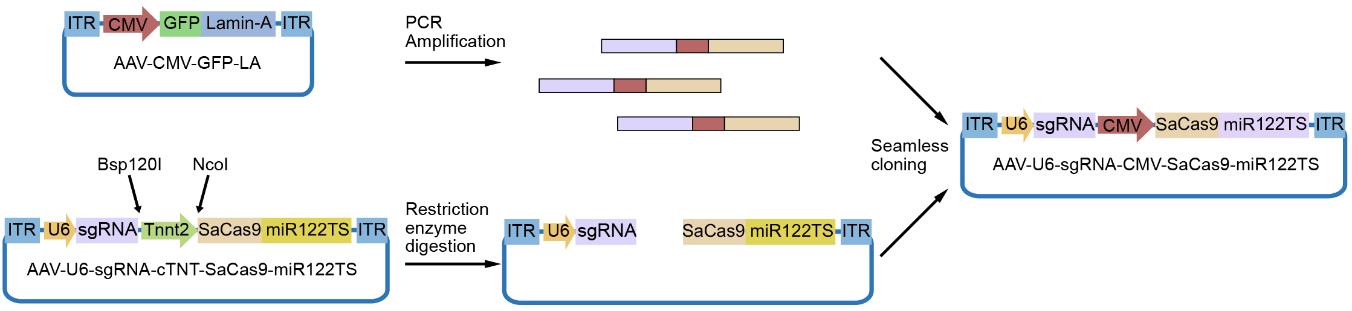
Figure 4. Workflow of plasmid cloning replacing the Tnnt2 promoter by the CMV promoter in the adeno-associated virus (AAV)-U6-sgRNA-Tnnt2-SaCas9-miR122TS plasmid. The CMV promoter fragment is amplified from the pAAV-CMV-GFP-LA plasmid and inserted into the AAV-U6-sgRNA-Tnnt2-SaCas9-miR122TS plasmid after Bsp120I and NcoI digestion.
8. The Tnnt2 promoter in the AAV-U6-sgRNA-Tnnt2-SaCas9-miR122TS plasmid is removed by digestion with Bsp120I and NcoI.
9. CMV promoter fragment can be obtained using pAAV-CMV-GFP-LA as the template and PCR-amplified using the following primers (Table 5):
Table 5. PCR primers for plasmid construction
| Primer name | Sequence |
|---|---|
| PCR-CMV-Primer-F | 5'-ggcgagatttttttctagagacattgattattgactagt-3' |
| PCR-CMV-Primer-R | 5'-ttccgcttcttctttggggcggcggcccgcgtgaaacta-3' |
10. The digested vector is ligated with the PCR product via seamless cloning to obtain the CMV promoter version of the AAV-U6-sgRNA-CMV-SaCas9- miR122TS vector.
C. In vitro sgRNA selection
1. Genome editing in Neuro2A cells
a. Culture Neuro2A cells at 37 °C with 5% CO2 until cells reach 60% confluency in 12-well plates. Replace fresh medium before transfection.
b. When cell confluency is at 60%–70%, perform plasmid transfection using the Lipo8000 transfection reagent. Gently mix 50 μL of Opti-MEM reduced serum medium, 1 μg of plasmid DNA, and 1.6 μL of Lipo8000 transfection reagent and incubate at room temperature for 15 min.
c. Gently drip the 50 μL mixture of transfection reagent into the medium and thoroughly mix with the culture medium.
d. Culture the cells at 37 °C with 5% CO2. Collect Neuro2A cells approximately 48 h post-transfection.
2. Amplicon sequencing
a. Extract genomic DNA from Neuro2A cells using a Genomic DNA Extraction kit.
b. Design three pairs of short primers to amplify the genomic fragments with the sgRNA-targeted sites in the middle. PCR fragments have approximately 250 bp, with primer sequences approximately 20 bp in length on both sides with an annealing temperature of approximately 50 °C. Verify the specificity of the primers on the NCBI website (https://www.ncbi.nlm.nih.gov/) using the BLAST—Primer Blast tool. Under Primer Parameters, enter the designed primer pair. In the database section, select Refseq representative genomes. In the organism section, choose Mus musculus and click on Get Primers. If the results show good specificity of the designed primers in the genome, i.e., no potential nonspecific bands around 250 bp, the primers are considered potentially suitable.
c. Use the three pairs of short primers to amplify the genomic DNA of Neuro2A cells to validate their specificity. Table 6 shows the PCR reaction mixtures, and Table 7 shows the PCR program.
Table 6. PCR mixtures
| Reagent | Volume |
|---|---|
| 2× Taq DNA polymerase | 10 μL |
| 10 μM forward primer | 1 μL |
| 10 μM reverse primer | 1 μL |
| Template gDNA | 800 ng |
| Nuclease-free H2O | To 20 μL |
Table 7. PCR program
| Step | Temperature | Duration |
|---|---|---|
| Hot start | 94 °C | 3 min |
| Denaturation | 94 °C | 20 s (35 cycles) |
| Annealing | 55 °C | 20 s (35 cycles) |
| Extension | 72 °C | 30 s (35 cycles) |
| Final extension | 72 °C | 3 min |
| Hold | 12 °C | ∞ |
d. After the reaction, subject the products to DNA gel electrophoresis. A single band at the expected size indicates good specificity of the short primers. Select one pair of specific short primers for subsequent library construction.
e. For amplicon sequencing, add high throughput sequencing-related fragments such as adaptor and index sequences to the 5' ends of the selected short primers. Table 8 shows the structure of the designed long primers.
Table 8. Structure of long primers
| Primer name | Sequence |
|---|---|
| ITR-Primer-F | 5'-aatgatacggcgaccaccgagatctacac-[i5]-acactctttccctacacgacgctcttccgatct-[upstream short primer sequence]-3' |
| ITR-Primer-R | 5'-caagcagaagacggcatacgagat-[i7]-gtgactggagttcagacgtgtgctcttccgatct-[downstream short primer sequence]-3' |
Note: Here, i5 and i7 are 8 bp sequences that differ in each set, allowing for the labeling of different groups and their distinction in subsequent Illumina sequencing. See the manual of the Illumina dual-index primer kit for more information about the structure of this sequencing library design.
f. Using the same PCR amplification conditions as for the short primers, amplify the target genomic DNA of Neuro2A cells using the designed long primers. Mix the amplified products from each group at a concentration of 20 ng/μL to construct the Illumina library.
g. High-throughput sequencing is performed on the Illumina NovaSeq 6000 platform at Novogene, China. The sequencing results are analyzed and processed using CRISPResso2 (see Data analysis section).
h. Based on amplicon sequencing analyses, choose two sgRNAs with the highest genome editing efficiency to further package into AAV and validate in vivo.
D. AAV packaging
1. Plasmid preparation
a. Freshly inoculate bacteria stock carrying the AAV vector, pRepCap2/9, and pHGTI plasmids on an LB plate.
b. Pick single bacteria clones and use them to inoculate liquid LB medium. Culture the bacteria at 37 °C at 200 rpm for 12–16 h.
c. Extract the pHGTI plasmids using the E.Z.N.ATM Endo-Free Plasmid Giga Prep Kit. Extract the AAV vector and pRepCap2/9 plasmids using the EndoFree Maxi Plasmid kit.
2. AAV production
Day 0–1: Thaw HEK293T cells
a. Thaw frozen cells rapidly (<1 min) in a 37 °C water bath.
b. Spin down the thawed cells at 1,000 rpm for 5 min.
c. Resuspend the cells in complete cell culture medium and transfer to a culture dish.
d. Incubate in a 37 °C, 5% CO2 incubator for 48 h. Change fresh medium daily.
Day 2–6: Passage and expand HEK293T cells to ten 15 cm dishes
a. Remove the culture medium and wash cells using PBS once.
b. Remove PBS and add 2 mL of 0.25% trypsin to cover the cell layer at room temperature for 2 min.
c. Add 4 mL of complete medium and dissociate the cells by pipetting several times.
d. Transfer the cells to a 15 mL tube and centrifuge at 1,100× g for 5 min.
e. Resuspend the cells with complete cell culture medium and split 1:3–1:5 into new dishes.
f. Culture cells for two days and passage again. Repeat this procedure to generate cells in ten 15 cm dishes with a confluency of ~70%.
Day 7–11: Cell transfection and AAV production
a. Change fresh cell culture medium at 2–5 h before transfection.
b. Prepare transfection complexes as shown in Table 9.
Table 9. Preparation of transfection mixtures
| Reagents | Quantity or Volume | Note |
| Opti-MEM | 20 mL | Prewarm to room temperature |
| AAV vector plasmid | 140 μg | The molar ratio of the three plasmids should be ~1:1:1. |
| pRepCap2/9 plasmid | 140 μg | |
| pHGTI plasmid | 320 μg | |
| 1 mg/mL PEI | 2.4 mL | Total DNA (μg): PEI (μg) = 1:4–1:5 |
Note: Add the first four components above by order and mix well by pipetting up and down gently. Mix immediately after adding PEI. Incubate at room temperature for 15–20 min.
c. Add 2 mL of transfection complexes dropwise to each plate and gently shake the culture plate to evenly distribute the transfection complexes.
d. After 12–18 h of transfection, change the medium to serum-free (SF) medium.
e. Incubate the transfected cells in a 37 °C, 5% CO2 incubator for another 60–72 h.
3. AAV purification
a. AAV extraction
i. Detach the cells with a cell scraper in cell culture medium.
ii. Centrifuge at 1,000× g for 5 min to separate the supernatant and the cell pellet.
iii. Mix one fraction of precipitation solution with four fractions of supernatant and keep on ice for 2 h.
iv. Use 7 mL of lysis buffer to resuspend the cell pellet from step 3a.ii and add 1 μL of benzonase.
v. Lyse cells via three freeze-thaw cycles. Freeze at -80 °C for 30 min and thaw at 37 °C for 30 min.
vi. Centrifuge the precipitant from step 3a.iii at 3,300× g for 30 min at 4 °C. Resuspend the precipitant with 1 mL of lysis buffer.
vii. Pool the cell lysate from step 3a.v and the suspension from step 3a.vi together. Leave the final mixture in -80 °C for purification.
b. AAV ultracentrifugation
i. Thaw the mixture at 37 °C and centrifuge at 4,500× g for 30 min at 4 °C.
ii. Transfer the supernatant to the optiseal ultracentrifugation tube.
iii. Gently add 5 mL of 17% iodixanol solution to the bottom of the tube through a long 16G blunt-end needle on a luer lock syringe. Then, change the needle and syringe.
iv. Gently add 5 mL of 25% iodixanol solution to the bottom of the tube through a long 16G blunt-end needle on a luer lock syringe. Then, change the needle and syringe.
v. Gently add 5 mL of 40% iodixanol solution to the bottom of the tube through a long 16G blunt-end needle on a luer lock syringe. Then, change the needle and syringe.
vi. Gently add 5 mL of 60% iodixanol solution to the bottom of the tube through a long 16G blunt-end needle on a luer lock syringe.
vii. Use lysis buffer for balance and centrifuge at 170,000× g for 160 min at 4 °C with a Type 70 TI rotor.
Note: Carefully move the tubes to maintain the density layers. Carefully balance the tubes to protect the centrifuge.
viii. After centrifugation finishes, insert the 1.2 mm diameter needle on a 5 mL syringe inside of the optiseal tube at 2 mm below the 40%/60% interface. Face the bevel of the needle upward. Remove the cap of the tube. Aspirate approximately 5 mL of solution containing AAV (Figure 5).
Note: Do not aspirate materials at the 25%/40% interface, which contains contaminants that will clog the centrifugal filter in the next step.
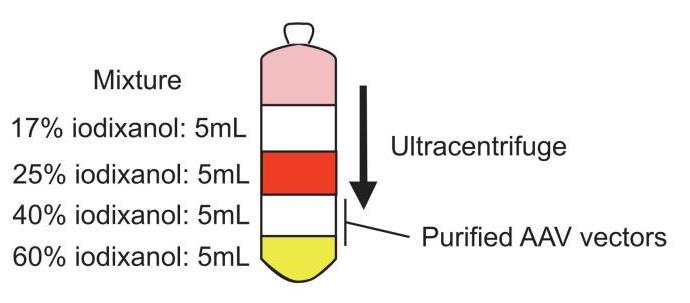
Figure 5. Diagram showing the ultracentrifugation tube with the density layers. The fraction containing adeno-associated virus (AAV) is indicated in the picture.
c. AAV cleaning and concentration
i. Inject solution smoothly into the ultra-centrifugal filter tube with 10 mL of AAV washing buffer. Centrifuge at 5,000× g for 30 min at 4 °C.
ii. Discard the flowthrough fluid and add 15 mL of new AAV washing buffer to the concentrated AAV solution. Centrifuge at 5,000× g for 30 min at 4 °C.
iii. Repeat step 3c.ii 3–4 times to fully remove iodixanol; the final AAV solution will become less viscous and appear indistinguishable from the washing buffer.
iv. Adjust the volume of AAV solution as desired, aliquot, and store at -80 °C.
4. AAV vector quantification
a. Prepare the AAV genome as follows (Table 10).
Table 10. Digestion procedures
| Reagents | Volume (μL) | Program |
| AAV vector | 1* | Incubate at 37 °C for 15 min Inactivate at 95 °C for 10 min |
| DNase I | 1 | |
| 10× reaction buffer | 5 | |
| ddH2O | 43 | |
| Digested product above | 50 | Incubate at 37 °C for 15 min Inactivate at 95 °C for 10 min |
| Proteinase K | 1 | |
| 10× reaction buffer | 5 | |
| ddH2O | 44 |
*Note: The AAV volume is adjustable. All reagents should be mixed on ice.
b. Prepare a gradient of DNA standards.
i. Adjust the pAAV-U6-sgRNA-Tnnt2-SaCas9-HA-miR122TS plasmid to 10 ng/μL.
ii. Serially dilute the plasmid to get the gradient DNA standard of 10 ng/μL, 1 ng/μL, 10-1 ng/μL, 10-2 ng/μL, 10-3 ng/μL, 10-4 ng/μL, 10-5 ng/μL, and 10-6 ng/μL.
c. qPCR reaction
i. Use the DNA standards and AAV genome to generate a real-time quantitative PCR (RT-qPCR) mix as follows (Table 11).
Table 11. Mixtures for the qPCR reaction
| Reagent | Volume |
|---|---|
| 2× qPCR SYBR master mix | 10 μL |
| 10 μM forward primer | 0.5 μL |
| 10 μM reverse primer | 0.5 μL |
| Template DNA | 1 μL |
| Nuclease-free H2O | To 20 μL |
Note: Primers are designed on inverted terminal repeats of the AAV vector.
The primer sequences are as follows (Table 12):
Table 12. Primer sequences for AAV vector quantification
| Primer name | Sequence |
|---|---|
| ITR-Primer-F | 5'-ggaacccctagtgatggagtt-3' |
| ITR-Primer-R | 5'-cggcctcagtgagcga-3' |
ii. Run qPCR using the following program (Table 13).
Table 13. Thermocycling program for the qPCR reaction
| Step | Temperature | Duration |
|---|---|---|
| Hot start | 95 °C | 2 min |
| Denaturation | 95 °C | 15 s (40 cycles) |
| Annealing | 60 °C | 20 s (40 cycles) |
| Extension | 72 °C | 15 s (40 cycles) |
| Melt curve stage | 95 °C | 1 min |
| Melt curve stage | 65 °C | 30 s |
| Melt curve stage | 95 °C | 30 s |
d. qPCR analysis
i. Average all three replicates of Cq from the standards and sample.
ii. Use the average Cq of standards and the concentration gradients as log10 (concentration, ng/μL) to draw a standard curve. Calculate the intercept u and slope k of the curve.
iii. Concentration of the sample (ng/μL) = 10 (Cq - u) /k.
iv. Concentration of the sample (nmol/μL) = concentration (ng/μL) / the molecular weight of plasmid pAAV-U6-sgRNA-Tnnt2-SaCas9-HA-miR122 TS.
v. AAV Concentration of the sample (vg/mL) = concentration (nmol/μL) × 6 × 1014 × 103 × 102.
Note: 102 is the dilution factor of the AAV vector in the AAV genome preparation step, which is adjustable according to the actual dilution of AAV.
E. In vivo genome editing validation
1. AAV administration and sample collection
a. Dilute AAV9 in PBS, reaching a final volume of 50 μL. AAV is administered at 5 × 1010 vg/g (vector genome per gram bodyweight) per animal.
b. Anesthetize the P1 mice with 2% isoflurane. Inject the 50 μL of AAV subcutaneously into the prethoracic region of mice with a 29 G needle syringe. See Figure 6.
c. At one week after AAV injection, euthanize the mice.
d. Lay the mice on their back on the operating table, exposing the chest area and collecting the heart and lungs.
e. Open the abdominal cavity and collect the liver, spleen, and kidney.
f. Tear apart the hindlimb skin, exposing the quadriceps and collecting the muscle.
g. Remove the skull to collect the brain.
h. Snap-freeze all tissues in liquid nitrogen and then transfer to -80 °C for the next experiment.
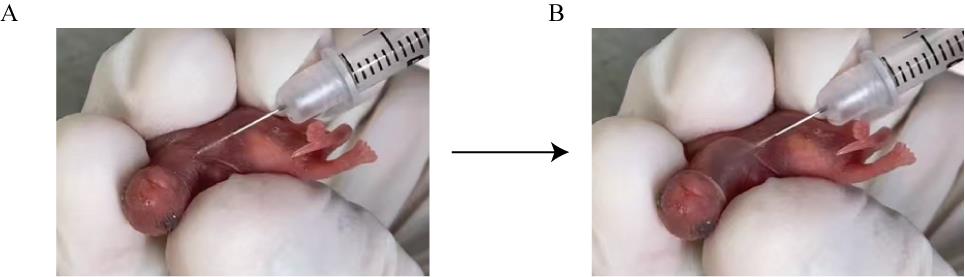
Figure 6. Images of adeno-associated virus (AAV) injection in a P1 mouse. The injection site is subcutaneous in the thoracic region (A), and an obvious bulge is formed after injection of 50 μL of AAV (B).
2. qPCR validation of AAV expression
a. Extract the total RNA of the heart and liver tissues using the TransZol Up Plus RNA kit. Extract genomic DNA (gDNA) with TIANamp Genomic DNA kit.
b. Assess total RNA integrity by 1.2% agarose gel electrophoresis at 110 V for 30 min.
c. Reverse-transcribe 1 μg of RNA to cDNA by HiScript III All-in-one RT SuperMix perfect for qPCR. Remove genomic DNA from RNA before reverse transcription.
d. Set up qPCR reaction as follows (Table 14):
Table 14. QPCR reaction mixtures
| Reagent | Volume |
|---|---|
| 2× qPCR SYBR master mix | 10 μL |
| 10 μM forward primer | 0.4 μL |
| 10 μM reverse primer | 0.4 μL |
| Template gDNA or cDNA | 500 ng or 1 μL |
| Nuclease-free H2O | To 20 μL |
e. Table 15 shows the primer sequences for RT-qPCR or qPCR.
Table 15. The sequences of AAV genome quantification experiment primers
| Primer name | Sequence |
|---|---|
| SaCas9-Primer-F | 5'-gctgtccaccaaagagcagatc-3' |
| SaCas9-Primer-R | 5'-tggtggtaggccttctgcac-3' |
| Gapdh(internal control)-Primer-F (for cDNA) | 5'-tgaccacagtccatgccatc-3' |
| Gapdh(internal control)-Primer-R (for cDNA) | 5'-gacggacacattgggggtag-3' |
| Tnni3(internal control)-Primer-F (for gDNA) | 5'-gccaggttgactgaagagacag-3' |
| Tnni3(internal control)-Primer-R (for gDNA) | 5'-ctggtttggagaggtttattctgc-3' |
f. Set up a qPCR program as follows (Table 16):
Table 16. QPCR program
| Step | Temperature | Duration |
|---|---|---|
| Hot start | 95 °C | 2 min |
| Denaturation | 95 °C | 15 s (40 cycles) |
| Annealing | 60 °C | 20 s (40 cycles) |
| Extension | 72 °C | 10 s (40 cycles) |
| Melt curve stage | 95 °C | 1 min |
| Melt curve stage | 65 °C | 30 s |
| Melt curve stage | 95 °C | 30 s |
g. Analyze RT-qPCR products by 1.5% agarose gel electrophoresis.
Note: Examine the specificity of the primers by checking the purity and size of the PCR product.
h. Quantify AAV expression by ΔCT calculations of gDNA and cDNA RT-qPCR performance.
3. Amplicon-sequencing assessment of gene editing
a. Extract genomic DNA from heart and liver tissues using the TIANamp Genomic DNA kit according to the manufacturer’s instructions.
b. After detecting the concentration and purity of gDNA, use 800 ng of DNA to amplify it by PCR. The size of PCR products should not exceed 300 bp.
Note: Design the primers in the sgRNA-targeted DNA locus. These primers are amplicon-sequencing short primers.
c. Set up a reaction as follows (Table 17). Table 18 shows the primer sequences for PCR.
Table 17. PCR reaction mixtures
| Reagent | Volume |
|---|---|
| 2× Taq PCR master mix | 10 μL |
| 10 μM forward primer | 1 μL |
| 10 μM reverse primer | 1 μL |
| Template gDNA | 800 ng |
| Nuclease-free H2O | To 20 μL |
Table 18. Sequences of Camk2d primer for amplicon-sequencing short primer
| Primer name | Sequence |
|---|---|
| Camk2d-Primer-F | 5'-ggttcatgaaaccgctcaga-3' |
| Camk2d-Primer-R | 5'-caccaaccattcacacaacaaatac-3' |
d. Set up a program as follows (Table 19):
Table 19. PCR program
| Step | Temperature | Duration |
|---|---|---|
| Hot start | 94 °C | 3 min |
| Denaturation | 94 °C | 30 s (33 cycles) |
| Annealing | 60 °C | 30 s (33 cycles) |
| Extension | 72 °C | 15 s (33 cycles) |
| Final Extension | 72 °C | 5 min |
| Hold | 12 °C | ∞ |
e. Analyze PCR products by 1.5% agarose gel electrophoresis.
f. After identifying good PCR short primers that produce a single specific band, add primers at their 5' with the adaptor sequences including index for multiplexed sequencing. According to the TansNGC Dual Index Primers Kit for Illumina, representative amplicon-sequencing long primer sequences are as follows (Table 20):
Table 20. Amplicon-sequencing long primer sequences
| Primer name | Sequence |
| amplicon-sequencing long primers, forward | 5′-aatgatacggcgaccaccgagatctacacgcctatcaacactctttccctacacgacgctcttccgatctggttcatgaaaccgctcaga-3′ |
| 5′-aatgatacggcgaccaccgagatctacaccttggatgacactctttccctacacgacgctcttccgatctggttcatgaaaccgctcaga-3′ | |
| 5′-aatgatacggcgaccaccgagatctacacagtctcacacactctttccctacacgacgctcttccgatctggttcatgaaaccgctcaga-3′ | |
| amplicon-sequencing long primers, reverse | 5′-caagcagaagacggcatacgagataatggtcggtgactggagttcagacgtgtgctcttccgatctcaccaaccattcacacaacaaatac-3′ |
| 5′-caagcagaagacggcatacgagattcgctatcgtgactggagttcagacgtgtgctcttccgatctcaccaaccattcacacaacaaatac-3′ | |
| 5′-caagcagaagacggcatacgagatcgtccattgtgactggagttcagacgtgtgctcttccgatctcaccaaccattcacacaacaaatac-3′ |
Note: Indices and Camk2d-matching sequences are inbold.
g. Amplify the sgRNA-targeted Camk2d locus using Taq PCR master mix.
h. Run the products on a 1.2% agarose gel.
i. Purify the PCR products using TIANgel Purification kit.
j. Mix 20 ng per product in a total of 20 µL volume to construct the sequencing library.
k. Perform the sequencing on an Illumina NovaSeq 6000 platform with 2× 150 bp pair-end reads at Novogene, China.
l. Use the CRISPResso2 to analyze the sequencing reads (see Data analysis section).
4. Western blot verification of gene knockout
a. Grind the heart tissues in RIPA buffer using a tissue grinder three times, 1 min each.
b. Lyse the tissues on ice for 30 min.
c. Centrifuge the lysates at 15,000× g for 10 min at 4 °C.
d. Dilute the supernatants into 4× SDS sample buffer and boil them for 10 min at 70 °C.
e. Run 30 μg of proteins per sample on a 4%–12% SurePAGETM gel in Tris-MOPS-SDS buffer at 120 V for 75 min.
f. Transfer the proteins to a PVDF membrane at 90 V for 90 min.
g. Incubate the membranes overnight at 4 °C with the primary antibodies (Rb-anti-CAMK2D, 1:2,500; Rb-anti-HA, 1:2,500; Ms-anti-GAPDH, 1:10,000).
h. Wash the membranes three times, 5 min each.
i. Incubate the membranes for 1 h at RT with the secondary antibodies (HRP-conjugated goat anti-rabbit IgG, 1:10,000; HRP-conjugated goat anti-mouse IgG, 1:10,000).
j. After washing the membranes, collect chemiluminescence signals using ECL western blotting substrate with a Bio-Rad imager. Figure 7 shows the representative western blot data.
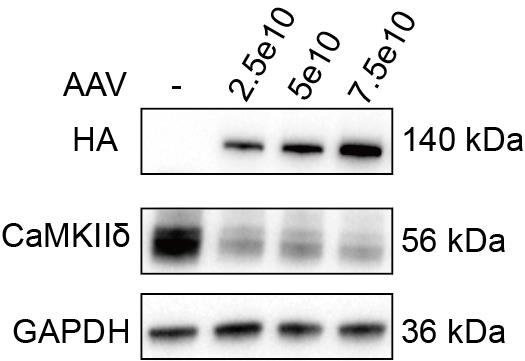
Figure 7. Western blot validation of AAV-Tnnt2-SaCas9-miR122TS in the heart. The adeno-associated virus (AAV) injection dose from channels 2–4 is 2.5 × 1010, 5 × 1010, and 7.5 × 1010 vg/g.
Data analysis
1. Sequencing data is trimmed by Novagene to generate the *.fq.gz file for the following analyses.
2. Amplicon sequencing data is first quality controlled using FastQC.
3. The sequences are next aligned to sgRNA-targeted Camk2d reference sequences, such as:
5'-GGTTCATGAAACCGCTCAGATTTGTGAGTTTGCAAGTGGATACTAGATCTGATTA TACTGACGACTTGCTAATTATTGGTTTCCAGGGGGGCGTTCTCAGTGGTGAGAAGATGTATGAAAATCCCTACTGGACAAGAGTATGCTGCCAAAATTATCAACACCAAAAAGCTTTCTGCTAGGGGTGGGTATTTTAAACCACATTTATTTATATATGTTAATGTTGTATTTGTTGTGTGAATGGTTGGTG-3'
4. CRISPResso2 analysis is performed using the following command:
CRISPResso \
-r1 *.clean.fq.gz \ (read 1)
-r2 *.clean.fq.gz \ (read 1)
-a NNNNNNNNNNNNNNNNNNNNNNNNNNNN \ (reference sequence)
-g NNNNNNNNNNN \ (sgRNA sequence)
-w 20 \
-wc -3 \
--ignore_substitutions \
--base_editor_output \
--write_cleaned_report \
--place_report_in_output_folder \
-o XXXXX \
5. Insertions/deletions in the amplicon sequencing libraries are quantified by CRISPResso2. Representative output data are as follows (Figure 8):
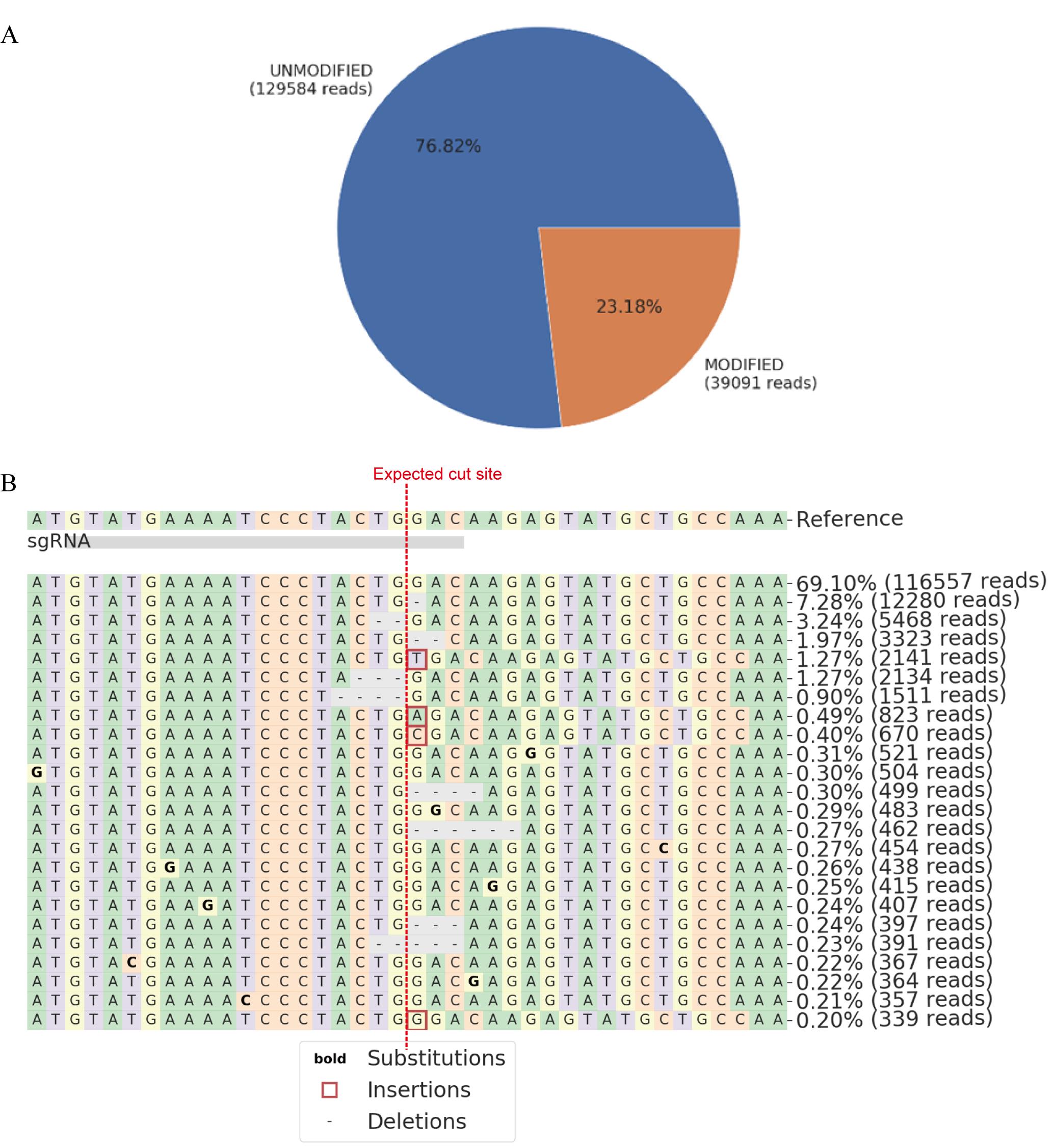
Figure 8. Representative amplicon-sequencing result generated by CRISPResso2. (A). Quantification of total edited counts. (B). Representative mutated reads and allele frequency at the sgRNA targeted site.
Validation of protocol
This protocol has been validated in the original publication:
Yang et al. [3]. MicroRNA-122–Mediated Liver Detargeting Enhances the Tissue Specificity of Cardiac Genome Editing. Circulation. 149(22): 1778–1781. DOI: 10.1161/CIRCULATIONAHA.123.065438
General notes and troubleshooting
Genome editing could generate high-variability outcomes. The following key steps should be carefully taken to enhance the chance of success in designing cardiac genome editing.
1. SgRNA design: The gene has different splicing variants, so the designed target sites of sgRNA need to be located on the common exons of different splicing variants of the gene.
2. In vitro sgRNA screening: Screening an array of candidate sgRNAs for a couple of sgRNAs with the best editing efficiency in vitro will significantly enhance the chance of success in vivo. It is possible to bypass this step to save time, but in vivo sgRNA screening by AAV is costly and mainly suitable for well-funded laboratories.
3. Application of Neuro2A cells: SgRNA screen could also be performed in other mouse cell lines, such as Hepa 1–6 cells, NIH/3T3 cells, or BV2 cells. These cell lines need to exhibit high and stable plasmid transfection efficiency to facilitate the evaluation of editing efficiency.
4. AAV packaging: The quality of AAV packaging largely depends on the HEK293T cells. If the AAV titer is low, the state of 293T cells should be checked. Subject to adequate funding, a CRO company can be commissioned to perform AAV packaging, which can increase the success rate and stability of AAV packaging.
5. AAV functional validation: AAV transduction could be assessed by AAV genome DNA qPCR. AAV-delivered SaCas9 expression could be assessed by AAV cDNA qPCR. It is recommended to perform these experiments to determine if AAV works at a similar level in every group to make sure AAV quality is not a major concern.
6. Amplicon sequencing: When the amplicon sequencing results cannot be split, the correctness of the index sequence and the sgRNA targeting sequence should be carefully checked.
7. This protocol describes AAV9 and SaCas9 experiments that are also applicable to other AAV serotypes and genome editors. With the continuous evolution of new AAV and editors, the performance of cardiac genome editing could also improve.
Acknowledgments
This work was funded by the National Key R&D Program of China (2022YFA1104800) to Y.G.
Competing interests
The authors declare that they have no competing interests.
References
- Wang, J. H., Gessler, D. J., Zhan, W., Gallagher, T. L. and Gao, G. (2024). Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduction Targeted Ther. 9(1): 78. https://doi.org/10.1038/s41392-024-01780-w
- Prasad, K. M., Xu, Y., Yang, Z., Acton, S. T. and French, B. A. (2010). Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 18(1): 43–52. https://doi.org/10.1038/gt.2010.105
- Yang, L., Liu, Z., Chen, G., Chen, Z., Guo, C., Ji, X., Cui, Q., Sun, Y., Hu, X., Zheng, Y., et al. (2024). MicroRNA-122–Mediated Liver Detargeting Enhances the Tissue Specificity of Cardiac Genome Editing. Circulation. 149(22): 1778–1781. https://doi.org/10.1161/circulationaha.123.065438
- Shang, R., Lee, S., Senavirathne, G. and Lai, E. C. (2023). microRNAs in action: biogenesis, function and regulation. Nat Rev Genet. 24(12): 816–833. https://doi.org/10.1038/s41576-023-00611-y
- Dhungel, B., Ramlogan-Steel, C. A. and Steel, J. C. (2018). MicroRNA-Regulated Gene Delivery Systems for Research and Therapeutic Purposes. Molecules. 23(7): 1500. https://doi.org/10.3390/molecules23071500
- Tsai, W. C., Hsu, S. D., Hsu, C. S., Lai, T. C., Chen, S. J., Shen, R., Huang, Y., Chen, H. C., Lee, C. H., Tsai, T. F., et al. (2012). MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 122(8): 2884–2897. https://doi.org/10.1172/jci63455
- Qiao, C., Yuan, Z., Li, J., He, B., Zheng, H., Mayer, C., Li, J. and Xiao, X. (2010). Liver-specific microRNA-122 target sequences incorporated in AAV vectors efficiently inhibits transgene expression in the liver. Gene Ther. 18(4): 403–410. https://doi.org/10.1038/gt.2010.157
- Geisler, A., Jungmann, A., Kurreck, J., Poller, W., Katus, H. A., Vetter, R., Fechner, H. and Müller, O. J. (2010). microRNA122-regulated transgene expression increases specificity of cardiac gene transfer upon intravenous delivery of AAV9 vectors. Gene Ther. 18(2): 199–209. https://doi.org/10.1038/gt.2010.141
Article Information
Publication history
Received: Dec 1, 2024
Accepted: Feb 4, 2025
Available online: Feb 25, 2025
Published: Mar 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Yang, L., Guo, C., Sun, Y. and Guo, Y. (2025). Cardiac-Specific Gene Editing via an AAV9-Tnnt2-SaCas9-miR122TS Vector. Bio-protocol 15(5): e5235. DOI: 10.21769/BioProtoc.5235.
Category
Developmental Biology > Genome editing > Targeted integration
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.