- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Protocol to Retrieve Unknown Flanking DNA Using Fork PCR for Genome Walking
Published: Vol 15, Iss 2, Jan 20, 2025 DOI: 10.21769/BioProtoc.5161 Views: 2618
Reviewed by: Alba BlesaFernando A Gonzales-ZubiateAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Sep 2023
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
PCR-based genome walking is one of the prevalent techniques implemented to acquire unknown flanking genomic DNAs. The worth of genome walking includes but is not limited to cloning full-length genes, mining new genes, and discovering regulatory regions of genes. Therefore, this technique has advanced molecular biology and related fields. However, the PCR amplification specificity of this technique needs to be further improved. Here, a practical protocol based on fork PCR is proposed for genome walking. This PCR uses a fork primer set of three arbitrary primers to execute walking amplification task, where the primary fork primer mediates walking by partially annealing to an unknown flank, and the fork-like structure formed between the three primers participates in inhibiting non-target amplification. In primary fork PCR, the low-annealing temperature (25 °C) cycle allows the primary fork primer to anneal to many sites of the genome, synthesizing a cluster of single-stranded DNAs; the subsequent 65 °C cycle processes the target single-strand into double-strand via the site-specific primer; then, the remaining 65 °C cycles selectively enrich this target DNA. However, any non-target single-stranded DNA formed in the 25 °C cycle cannot be further processed in the following 65 °C cycles because it lacks an exact binding site for any primer. Secondary, or even tertiary nested fork PCR further selectively enriches the target DNA. The practicability of fork PCR was validated by walking three genes in Levilactobacillus brevis CD0817 and one gene in Oryza sativa. The results indicated that the proposed protocol can serve as a supplement to the existing genome walking protocols.
Key features
• This protocol builds upon the method developed by Pan et al. [1], which is applicable to genome-walking for any species.
• The developed protocol is a random priming PCR-based genome-walking scheme.
• Two rounds of nested fork PCR amplifications suffice to release a positive walking result.
Keywords: Genome walkingGraphical overview
Background
Genome walking refers to a strategy used to mine unknown genomic regions flanking known DNAs. Genome walking has promoted the development of molecular biology–related fields by being widely applied, such as in cloning full-length genes, uncovering transgenic sites, and acquiring regulatory regions of genes. To date, many PCR-based genome walking techniques have been developed [2–6]. These techniques fall into two clusters according to the involved methodological principles: cluster I, genome pre-processing-dependent PCR [7–10], and cluster II, random priming PCR [11–13].
A random priming PCR method omits the genome pre-processing prior to PCR, having thus attracted increasing interest from researchers [14–16]. Over the past decades, about a dozen random PCR methods have been constructed, such as wristwatch PCR [4,17], racket PCR [3,5], and thermal asymmetric interlaced PCR [18,19]. In such a PCR, an arbitrary primer mediates genome walking by partially hybridizing to an unknown flank genomic region in the low-temperature cycle, while a nested sequence-specific primer(s) (NSP) ensures PCR amplification specificity. In general, this method requires at least two rounds of nested amplifications to obtain a target amplicon(s) but still suffers from a non-target background arising from the arbitrary primer [4,14]. Therefore, developing a random PCR genome walking method with satisfactory amplification specificity remains of interest.
Recently, we have developed a new genome-walking technique called fork PCR. The fork PCR depends on the partial overlaps between the three arbitrary primers [primary fork primer (PFP), secondary fork primer (SFP), and branch primer (BP)] in a fork primer set. PFP and SFP share a 3' part (stem), while their 5' parts (branches) are heterologous to each other. BP corresponds to the 5' branch of SFP. Therefore, PFP, SFP, and BP form a fork-like structure (Figure 1). The fork PCR has shown satisfactory amplification specificity mainly due to the following two facts: First, all thermal cycles in secondary/tertiary fork PCR are stringent (annealing temperature 65 °C). Second, the non-target product defined by PFP is hard to amplify in secondary/tertiary PCR as it tends to form a hairpin via intra-strand annealing mediated by the inverted PFP terminal repeats, rather than being annealed by SFP or BP. The feasibility of fork PCR has been verified by walking several selected genetic sites [1].
Materials and reagents
Biological materials
1. Genomic DNA of Levilactobacillus brevis CD0817 [20–23], prepared by our lab at Nanchang University (Nanchang, China)
2. Genomic DNA of Oryza sativa, obtained from the Lab of Dr. Xiaojue Peng at Nanchang University (Nanchang, China)
Reagents
1. LA Taq polymerase (hot-start version) (Takara, catalog number: RR042A)
2. dNTP mixture (Takara, catalog number: RR042A)
3. 10× LA PCR buffer (Mg2+ plus) (Takara, catalog number: RR042A)
4. 6× Loading buffer (Takara, catalog number: 9156)
5. DL 5,000 DNA marker (Takara, catalog number: 3428Q)
6. 1× TE buffer (Sangon, catalog number: B548106)
7. Agarose (Sangon, catalog number: A620014)
8. 1 M NaOH (Yuanye, catalog number: B28412)
9. Green fluorescent nucleic acid dye (10,000×) (Solarbio, catalog number: G8140)
10. 0.5 M EDTA (Solarbio, catalog number: B540625)
11. Boric acid (Solarbio, catalog number: B8110)
12. Tris (Solarbio, catalog number: T8060)
13. DiaSpin DNA Gel Extraction kit (Sangon, catalog number: B110092)
14. Primers (Sangon)
PFP1: 5'-ACGCGTAATAGCTCGGGATGATGCTGCTCGTGGATGACTCT-3'
SFP1: 5'-CCTGACCGCCTTCTACACCTATGCTGCTCGTGGATGACTCT-3'
PFP2: 5'-ATCCGCCCATAGCCTTCAGTGACTACGCTGCCTTGCTACTT-3'
SFP2: 5'-CCTGACCGCCTTCTACACCTGACTACGCTGCCTTGCTACTT-3'
BP: 5'-CCTGACCGCCTTCTACACCT-3'
oNSP-gadA: 5'-GTTTCTGGTCACAAGTACGGCATGG-3'
mNSP-gadA: 5'-TGCTGATACGCTGCCAGAAGAAATG-3'
iNSP-gadA: 5'-ACGGTTGACTCCATTGCCATTAACT-3'
oNSP-gadR: 5'-TCCTTCGTTCTTGATTCCATACCCT-3'
mNSP-gadR: 5'-CCATTTCCATAGGTTGCTCCAAGG-3'
iNSP-gadR: 5'-GGATACTGGCTAAAATGAATTAACTCGGATAA-3'
oNSP-pct: 5'-TCTTGTTCTTCAACAGTGGTGGGTA-3'
mNSP-pct: 5'-TCGTCTTTCGTGTAAGTGTTGGTGT-3'
iNSP-pct: 5'-AGGAAATATGCACTCTTGGGAAGCG-3'
oNSP-hyg: 5'-ACGGCAATTTCGATGATGCAGCTTG-3'
mNSP-hyg: 5'-GGGACTGTCGGGCGTACACAA-3'
iNSP-hyg: 5'-CTGGACCGATGGCTGTGTAGAAG-3'
Solutions
1. 2.5× TBE buffer (see Recipes)
2. 0.5× TBE buffer (see Recipes)
3. 100 μM primer (see Recipes)
4. 10 μM primer (see Recipes)
5. 1% agarose gel (see Recipes)
Recipes
1. 2.5× TBE buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 0.5 M EDTA solution | 5 mM | 10 mL |
| Tris | 225 mM | 27 g |
| Boric acid | 225 mM | 13.75 g |
| ddH2O | n/a | 950 mL |
| Total | n/a | 1,000 mL |
Adjust pH to 8.3 with 1 M NaOH and then top the solution to 1,000 mL with ddH2O.
2. 0.5× TBE buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 2.5× TBE buffer | 0.5× | 200 mL |
| ddH2O | n/a | 800 mL |
| Total | n/a | 1,000 mL |
3. 100 μM primer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Powdery primer | 100 μM | n/a |
| 1× TE buffer | 1× | Volume specified in the sheet of primer synthesis |
| Total | n/a | Volume specified in the sheet of primer synthesis |
Note: Dilute a portion of the 100 μM primer to prepare 10 μM primer and store the remaining portion at -80 °C.
4. 10 μM primer
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| 100μM primer | 10 μM | 1 μL |
| 1× TE buffer | 1× | 9 μL |
| Total | n/a | 10 μL |
Note: Prepare extra volume of a 10 μM primer and pipette it to multiple 1.5 mL microcentrifuge tubes. Then, store the tubes at -80 °C. Take one tube at a time and store it at -20 °C after use.
5. 1% agarose gel
| Reagent | Final concentration | Quantity or Volume |
|---|---|---|
| Agarose | 1% | 1 g |
| 0.5× TBE buffer | 0.5× | 100 mL |
| Green fluorescent nucleic acid dye (10,000×) | 1× | 10 μL |
| Total | n/a | 100 mL |
Laboratory supplies
1. 0.2 mL thin-wall PCR tubes (Kirgen, catalog number: KG2311)
2. 10 μL pipette tips (Sangon, catalog number: F600215)
3. 200 μL pipette tips (Sangon, catalog number: F600227)
4. 1,000 μL pipette tips (Sangon, catalog number: F630101)
5. 1.5 mL microcentrifuge tubes (Labselect, catalog number: MCT-001-150)
Equipment
1. PCR apparatus (Analtytikjena, model: Biometra TAdvanced)
2. Microcentrifuge (Tiangen, model: TGear)
3. Electrophoresis apparatus (Beijing Liuyi, model: DYY-6C)
4. Gel imaging system (Bio-Rad, model: ChemiDoc XRS+)
Software and datasets
1. Oligo 7 software (Molecular Biology Insights, Inc., USA)
2. DNASTAR Lasergene software (DNASTAR, Inc.)
Procedure
A. Design of primers
1. Select three NSPs—outmost NSP (oNSP), middle NSP (mNSP), and innermost NSP (iNSP)—from a known DNA.
Critical: The Tm values of NSPs are from 60 to 65 °C. An NSP itself should avoid forming a hairpin structure with a Tm value exceeding 40 °C.
2. Design two sets of fork primers. The three primers (PFP, SFP, and BP) in each set form a fork-like structure (Figure 1).

Figure 1. Fork-like structures of the two walking primer sets. PFP or SFP consists of 5' branch and 3' stem. The stems of the two primers are homologous to each other, while the branches are heterologous to each other. BP corresponds to the branch of SFP. The melting temperatures of PFP1, SFP1, PFP2, SFP2, and BP are 73.6, 74.2, 74.6, 74.5, and 60.7 °C, respectively. BP is universal to the two fork primer sets. PFP: primary fork primer, SFP: secondary fork primer, and BP: branch primer.
Critical: The sequence of a fork primer (PFP, SFP, or BP) is completely arbitrary, with the four bases adenine (A), thymine (T), cytosine (C), and guanine (G) being evenly distributed. Meanwhile, a fork primer itself should avoid forming a severe hairpin or dimer structure and meet the criteria shown in Table 1. Simultaneously designing more than one fork primer set is suggested to execute parallel fork PCRs in a walking cycle.
Table 1. Key criteria for designing fork primer
| Primer | Length (nt) | G+C content (%) | Melting temperature (°C) |
|---|---|---|---|
| PFP | 41 | 40–60 | ~75 |
| SFP | 41 | 40–60 | ~75 |
| BP | 20 | 40–60 | 60–65 |
PFP: primary fork primer; SFP: secondary fork primer; BP: branch primer; G: guanine; C: cytosine.
Note: Use the Oligo 7 software to devise and assess primers.
B. Fork PCR amplifications
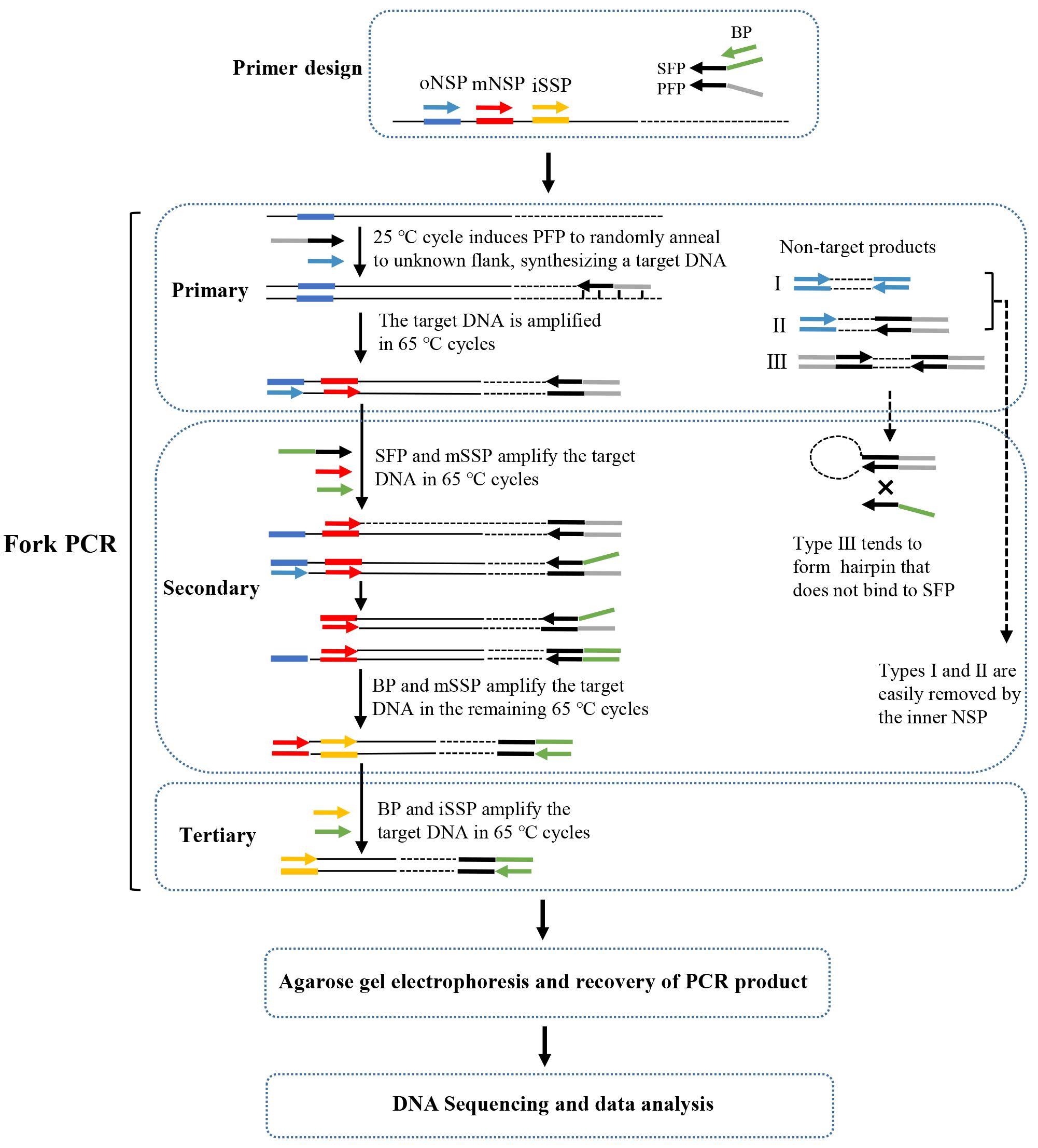
A fork PCR set comprises three rounds of nested amplifications. Figure 2 describes the process of fork PCR.
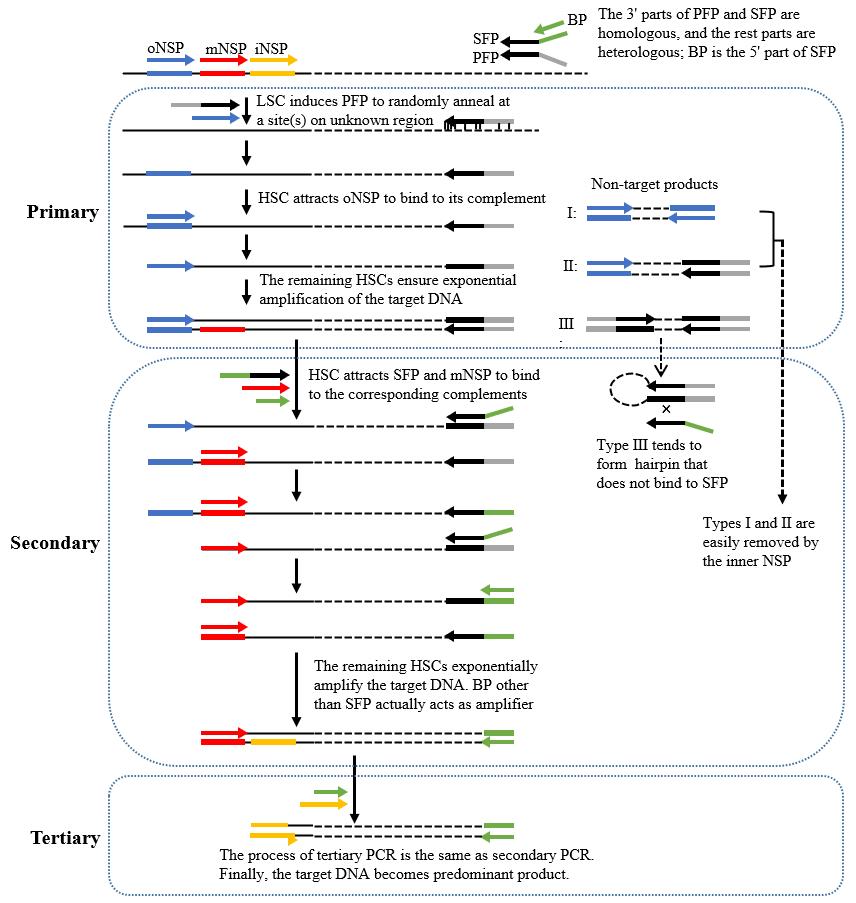
Figure 2. Schematic diagram of fork PCR. oNSP: outmost nested site-specific primer; mNSP: middle nested site-specific primer; iNSP: innermost nested site-specific primer; PFP: primary fork primer; SFP: secondary fork primer; BP: branch primer; HSC: high-stringency cycle; LSC: low-stringency cycle. Thin solid line: known DNA; dotted line: unknown DNA; arrows: primers; thick solid lines: primer complements.
Critical: The working concentration of SFP is 10% of that of BP or mNSP.
Note: In secondary fork PCR, types I (defined by oNSP) and II (defined by oNSP and PFP) non-target products are readily removed because they lack an exact binding site for mNSP. Type III non-target product (defined by PFP) tends to form hairpin via the PFP termini rather than binding with SFP, because PFP has a higher Tm value than the overlap between PFP and SFP. Clearly, the amplification of type III non-target product is also inhibited. However, the target product can be exponentially enriched once SFP integrates into the PFP site. Tertiary fork PCR further selectively enriches the target product.
1. Primary fork PCR
a. Pipette primary PCR components (Table 2) into a 0.2 mL PCR tube.
Table 2. Primary fork PCR mix
| Reagent | Final concentration | Amount (μL) |
|---|---|---|
| Genomic DNA | Microbe, 0.2–2 ng/μL; plant or animal, 2–20 ng/μL | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| PFP (10 μM) | 0.2 μM | 1 |
| oNSP (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (Mg2+ plus) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| ddH2O | n/a | 33.5 |
| Total | n/a | 50 |
b. Completely mix the components with a pipette.
c. Centrifuge for 10–20 s with a microcentrifuge to gather the mixture.
d. Run the amplification in the PCR apparatus (Table 3).
Table 3. Primary fork PCR cycling conditions
| Step | Temperature (°C) | Duration | Cycle |
| Initial denaturation | 95 | 2 min | 1 |
| Denaturation | 95 | 10 s | 1 |
| Annealing | 25 | 30 s | |
| Extension | 72 | 2 min | |
| Denaturation | 95 | 10 s | 30 |
| Annealing | 65 | 30 s | |
| Extension | 72 | 2 min | |
| Final extension | 72 | 5 min | 1 |
| Hold | 4 | forever | |
e. Put the PCR product onto ice.
f. Take 1 μL of the product as the template of secondary fork PCR.
g. Store the remaining product at -20 °C for future assays.
2. Secondary fork PCR
a. Pipette secondary PCR components (Table 4) into a 0.2 mL PCR tube.
Table 4. Secondary fork PCR mix
| Reagent | Final concentration | Amount (μL) |
|---|---|---|
| Primary PCR product | n/a | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| BP (10 μM) | 0.2 μM | 1 |
| SFP (1 μM) | 0.02 μM | 1 |
| mNSP (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (Mg2+ plus) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| ddH2O | n/a | 32.5 |
| Total | n/a | 50 |
Note: The working concentration of SFP is 10% of that of BP or mNSP.
Critical: Dilute primary PCR product 10–1,000 fold if necessary.
b. Completely mix the components with a pipette.
c. Centrifuge for 10–20 s with a microcentrifuge to gather the mixture.
d. Run the amplification in the PCR apparatus (Table 5).
Table 5. Secondary fork PCR cycling conditions
| Step | Temperature (°C) | Duration | Cycle |
| Initial denaturation | 95 | 2 min | 1 |
| Denaturation | 95 | 10 s | 30 |
| Annealing | 65 | 30 s | |
| Extension | 72 | 2 min | |
| Final extension | 72 | 5 min | 1 |
| Hold | 4 | forever | |
e. Put the PCR product onto ice.
f. Take 1 μL of the product as the template of tertiary fork PCR.
g. Store the remaining product at -20 °C for future assays.
3. Tertiary fork PCR
a. Pipette tertiary amplification components (Table 6) into a 0.2 mL PCR tube.
Table 6. Tertiary fork PCR mix
| Reagent | Final concentration | Amount (μL) |
|---|---|---|
| Secondary PCR product | n/a | 1 |
| LA Taq polymerase (5 U/μL) | 0.05 U/μL | 0.5 |
| BP (10 μM) | 0.2 μM | 1 |
| iNSP (10 μM) | 0.2 μM | 1 |
| 10× LA PCR buffer II (Mg2+ plus) | 1× | 5 |
| dNTP mixture (2.5 mM each) | 0.4 mM each | 8 |
| ddH2O | n/a | 33.5 |
| Total | n/a | 50 |
Critical: Dilute secondary PCR product 10–1,000 fold if necessary.
b. Completely mix the components with a pipette.
c. Centrifuge for 10–20 s with a microcentrifuge to gather the mixture.
d. Run the amplification in the PCR apparatus (Table 7).
Table 7. Tertiary fork PCR cycling conditions
| Step | Temperature (°C) | Duration | Cycle |
| Initial denaturation | 95 | 2 min | 1 |
| Denaturation | 95 | 10 s | 30 |
| Annealing | 65 | 30 s | |
| Extension | 72 | 2 min | |
| Final extension | 72 | 5 min | 1 |
| Hold | 4 | forever | |
e. Store the PCR product at -20 °C for future assays.
C. Gel electrophoresis
1. Completely mix 5 μL of PCR product and 1 μL of 6× loading buffer.
2. Load the mixture into 1% agarose gel supplemented with 1× green fluorescent nucleic acid dye.
3. Set the electrophoresis apparatus to a voltage of 150 V (the distance between the two electrodes is 30 cm).
4. Check the PCR product using the ChemiDoc XRS+ imaging system after approximately 25 min of electrophoresis (Figure 3).
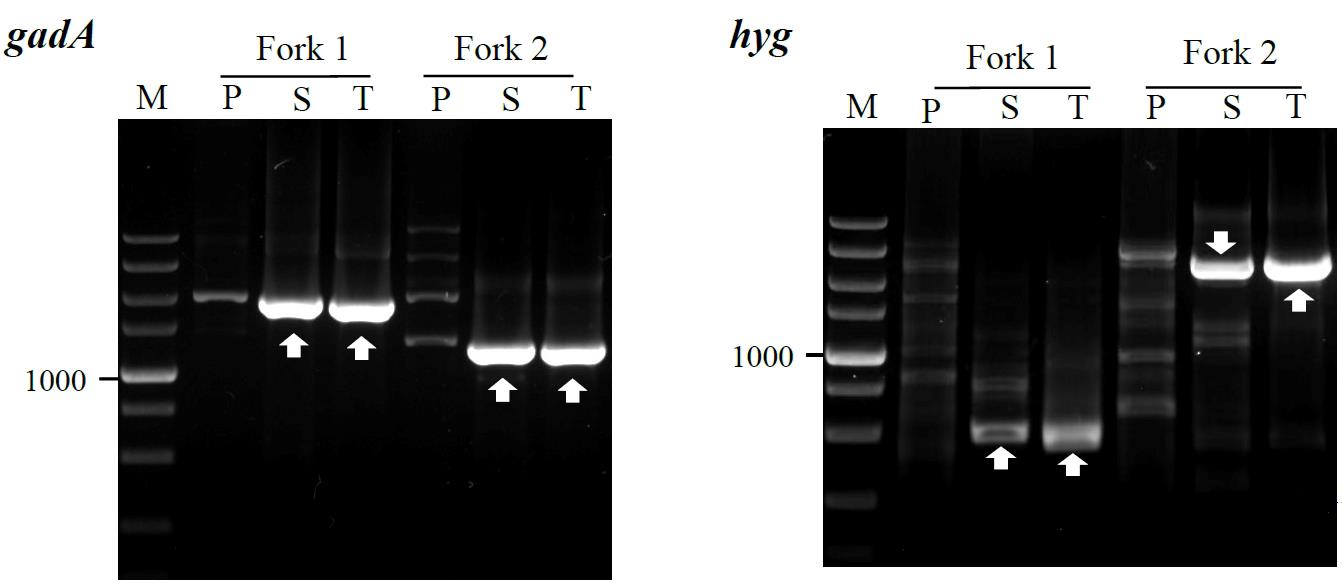
Figure 3. Fork PCR to genes gadA and hyg. Forks 1 and 2 denote the two parallel fork PCR sets in the walking experiment. The fragments marked with white arrows indicate the target products. Lanes P, S, and T show primary, secondary, and tertiary PCR, respectively. Lane M: TaKaRa DL5000 Marker.
D. Recovery of PCR product
1. Completely mix 40 μL of secondary/tertiary fork PCR product and 8 μL of 6× loading buffer.
2. Load the mixture into 1% agarose gel supplemented with 1× green fluorescent nucleic acid dye.
3. Set the electrophoresis apparatus to a voltage of 150 V (the distance between the two electrodes is 30 cm).
4. Visualize the PCR product using the ChemiDoc XRS+ imaging system. Subsequently, cut out clear DNA band(s) using a knife.
5. Extract DNA from the cut gel using the DiaSpin DNA Gel Extraction kit.
6. Confirm the extracted DNA with 1% agarose gel electrophoresis.
E. DNA sequencing
Directly sequence the extracted DNA at Sangon Biotech Co., Ltd.
Data analysis
The correctness of obtained DNA is tested by sequence alignment performed by the “By Clustal W Method” function in MegAlign software. The walking is regarded as correct if one end of the obtained DNA sequence overlaps with one end of the known DNA [24,25].
1. Open the MegAlign software, click File, and then click Enter Sequences (Figure 4).
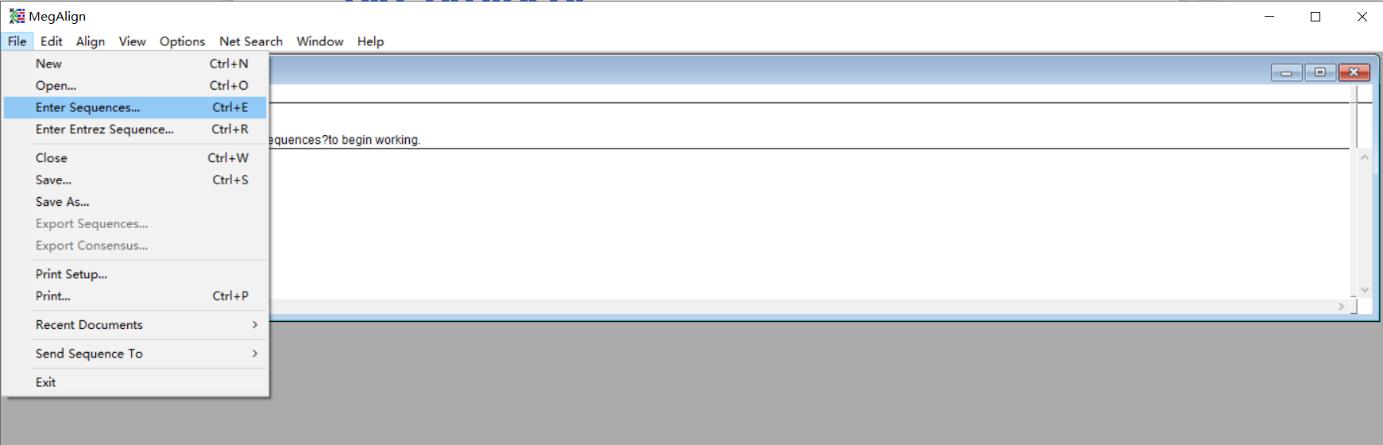
Figure 4. Screenshot of the MegAlign software showing the location of the Enter Sequences under the File tab
2. Input a DNA sequence walked by fork PCR and the corresponding known DNA segment between iNSP and unknown flank.
3. Click Align, and then click the By Clustal W Method (Figure 5) to output the alignment result (Figure 6).
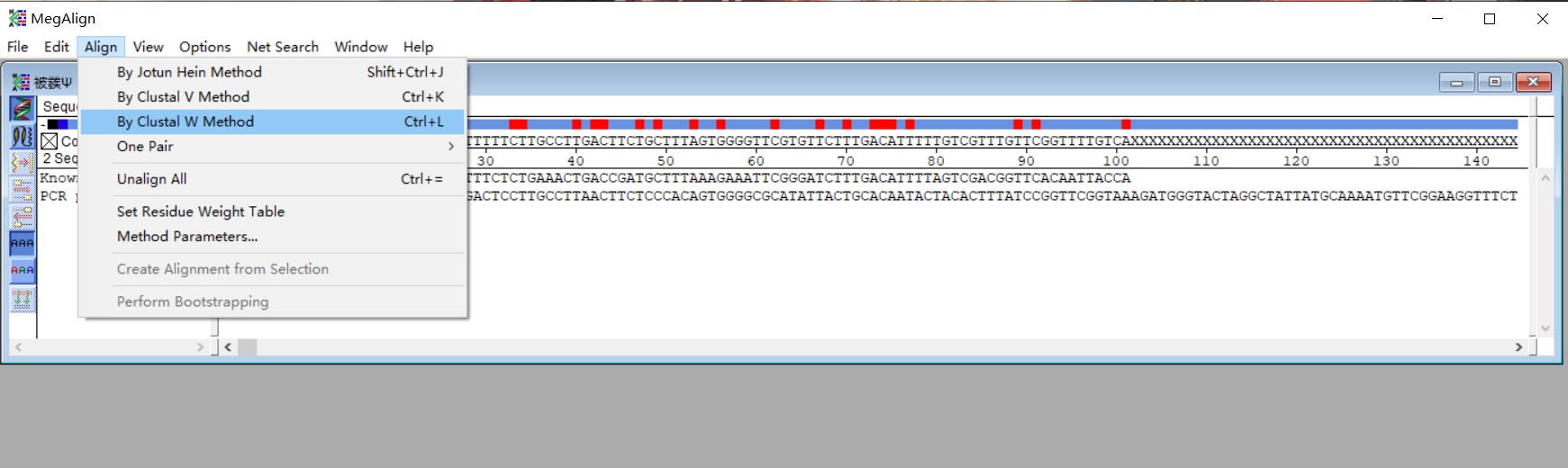
Figure 5. Screenshot of the MegAlign software displaying the input DNA sequences
Figure 6. Screenshot of the MegAlign software displaying the alignment result
Note: The walking is considered successful if the iNSP-sided part of the PCR product overlaps the known DNA.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article:
Pan et al. [1] Fork PCR: a universal and efficient genome-walking tool. Frontiers in Microbiology (Figure 3).
General notes and troubleshooting
General notes
1. Three primers, SFP, BP, and mNSP, are used in secondary fork PCR. However, the working concentration of SFP is only 10% of that of BP or mNSP. The major role of SFP is to integrate the BP sequence into the PFP site, and the real amplifiers are BP and mNSP.
2. Primary fork PCR product can be exempted from testing.
3. After secondary fork PCR, the PCR product is checked using agarose gel electrophoresis to determine whether a clear DNA band(s) appears. If a clear DNA band(s) appears, it is recovered and sequenced. There is no need to perform tertiary PCR if this DNA band(s) is correct.
4. Like the other PCR-based walking protocols, the current protocol also suffers from the multiple-band phenomenon. In general, only the largest DNA band needs to be analyzed if multiple DNA bands appear.
5. Simultaneously performing parallel fork PCRs in a walking cycle will improve walking success and efficiency.
6. The two fork primer sets provided here are universal to any species. A researcher needs only to design an NSP set in a walking experiment. An NSP itself should avoid forming a severe hairpin or dimer; in addition, it should avoid forming a severe dimer with the paired fork primer.
7. Researchers can also design more fork primer sets as they wish, provided that they meet the criteria mentioned above. N fork primer sets require n PFPs, n SFP, and one BP. The BP is universal to the N fork primer sets.
Troubleshooting
Problem 1: No clear DNA band(s) appears in secondary or even tertiary fork PCR.
Possible cause: In primary fork PCR, non-target amplification is very strong, while target amplification is very weak.
Solution: Dilute primary PCR product 10–1,000 times; then, use 1 μL dilutions as templates for secondary fork PCRs. Afterward, perform the corresponding tertiary PCRs. If still no clear DNA(s) appears in any secondary/tertiary PCR, redesign an NSP set with a different position from the previous one.
Problem 2: The multi-band phenomenon is serious, which affects the cutting and recovery of target PCR products from agarose gel.
Possible cause: In primary fork PCR, PFP randomly anneals to many sites on unknown flanking DNA.
Solution: Dilute the primary PCR product 10–1,000 times and use 1 μL dilutions as templates for secondary fork PCRs. Then, select a satisfactory secondary PCR. In fact, the multi-band phenomenon is common in random priming PCR-based genome-walking methods but, generally, only the largest DNA band needs to be considered.
Problem 3: Direct sequencing of fork PCR product is not smooth.
Possible cause: There is interference from the non-target background.
Solution: Clone the product and then sequence it.
Problem 4: Clear DNA band(s) is the non-target product.
Possible cause: Genomic DNA template may be contaminated.
Solution: Use an uncontaminated genomic DNA template, use dedicated sterile consumables, and perform PCR in a separate and clean region.
Acknowledgments
This study was supported by the Jiangxi Provincial Department of Science and Technology (Grant no. 20225BCJ22023), China and the National Natural Science Foundation of China (Grant no. 32160014), China. This fork PCR-based genome-walking protocol has been originally described and validated in Frontiers in Microbiology [1].
Competing interests
The authors declare no competing interests.
References
- Pan, H., Guo, X., Pan, Z., Wang, R., Tian, B. and Li, H. (2023). Fork PCR: a universal and efficient genome-walking tool. Front Microbiol. 14: e1265580.
- Kotik, M. (2009). Novel genes retrieved from environmental DNA by polymerase chain reaction: Current genome-walking techniques for future metagenome applications. J Biotechnol. 144(2): 75–82.
- Sun, T., Jia, M., Wang, L., Li, Z., Lin, Z., Wei, C., Pei, J. and Li, H. (2022). DAR-PCR: a new tool for efficient retrieval of unknown flanking genomic DNA. AMB Express. 12(1): 131.
- Wang, L., Jia, M., Li, Z., Liu, X., Sun, T., Pei, J., Wei, C., Lin, Z. and Li, H. (2022). Wristwatch PCR: A Versatile and Efficient Genome Walking Strategy. Front Bioeng Biotechnol. 10: e792848.
- Pei, J., Sun, T., Wang, L., Pan, Z., Guo, X. and Li, H. (2022). Fusion primer driven racket PCR: A novel tool for genome walking. Front Genet. 13: e969840.
- Wang, R., Gu, Y., Chen, H., Tian, B. and Li, H. (2025). Uracil base PCR implemented for reliable DNA walking. Anal Biochem. 696: 115697.
- Yik, M. Y., Lo, Y. T., Lin, X., Sun, W., Chan, T. F. and Shaw, P. C. (2021). Authentication of Hedyotis products by adaptor ligation-mediated PCR and metabarcoding. J Pharm Biomed Anal. 196: 113920.
- Uchiyama, T. and Watanabe, K. (2006). Improved inverse PCR scheme for metagenome walking. Biotechniques. 41(2): 183–188.
- Alquezar‐Planas, D. E., Löber, U., Cui, P., Quedenau, C., Chen, W. and Greenwood, A. D. (2020). DNA sonication inverse PCR for genome scale analysis of uncharacterized flanking sequences. Methods Ecol Evol. 12(1): 182–195.
- Trinh, Q., Shi, H., Xu, W., Hao, J., Luo, Y. and Huang, K. (2012). Loop-linker PCR: An advanced PCR technique for genome walking. IUBMB Life. 64(10): 841–845.
- Lin, Z., Wei, C., Pei, J. and Li, H. (2023). Bridging PCR: An efficient and reliable scheme implemented for genome-walking. Curr Issues Mol Biol. 45(1): 501–511.
- Wei, C., Lin, Z., Pei, J., Pan, H. and Li, H. (2023). Semi-site-specific primer PCR: A simple but reliable genome-walking tool. Curr Issues Mol Biol. 45(1): 512–523.
- Chen, H., Wei, C., Lin, Z., Pei, J., Pan, H. and Li, H. (2024). Protocol to retrieve unknown flanking DNA sequences using semi-site-specific PCR-based genome walking. STAR Protoc. 5(1): 102864.
- Chang, K., Wang, Q., Shi, X., Wang, S., Wu, H., Nie, L. and Li, H. (2018). Stepwise partially overlapping primer-based PCR for genome walking. AMB Express. 8(1): 77.
- Li, H., Lin, Z., Guo, X., Pan, Z., Pan, H. and Wang, D. (2024). Primer extension refractory PCR: an efficient and reliable genome walking method. Mol Genet Genomics. 299(1): 27.
- Guo, X., Zhu, Y., Pan, Z., Pan, H. and Li, H. (2024). Single primer site-specific nested PCR for accurate and rapid genome-walking. J Microbiol Methods. 220: 106926.
- Wang, L., Jia, M., Li, Z., Liu, X., Sun, T., Pei, J., Wei, C., Lin, Z. and Li, H. (2023). Protocol to access unknown flanking DNA sequences using Wristwatch-PCR for genome-walking. STAR Protoc. 4(1): 102037.
- Liu, Y. G. and Whittier, R. F. (1995). Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking. Genomics. 25(3): 674–681.
- Foster, J. M., Christodoulou, Z., Cowan, G. M. and Newbold, C. I. (1999). Thermal asymmetric interlaced PCR amplification of YAC insert end fragments for chromosome walking in Plasmodium falciparum and other A/T-rich genomes. Biotechniques. 27(2): 240–248.
- Gao, D., Chang, K., Ding, G., Wu, H., Chen, Y., Jia, M., Liu, X., Wang, S., Jin, Y., Pan, H., et al. (2019).Genomic insights into a robust gamma-aminobutyric acid-producer Lactobacillus brevis CD0817. AMB Express. 9(1): 72.
- Jia, M., Zhu, Y., Wang, L., Sun, T., Pan, H. and Li, H. (2022). pH auto-sustain-based fermentation supports efficient gamma-aminobutyric acid production by Lactobacillus brevis CD0817. Fermentation. 8(5): 208.
- Li, H., Sun, T., Jia, M., Wang, L., Wei, C., Pei, J., Lin, Z. and Wang, S. (2022). Production of gamma-aminobutyric acid by Levilactobacillus brevis CD0817 by coupling fermentation with self-buffered whole-cell catalysis. Fermentation. 8(7): 321.
- Wang, L., Jia, M., Gao, D. and Li, H. (2024). Hybrid substrate-based pH autobuffering GABA fermentation by Levilactobacillus brevis CD0817. Bioprocess Biosyst Eng. 47(12): 2101–2110.
- Li, H., Ding, D., Cao, Y., Yu, B., Guo, L. and Liu, X. (2015). Partially overlapping primer-based PCR for genome walking. PLoS One. 10(3): e0120139.
- Tian, B., Wu, H., Wang, R., Chen, H. and Li, H. (2024). N7-ended walker PCR: An efficient genome-walking tool. Biochem Genet. doi.org/10.1007/s10528-024-10896-1.
Article Information
Publication history
Received: Sep 14, 2024
Accepted: Nov 17, 2024
Available online: Nov 27, 2024
Published: Jan 20, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Wu, H., Pan, H. and Li, H. (2025). Protocol to Retrieve Unknown Flanking DNA Using Fork PCR for Genome Walking. Bio-protocol 15(2): e5161. DOI: 10.21769/BioProtoc.5161.
Category
Molecular Biology > DNA > PCR
Microbiology > Microbial genetics > DNA > PCR
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.