- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Using the Sleeping Beauty Transposon System for Doxycycline-inducible Gene Expression in RAW264.7 Macrophage Cells to Study Phagocytosis
Published: Vol 15, Iss 3, Feb 5, 2025 DOI: 10.21769/BioProtoc.5178 Views: 2267
Reviewed by: Paurvi ShindeChiara Ambrogio
Original research article
The authors used this protocol in:

Sep 2022

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
Macrophages are known for engulfing and digesting pathogens and dead cells through a specialized form of endocytosis called phagocytosis. Unfortunately, many macrophage cell lines are refractory to most reagents used for transient transfections. Alternative transient approaches, such as electroporation or transduction with lentiviral vectors, typically cause cell death (electroporation) or can be time-consuming to generate numerous lentivirus when using different genes of interest. Therefore, we use the Sleeping Beauty system to generate stably transfected cells. The system uses a “resurrected” transposase gene named Sleeping Beauty found in salmonid fish. Experimentally, the system introduces two plasmids: one carrying the Sleeping Beauty transposase and the other with an integration cassette carrying the gene of interest, a reverse-doxycycline controlled repressor gene, and an antibiotic resistance gene. The construct used in this protocol provides puromycin resistance. Stable integrations are selected by culturing the cells in the presence of puromycin, and further enrichment can be obtained using fluorescence-activated cell sorting (FACS). In this protocol, we use the Sleeping Beauty transposon system to generate RAW264.7 cells with doxycycline-inducible inositol polyphosphate 4-phosphatase B containing a C-terminal CaaX motif (INPP4B-CaaX). INPP4B-CaaX dephosphorylates the D-4 position of phosphatidylinositol 3,4-bisphosphate and inhibits phagocytosis. One benefit is that generating stable cell lines is substantially faster than selecting for random integrations. Without FACS, the method typically gives ~50% of the cells that are transfected; with sorting, this approaches 100%. This makes phagocytosis experiments easier since more cells can be analyzed per experiment, allowing for population-based measurements where a ~10% transient transfection rate is insufficient. Finally, using the doxycycline-promoter allows for low near endogenous expression of proteins or robust overexpression.
Key features
• This protocol builds on the protocols and reagents developed by Kowarz et al. [1] and extends it to using RAW macrophages.
• Allows for the rapid generation of stably induced cell lines.
• This protocol also determines the phagocytic index and efficiency.
Keywords: PhagocytosisGraphical overview
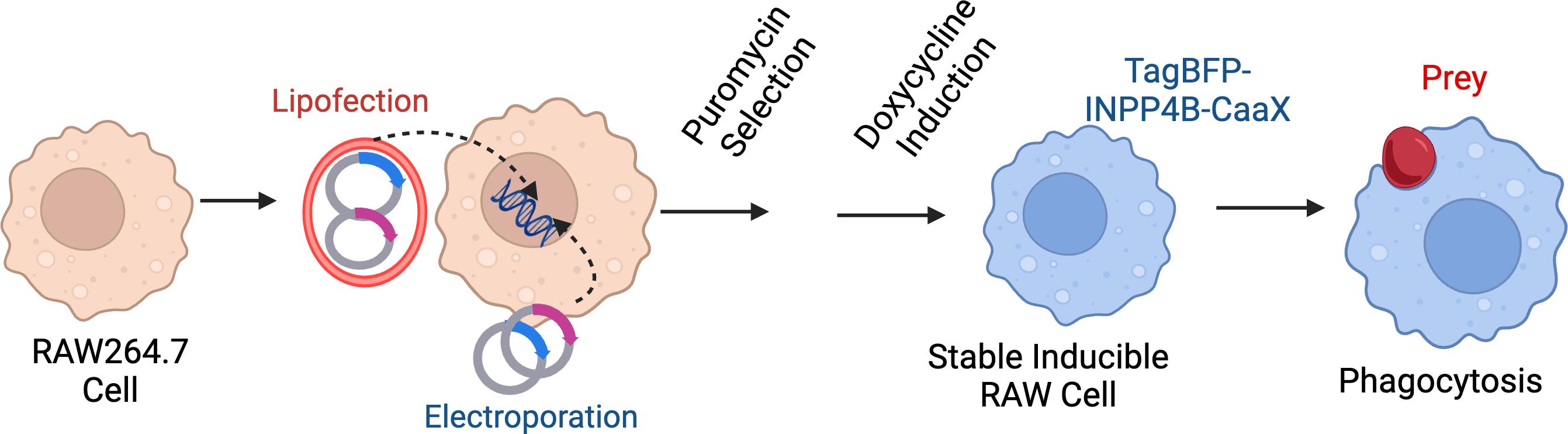
Protocol overview. Through electroporation or lipofection, RAW 264.7 cells are transfected with two plasmids: one carrying the Sleeping Beauty transposase and the other with an integration cassette carrying the gene of interest, reverse doxycycline-controlled repressor gene, and a puromycin-resistance gene. Transfected cells are then selected with 2.5 μg/μL of puromycin for at least 5 days to select for a polyclonal mix of stably transfected cells. For experiments, cells are treated with doxycycline to induce the TagBFP-INPP4B-CAAX or the inactive control TagBFP-INPP4BC842A-CAAX. Cells are exposed to opsonized particles to induce phagocytosis.
Background
The introduction of plasmid DNA into mammalian cells has been an essential method to support cellular and molecular biology investigations. Typically, the introduction of plasmid DNA can be mediated by a few approaches, including electroporation, chemical transformation, polyfection using polymer-based transfection reagents, microinjection, and viral transduction. These methods vary in efficiency depending on cell type and scale. Another factor to consider is whether the researcher wants transient over-expression of a gene of interest or generation of stably transfected cell lines. The generation of a stable cell line is typically more time-consuming. Still, it can be advantageous if cells are refractory to lipofection or if the same cell lines will be used for several experiments. In our lab, we use the Sleeping Beauty transposon system to help facilitate the integration of DNA into the genome to support the generation of stable cell lines [2–4]. This transposon system has been widely used as a genetic engineering tool for the last decade. Indeed, we have used this system to generate RAW264.7 cells expressing the reverse tetracycline repressor (rTetR) for controllable induction of our protein of interest, TagBFP-INPP4B-CaaX [5]. As these cells are generally challenging to transfect transiently, this approach allows for the rapid creation of cell lines.
The protocol described here is an adaptation of Kowarz et al. [1]. Briefly, this system utilizes a plasmid (SB100x) encoding the Sleeping Beauty transposase and a second plasmid containing an integration cassette with a gene of interest, a selectable marker, flanked by inverted terminal repeats. Kowarz et al. [1] generated a variety of plasmids, available from Addgene, to allow researchers to create cells with constitutive or doxycycline-inducible genes, an array of drug-resistant markers (e.g., puromycin or hygromycin) and the option also to deliver the reverse tetracycline repressor.
Phagocytosis is an actin-driven process used when cells ingest particles greater than 0.5 mm, such as pathogens and apoptotic bodies [6–8]. Phagocytosis entails the extension of actin-rich pseudopods to surround the target and pull the particle into the cell, forming a nascent phagosome [9]. The extensive actin remodeling, polymerization, bundling, and depolymerization must be exquisitely controlled and temporally coordinated. Major regulators in this process are the metabolism and interconversion of phosphoinositides lipids [10]. For instance, the generation of phosphatidylinositol (3,4)-bisphosphate (PtdIns3,4P2) at the engagement site is crucial for extending pseudopods to envelop prey [5]. Mammalian cells contain at least two pathways and multiple enzymes capable of synthesizing PtdIns3,4P2; thus, the knockdown of individual enzymes or specific inhibitors often generates unsatisfying results [11]. Instead, we used a synthetic plasmid-borne enzyme inositol polyphosphate 4-phosphatase type IIB (INPP4B) produced as a chimera with TagBFP for visualization, as well as a C-terminal CaaX motif for prenylation and membrane targeting [12]. This chimeric protein catalyzes the dephosphorylation of the D-4 position of PtdIns3,4P2 in the plasma membrane regardless of the biosynthetic route [12].
We generate doxycycline-inducible RAW264.7 cells expressing BFP-INPP4B-CaaX and a second cell line expressing the catalytically inactive BFP-INPP4BC842A-CaaX that serves as a control. Following overnight treatment with doxycycline to induce the INPP4B-containing constructs, we conduct phagocytosis assays to determine the role of PtdIns3,4P2 [5]. Typically, our phagocytosis assays use IgG-opsonized sheep red blood cells or IgG-coated polystyrene beads as prey to assess Fcg receptor-mediated phagocytosis [13].
Many approaches are available to assess phagocytosis; most rely on fluorescent labels on the prey to quantify internalization and phagosome maturation. Flow cytometry analysis determines the overall ingestion of particles by measuring total fluorescence intensity/cell as a parameter for particle uptake [14]. Other methods use pH-sensitive fluorescent particles that become brighter in acidic conditions, such as in the phagolysosome, that measure particle uptake and phagosome maturation [15]. However, both these approaches are limited in defining particle engagement vs. complete engulfment. Instead, we prefer a high-resolution confocal microscope and an antibody staining approach to distinguish bound vs. fully internalized particles (Figure 1).
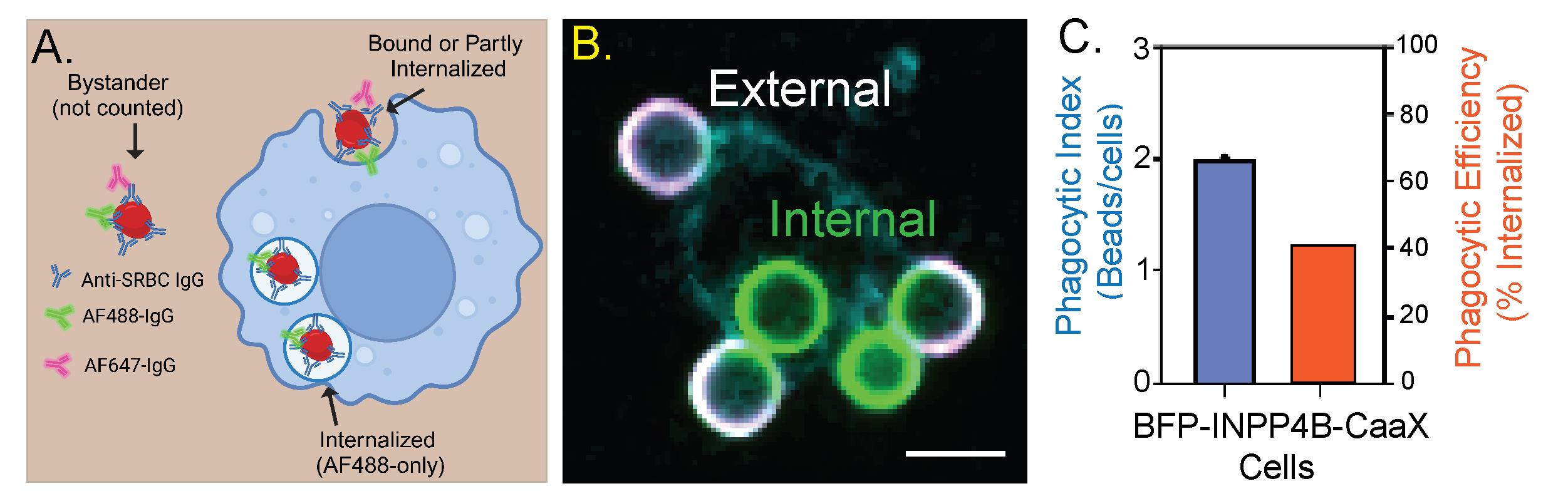
Figure 1. INPP4B-CaaX inhibits the internalization of IgG opsonized particles. A. Representation of the assay. All sheep red blood cells (SRBCs) are labeled with the AF488-conjugated antibody, whereas only extracellular SRBCs are stained with the AF647 antibody at the end of the incubation period. B. Representative image. Fluorescence markers are pseudocolored: AF488 (green), AF647 (magenta), and BFP (cyan). Scale bar = 5 μm. C. A double y-axis graph depicting the results of panel B. The phagocytic index represents the number of beads internalized by the cells over a fixed period. Phagocytic efficiency represents the percentage of engaged particles that are internalized.
Materials and reagents
Biological materials
1. RAW264.7 cell line (American Type Culture Collection, catalog number: TIB-71)
2. Sheep red blood cells, 10% suspension (MP Biomedicals, catalog number: 55876), stored at 2–8 °C
3. pCMV(CAT)-T7-SB100X plasmid (Addgene, plasmid #34879)
4. pSBtet-Pur plasmid (Addgene, plasmid #60507)
5. Primary antibodies (store at -20 °C):
a. Anti-sheep red blood cell rabbit IgG antibody (85 mg/mL) (Rockland, catalog number: 113-4139)
b. Total human serum IgG antibody (50 mg/mL) (Millipore Sigma, catalog number: I4381)
6. Secondary antibodies (store at -20 °C):
a. Goat anti-rabbit IgG (H+L) antibody-AlexaFluor647 (Jackson ImmunoResearch, catalog number: 111-605-144)
b. Goat anti-rabbit IgG (H+L) antibody-AlexaFluor488 (Jackson ImmunoResearch, catalog number: 111-545-144)
c. Donkey anti-human IgG (H+L) antibody-AlexaFluor647 (Jackson ImmunoResearch, catalog number: 709-605-149)
d. Donkey anti-human IgG (H+L) antibody-AlexaFluor488 (Jackson ImmunoResearch, catalog number: 709-545-149)
Reagents
1. Paraformaldehyde 16% wt/vol (PFA) (Electron Microscopy Sciences, catalog number: 15700)
2. Doxycycline hyclate (Sigma-Aldrich, catalog number: D9891)
3. Puromycin (Sigma-Aldrich, catalog number: P8833
4. Roswell Park Memorial Institute Medium (RPMI) 1640 with L-glutamine and sodium pyruvate (Wisent Bioproducts, catalog number: 350-015-CL)
5. Phosphate buffered saline (PBS) without calcium and magnesium (Wisent bioproducts, catalog number: 311-013-CL)
6. Phosphate buffered saline with calcium and magnesium (PBS+/+) (Wisent bioproducts, catalog number: 311-011-CL)
7. Fetal bovine serum (FBS) (Wisent Bioproducts, catalog number: 080-150)
8. 4.2 mm Polystyrene beads with 2% divinylbenzene (Bangs Laboratories, Inc., catalog number: PS06005), stored at 2–8 °C
9. Fugene HD (Promega, catalog number: E2311)
10. Electrolytic buffer E2 (Invitrogen, catalog number: MPK10096E)
11. Resuspension buffer R (Invitrogen, catalog number: MPK10096R)
Solutions
1. 4% PFA (see Recipes)
2. Selection medium (see Recipes)
3. Induction medium (see Recipes)
Recipes
1. 4% PFA
| Reagent | Final concentration | Amount |
|---|---|---|
| 16% PFA | 4% | 1 mL |
| PBS+/+ | n/a | 3 mL |
| Total | n/a | 4 mL |
2. Selection medium (2.5 μg/mL of puromycin in RPMI)
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 | n/a | ~10 mL |
| Puromycin (10 mg/mL) | 2.5 μg/mL | 2.5 μL |
| Total | n/a | 10 mL |
3. Induction medium (1 μg/mL of doxycycline in RPMI)
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 | n/a | ~10 mL |
| Doxycycline hyclate (10 mg/mL) | 1 μg/mL | 2.5 μL |
| Total | n/a | 10 mL |
Laboratory supplies
1. T-25 (Sarstedt, catalog number: 83.3910)
2. T-75 (Sarstedt, catalog number: 83.3911)
3. Cell scraper (Wuxi NEST Biotechnology, catalog number: 710001)
4. 6-well plates (Sarstedt, catalog number: 83.3920)
5. 12-well plates (Sarstedt, catalog number: 83.3921)
6. 18 mm circular cover glass, #1½ (Electron Microscopy Sciences, catalog number: 72222-01
7. Kimwipes (Kimberly-Clark ProfessionalTM 34155, catalog number: 06-666A)
8. 1.5 mL Eppendorf tubes (Froggabio, catalog number: LMCT1.7B)
9. 15 mL tubes (Fisher Scientific, catalog number: 14-959-53A)
Equipment
1. Incubator (Eppendorf, model: CellXpert C170, catalog number: 6734)
2. Tabletop centrifuge (Thermo Scientific, model: Pico 21, catalog number: 75002553)
3. Plate centrifuge (Thermo Scientific, model: Sorvall Legend RT, catalog number: 75004377)
4. Tube revolver (Thermo Scientific, catalog number: 88881001)
5. Vortex (Scientific Industries, catalog number: SI-0236)
6. Neon Invitrogen transfection system (Thermo Fisher Scientific, Invitrogen, catalog number: MPK5000)
7. Hemocytometer (NanoEntek, catalog number: EVE-MC)
8. 3i MarianasTM spinning-disc confocal microscopy, based on the Zeiss Axio Observer 7 Advanced Microscope with Definite Focus 3 [Intelligent Imaging Innovation (3i), custom-built]
Software and datasets
The microscope and image acquisition were controlled with SlideBook 2024 (3i). TIFF images were exported and analyzed in ImageJ2 Version 2.14.0/1.54f [16]. Representative images were chosen based on a good signal-to-noise ratio. Merging and cropping fluorescent channels were performed in ImageJ2. To aid visualization, linear adjustments were made to brightness and contrast across the entire image. To help with data accessibility and enhance the presentation of micrographs, we switched the default red lookup table to magenta. Graphical overview was generated using BioRender.com. Figure 1 was assembled using Adobe Illustrator 2024 and GraphPad Prism V9.
Procedure
A. Cell culture
1. Grow RAW 264.7 cells in tissue culture flasks (T-25 or T-75) in 10 mL or 20 mL of RPMI 1640 supplemented with 5% heat-inactivated FBS in an incubator at 37 °C under 5% CO2.
2. Wash cells twice with 3–5 mL of prewarmed PBS.
3. Aspirate PBS and gently scrape cells with a sterile cell scraper.
4. Add 5 mL of RPMI 1640 + 10% FBS and gently pipette cells into a homogenous suspension.
5. Transfer cells into a 15 mL conical tube and centrifuge at 500× g for 5 min.
6. Aspirate the medium without disturbing the pellet.
7. Resuspend in 10 mL of RPMI 1640 + 5% FBS and pipette gently to break cell clumps.
8. Every three days, split in 1:10 dilution.
B. Electroporation (Neon transfection system)
1. Prepare a 6-well plate with 2 mL of RPMI without antibiotics. Use one well per electroporation (e.g., 1 for INPP4B-CaaX and 1 for the catalytically inactive INPP4BC842A-CaaX).
2. Place in an incubator at 37 °C under 5% CO2 for at least 30 min.
3. Transfer grown cells (5–10 mL) into a 15 mL conical tube and count them using a hemocytometer. Ideally, there should be 1–2 × 106 cells per milliliter.
4. Centrifuge the cell suspension in a 15 mL conical tube at 500× g for 5 min.
5. Aspirate media and wash with 1 mL of PBS.
6. Centrifuge at 500× g for 5 min.
7. Set up a Neon tube with 3 mL of electrolytic buffer E2.
8. Set the pulse condition at 1,700 V for 20 ms on the Neon system.
9. Aspirate the PBS and resuspend the cell pellet in resuspension buffer R to reach a final density of ~1.0 × 107 cells per milliliter.
10. Transfer 100 μL of cells in resuspension buffer R to a new Eppendorf tube.
11. Add 1 μg of pCMV(CAT)-T7-SB100 and 10 μg of pSBtet-Pur-TagBFP2-INPP4B-CAAX plasmids into the Eppendorf tube containing cells resuspended in resuspension buffer R. Gently mix by pipetting. Ideally, the volume of DNA should be no more than 10 μL.
12. Remove the pre-incubated 6-well plate and place it in the biosafety cabinet.
13. Using a 100 μL Neon tip, draw from the DNA–cell solution and dock it appropriately in the Neon transfection system.
14. Run the program. After receiving the “Complete” message, immediately transfer the contents of the 100 μL tip to one well of the 6-well plate.
15. Place the plate overnight in the incubator at 37 °C under 5% CO2.
C. Transfection (Fugene HD)
1. Seed approximately 5 × 104 cells from Section A in each well of a 12-well plate overnight.
2. Add 1 μg of DNA in 100 μL of serum-free RPMI.
3. Add 3 μL of Fugene HD into the DNA–DMEM suspension and incubate for 15 min.
4. Distribute the mixture evenly into two wells (50 μL in each) and place it in an incubator overnight.
D. Stable cell line generation with the Sleeping Beauty transposon system
1. Electroporate or transfect cells with pCMV(CAT)-T7-SB100 and pSBtet-Pur-TagBFP2-INPP4B-CAAX or pSBtet-Pur-TagBFP2-INPP4B(C842A)-CAAX plasmids in a 1:10 ratio (at least 1 μg:10 μg for electroporation)
2. Replace media with 2 mL of selection media per sample for at least 5 days.
a. To enhance selection, replace old media with fresh puromycin-RPMI media after 2 days.
3. Transfer to T-25 in fresh media without puromycin to grow.
E. Sheep erythrocyte opsonization
1. Gently vortex the 10% red blood cell suspension.
2. Draw 100–200 μL of the suspension into an Eppendorf tube.
3. Centrifuge cells at 3,000× g for 30 s and aspirate the supernatant.
4. Wash with 1 mL of PBS.
5. Centrifuge cells at 3,000× g for 30 s and aspirate the supernatant.
6. Resuspend in 100 μL of PBS with 3 μL of anti-sheep red blood cell rabbit antibody (see Troubleshooting).
7. Incubate at 37 °C for 1 h in a shaking block heater at 300–400 rpm.
F. Polystyrene bead opsonization
1. Add 100 μL of PBS in an Eppendorf tube.
2. Gently vortex the polystyrene bead solution.
3. Transfer 20 μL of beads into the Eppendorf tube with PBS.
4. Add 20 μL of human IgG (50 mg/mL) to the bead–PBS solution.
5. Incubate at 37 °C for 1 h in an end-over-end rotator.
G. Phagocytosis assay
1. Seed ~5 × 104 of stable cells (expressing INPP4B-CAAX or INPP4B(C842A)-CAAX) on 18 mm coverslips in a 12-well plate.
2. Once adhered, add 1 mL of induction media to stable cells and incubate at 37 °C under 5% CO2 overnight.
3. Label IgG opsonized particles with 2 μL of the AF488-fluorescent antibody (goat anti-rabbit for red blood cells and donkey anti-human for polystyrene beads).
4. Incubate for 5 min at room temperature in an end-over-end rotator.
5. Centrifuge at 3,000× g for 2 min and aspirate the supernatant.
6. Wash with 1 mL of PBS and centrifuge at 3,000× g for 2 min.
7. Aspirate supernatant and resuspend with 100 μL of PBS.
8. Add 10, 20, or 40 μL (1×, 2×, 4×) of the opsonized particles to cells on a 12-well plate.
9. Centrifuge the 12-well plate at 1,000× g for 1 min.
10. Incubate at 37 °C under 5% CO2 for 10–15 min.
11. Aspirate media and wash with 500 μL of ice-cold PBS+/+.
12. Aspirate and add 500 μL of PBS with 1 μL of AF647-fluorescent antibody to label non-internalized particles.
13. Mix gently and incubate for 5–10 min.
14. Aspirate and wash with ice-cold PBS+/+.
15. Add 800 μL of 4% PFA and cover the plate with foil for 30 min to fix cells on the coverslip.
16. Wash fixed cells twice with 500 μL of PBS.
17. The plate can be stored at 4 °C for five days before microscopy.
H. Microscopy
The Fairn lab spinning-disc confocal microscope is provided by Intelligent Imaging Innovation (3i), Denver, Colorado. The 3i MarianasTM spinning-disc confocal microscopy is based on the Zeiss Axio Observer 7 Advanced Microscope with Definite Focus 3. Epifluorescence viewing and imaging of samples used pE-340Fura Illumination System (CoolLED) with GFP or mCherry filter cubes (Chroma). The system incorporates a Yokogawa CSU-W1 T2 super-resolution spinning disk confocal, 50 μm and SoRa disks with a magnification changer (1×, 2.8×, and 4×) for confocal and super-resolution imagining. The system has a Plan-Apochromat 20×/0.8 NA and a C Plan-Apochromat 63×/1.4 NA oil objective (Zeiss). The system uses four lasers controlled by a LaserStack v4 with single-mode optical fibers for 405 nm (150 mW), 488 nm (200 mW), 561 nm (140 mW), and 638 nm (200 mW). Samples were collected using appropriate single bandpass filters or a quad-band filter (440/521/607/700 nm). Images were acquired with a Hamamatsu ORCA Fusion BT sCMOS camera with 2,304 × 2,304 pixels and a pixel size of 6.5 μm controlled with SlideBook software (3i). Acquisition settings and capture were controlled by SlideBook Software v2024 (Intelligent Imaging Innovations).
Data analysis
Post-acquisition images were analyzed, and the number of internal (single color) and external/bound particles were counted. Typical results are illustrated in Figure 1. For experiments, we usually compare uninduced to induced samples or, where possible, active enzyme vs. inactive enzyme. The results are presented as the mean ± standard error of n = 3–5 experiments, with 100 technical experiments (cells) per individual condition. Student’s t-test (2 groups) or Analysis of Variance (ANOVA) (> 2 groups) with an appropriate post-hoc test such as Tukey’s honest significance difference are used to test significance.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
• Montaño-Rendón et al. [5]. PtdIns(3,4)P2, Lamellipodin, and VASP coordinate actin dynamics during phagocytosis in macrophages. Journal of Cell Biology (Figure 3A and B).]
General notes and troubleshooting
General notes
1. Use early passage cells when making stable cell lines.
2. Determine the optimal doxycycline concentration for the desired level of expression. If robust over-expression is required, 1 mg/mL of doxycycline for 18 h is typically a good starting point. However, if the goal is to express genes of interest at near endogenous levels, we suggest using 100 ng/mL.
3. The protocol described is for RAW264.7 cells; however, we have also used this approach with other cell types, such as HeLa [17], HCT116 [18], and ARPE19 [19].
4. The protocol generates a polyclonal cell population. We prefer this approach in most cases since it prevents potential issues with expanding individual clones. However, following the introduction of the plasmids into the RAW264.7 cells, they can be diluted, and single cells can be aliquoted in each well of a 96-well plate for selection and expansion. RAW cells grow better in these situations when fresh media is supplemented with 2-day-old conditioned media (50:50).
5. In this example, we used spinning disc confocal microscopy. However, this assay can be done with less sophisticated systems. In our lab, we also use an EVOS M5000 widefield imaging system, which illuminates samples with three specific LEDs.
Troubleshooting
Opsonization of sheep red blood cells may result in agglutination, which can be observed as clumps after the incubation. To avoid agglutination, each vial of rabbit anti-sheep red blood cell antibody should be titrated (2–5 mL per milliliter).
Bangs Laboratories also sells latex beads without 2% DVB. In our experience, the human IgG does not adhere to these particles.
Phagocytosis is temperature-dependent. Ensure the media is prewarmed and cells are maintained at 37 °C during the assay.
Acknowledgments
This work was supported by a Project Grant from the Canadian Institutes of Health Research (PJT165968) to G.D.F. This bio-protocol is based on a previous publication from the laboratory, Montaño-Rendón et al. [5] Journal of Cell Biology (2022) 221(11): e202207042 and Cabral-Dias et al. [19] Journal of Cell Biology (2022) 221(4): e201808181.
Competing interests
The authors declare no competing interests.
References
- Kowarz, E., Löscher, D. and Marschalek, R. (2015). Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol J. 10(4): 647–653.
- Ivics, Z., Kaufman, C. D., Zayed, H., Miskey, C., Walisko, O. and Izsvak, Z. (2004). The Sleeping Beauty transposable element: evolution, regulation and genetic applications. Curr Issues Mol Biol. 6(1): 43–55.
- Ivics, Z., Hackett, P. B., Plasterk, R. H. and Izsvák, Z. (1997). Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell. 91(4): 501–510.
- Izsvák, Z., Ivics, Z. and Plasterk, R. H.(2000).Sleeping Beauty , a wide host-range transposon vector for genetic transformation in vertebrates 1 1Edited by J. Karn. J Mol Biol. 302(1): 93–102.
- Montaño-Rendón, F., Walpole, G. F., Krause, M., Hammond, G. R., Grinstein, S. and Fairn, G. D. (2022). PtdIns(3,4)P2, Lamellipodin, and VASP coordinate actin dynamics during phagocytosis in macrophages. J Cell Biol. 221(11): e202207042.
- Allison, A. C., Davies, P. and De Petris, S. (1971). Role of Contractile Microfilaments in Macrophage Movement and Endocytosis. Nat New Biol. 232(31): 153–155.
- Stossel, T. P. and Hartwig, J. H.(1976).Interactions of actin, myosin, and a new actin-binding protein of rabbit pulmonary macrophages. II. Role in cytoplasmic movement and phagocytosis. J Cell Biol. 68(3): 602–619.
- Greenberg, S. (1999). Modular components of phagocytosis. J Leukocyte Biol. 66(5): 712–717.
- Freeman, S. A. and Grinstein, S.(2014).Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. 262(1): 193–215.
- Levin, R., Grinstein, S. and Schlam, D.(2015).Phosphoinositides in phagocytosis and macropinocytosis. Biochim Biophys Acta Mol Cell Biol Lipids. 1851(6): 805–823.
- Ray, J., Sapp, D. G. and Fairn, G. D.(2024).Phosphatidylinositol 3,4-bisphosphate: Out of the shadows and into the spotlight. Curr Opin Cell Biol. 88: 102372.
- Goulden, B. D., Pacheco, J., Dull, A., Zewe, J. P., Deiters, A. and Hammond, G. R. (2018). A high-avidity biosensor reveals plasma membrane PI(3,4)P2 is predominantly a class I PI3K signaling product. J Cell Biol. 218(3): 1066–1079.
- Lu, S. M., Grinstein, S. and Fairn, G. D. (2016). Quantitative Live-Cell Fluorescence Microscopy During Phagocytosis. Methods Mol Biol. 1519: 79–91.
- Liu, S. Y., Mulugeta, N., Dougan, S. K. and Qiang, L. (2023). In vitro flow cytometry assay to assess primary human and mouse macrophage phagocytosis of live cells. STAR Protoc. 4(2): 102240.
- Lindner, B., Burkard, T. and Schuler, M.(2020).Phagocytosis Assays with Different pH-Sensitive Fluorescent Particles and Various Readouts. Biotechniques. 68(5): 245–250.
- Rueden, C. T., Schindelin, J., Hiner, M. C., DeZonia, B. E., Walter, A. E., Arena, E. T. and Eliceiri, K. W.(2017). ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinf. 18(1): 529.
- Walpole, G. F. W., Pacheco, J., Chauhan, N., Clark, J., Anderson, K. E., Abbas, Y. M., Brabant-Kirwan, D., Montaño-Rendón, F., Liu, Z., Zhu, H., et al. (2022). Kinase-independent synthesis of 3-phosphorylated phosphoinositides by a phosphotransferase. Nat Cell Biol. 24(5): 708–722.
- Dixon, C. L., Martin, N. R., Niphakis, M. J., Cravatt, B. F. and Fairn, G. D. (2023). Attenuating ABHD17 enhancesS-palmitoylation, membrane localization and signal transduction of NOD2 and Crohn’s disease-associated variants. bioRxiv. doi.org/10.1101/2023.12.20.572362.
- Cabral-Dias, R., Lucarelli, S., Zak, K., Rahmani, S., Judge, G., Abousawan, J., DiGiovanni, L. F., Vural, D., Anderson, K. E., Sugiyama, M. G., et al. (2022). Fyn and TOM1L1 are recruited to clathrin-coated pits and regulate Akt signaling. J Cell Biol. 221(4): e201808181.
Article Information
Publication history
Received: Sep 30, 2024
Accepted: Dec 2, 2024
Available online: Dec 26, 2024
Published: Feb 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Kamali, P. and Fairn, G. D. (2025). Using the Sleeping Beauty Transposon System for Doxycycline-inducible Gene Expression in RAW264.7 Macrophage Cells to Study Phagocytosis. Bio-protocol 15(3): e5178. DOI: 10.21769/BioProtoc.5178.
- Montaño-Rendón, F., Walpole, G. F., Krause, M., Hammond, G. R., Grinstein, S. and Fairn, G. D. (2022). PtdIns(3,4)P2, Lamellipodin, and VASP coordinate actin dynamics during phagocytosis in macrophages. J Cell Biol. 221(11): e202207042.
Category
Immunology > Immune cell function > Macrophage
Cell Biology > Cell-based analysis > Gene expression
Immunology > Immune mechanisms
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.