- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Simple Staining Method Using Pyronin Y for Laser Scanning Confocal Microscopy to Evaluate Gelatin Cryogels
Published: Vol 14, Iss 22, Nov 20, 2024 DOI: 10.21769/BioProtoc.5115 Views: 2112
Reviewed by: Hélène LégerHsih-Yin TanAnonymous reviewer(s)
Advertisement



Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
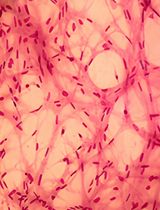
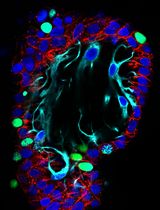
Abstract
This study explores the novel application of pyronin Y for fluorescently labeling extracellular matrices (ECMs) and gelatin cryogels, providing a simple and reliable method for laser scanning confocal microscopy. Pyronin Y exhibited remarkable staining ability of the porous structures of gelatin cryogels, indicating its potential as a reliable tool for evaluating such biomaterials. Confocal imaging of pyronin Y–stained cryogels produced high signal-to-noise ratio images suitable for quantifying pores using Fiji/Image J. Importantly, pyronin Y enabled effective dual-color imaging of cryogel-labeled mesenchymal stem cells, expanding its utility beyond traditional RNA assays. Traditional staining methods like Mason’s trichrome and Sirius Red have limitations in cryogel applications. Pyronin Y emerges as a powerful alternative due to its water solubility, minimal toxicity, and stability. Our results demonstrate pyronin Y’s ability to specifically stain gelatin cryogel's porous structures, surpassing its weak staining of ECMs in 2D. Confocal imaging revealed enduring staining even under rigorous scanning, with no notable photobleaching observed. Furthermore, pyronin Y's combination with Alexa Fluor 647 for dual-color imaging showed promising results, validating its versatility in fluorescence microscopy. In conclusion, this study establishes pyronin Y as a cost-effective and rapid option for fluorescent staining of gelatin cryogels. Its simplicity, efficacy, and compatibility with confocal microscopy make it a valuable tool for characterizing and evaluating gelatin-based biomaterials, contributing significantly to the field of cryogel imaging. The study opens new avenues for dual-color imaging in biomaterial research and tissue engineering, advancing our understanding of cellular interactions within scaffolds.
Key features
• Fluorescent staining of gelatin-based cryogels with an inexpensive yet less time-consuming protocol.
• Pyronin Y staining is suitable for dual-color confocal imaging by combining with far-red fluorophores (such as Alexa Fluor 647).
• The method is conducted routinely.
Keywords: Pyronin YGraphical overview
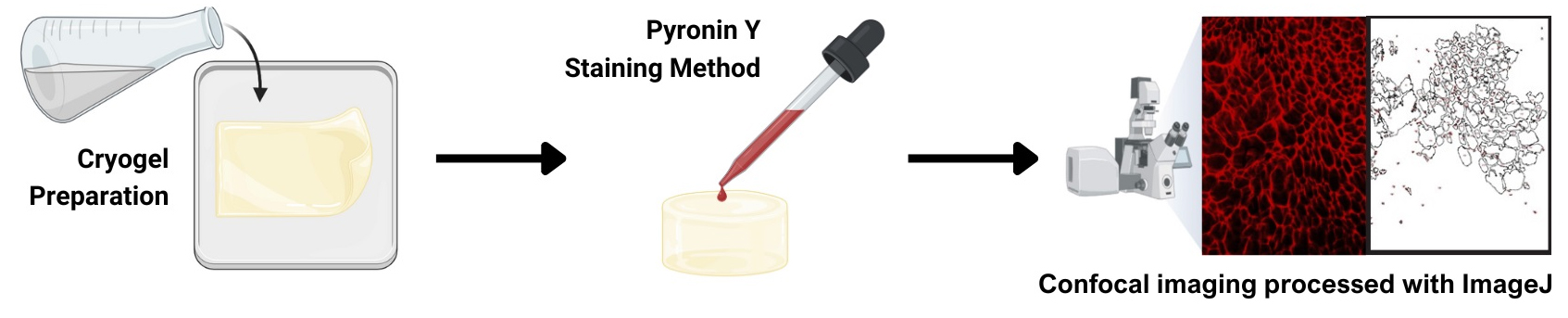
Gelatin cryogel staining using pyronin Y
Background
In this study, we report the novel application of pyronin Y dye in cryogel imaging combined with confocal microscopy, focusing on the detailed visualization of the cryogel’s porous structure. To our knowledge, this is the first protocol demonstrating a very fast yet reliable fluorescent staining of gelatin-based cryogels. We also show its compatibility with dual-color imaging.
The lack of a straightforward, rapid, and reliable fluorescent labeling method for cryogels is one obstacle in the field. Traditional methods like Mason’s trichrome for collagen [1] and the anionic dye, Sirius Red, for polarized microscopy [2] have limitations in cryogel applications. Mason’s trichrome staining is optimized to stain collagen fibers of tissue sections, not biomaterials [3]. While immunofluorescent staining of cryogels is possible using anti-gelatin antibodies, it is cost-prohibitive and not used for cryogels. Covalent conjugation of fluorescent dyes to gelatin is effective in visualizing the architecture of gelatin-based cryogels [4]; however, it is time-consuming.
Initially, we evaluated the dye’s potential for staining extracellular matrix (ECM)-coated glass surfaces in 2D cultures and discovered that pyronin Y can specifically stain 3D porous structures. Traditionally, pyronin Y has been a staple in RNA analysis [5–7]. Cryogels are unique porous scaffolds formed at sub-zero temperatures (around -20 °C) through a freeze-thaw process resulting in the formation of a porous network of polymer strands, post-thawing, due to the sublimation of ice crystals [8,9]. The interconnected porosity of cryogels allows efficient diffusion of nutrients and oxygen, rendering them advantageous for a variety of applications in tissue engineering, including superior cell growth [10].
Pyronin Y (C17H19CIN2O) features a dimethylamino group [N(CH3)2] and a xanthene-3-iminium group, which render the dye its cationic nature and distinctive red fluorescence. Additionally, pyronin Y is water-soluble and therefore easy to apply to various biological applications. Furthermore, it is considered to be safe for use in biological research, due to its relatively low toxicity. Lastly, pyronin Y is a stable dye that can be stored for extended periods without significant degradation or loss of fluorescence intensity. Past studies [5,7,11] have demonstrated pyronin Y’s efficacy in RNA analysis, owing to its ability to emit fluorescent light upon binding to RNA through ionic interactions. Thus, pyronin Y is established as a crucial component in a variety of RNA-based assays, such as in situ hybridization, [11], flow cytometry [5], and real-time RNA imaging [7]. However, no studies explored pyronin Y's potential for the imaging of ECMs, as shown here.
The properties of pyronin Y, such as its vivid red fluorescence, positive charge, water solubility, minimal toxicity, and chemical stability, render it a highly adaptable dye for the analysis of various extracellular matrices. Gelatin, which primarily constitutes heterogeneous peptides, is a predominantly negatively charged protein in alkaline pH [10]. This protocol explores the efficacy of pyronin Y in staining gelatin-based cryogels.
Materials and reagents
Biological materials
StemProTM BM mesenchymal stem cell (Gibco, Thermo Fisher Scientific, catalog number: A15652)
Note: Other mesenchymal stem cells (MSCs) may be used, such as those isolated from the umbilical cord [12] or from other sources.
Reagents
Pyronin Y (Alfa Aesar, Brand, catalog number: J61068, lot F18Z037), stable at room temperature
Glutaraldehyde 25% solution (Sigma-Aldrich, catalog number: G6257); once opened, you may store it in a refrigerator, tightly sealing the container
Sodium borohydride (Sigma-Aldrich, catalog number: 452882)
Sodium carbonate (Fisher Scientific, catalog number: S64-500) (any equivalent or higher quality of the same chemicals should work fine)
Hydrochloric acid (for pH adjustment) (Fisher Scientific, catalog number: A1445-500) (any equivalent or higher quality of the same chemicals should work fine)
16% paraformaldehyde (Electron Microscopic Sciences, catalog number: 15700); once opened, you may store it in a refrigerator, tightly sealing the container
Triton X-100 (Sigma-Aldrich, catalog number: T8787)
Fish skin gelatin (Sigma-Aldrich, catalog number: G7041)
Alexa Fluor 647 phalloidin (Invitrogen, Thermo Fisher Scientific, catalog number: A22287)
Dulbecco’s modified Eagle medium (Gibco, Thermo Fisher Scientific, catalog number: 11995), store at 4 °C
Fetal bovine serum (premium) (Gibco, Thermo Fisher Scientific, catalog number: A5670701)
Dulbecco’s phosphate-buffered saline (PBS) without calcium and magnesium (Gibco, Thermo Fisher Scientific, catalog number: 14190144)
Solutions
Gelatin cryogel solution (see Recipes)
0.1 M sodium bicarbonate buffer (see Recipes)
Pyronin Y solution (see Recipes)
4% Paraformaldehyde solution (see Recipes)
0.5% Triton X-100 solution (see Recipes)
Recipes
Gelatin cryogel solution
Reagent Final concentration Amount (per 1.5 mm thick gel) Gelatin (8.6%) 4.3% 3,000 μL Glutaraldehyde (25%) * 0.3% 72 μL H2O n/a 2,928 μL Total n/a 6,000 μL *Add after mixing other parts.
0.1 M sodium carbonate buffer (pH 9.2)
Reagent Final concentration Amount Sodium carbonate 0.1 M 10.6 g (when anhydrous is used) HCl n/a (add dropwise to adjust the pH) Total with H2O n/a 1,000 mL Pyronin Y solution
Reagent Final concentration Amount Pyronin Y 1 mg/mL 1 mg ddH2O n/a 1 mL Total n/a 1 mL It is highly recommended to spin briefly (~30 s) at 10,000× g to remove any insoluble pyronin Y particles after dissolving the pyronin Y powder.
4% paraformaldehyde solution
Reagent Final concentration Amount 16% paraformaldehyde solution 4% 2.5 mL PBS n/a 7.5 mL Total n/a 10 mL You can downscale the volume.
0.1% Triton X-100 solution
Reagent Final concentration Amount Triton X-100 0.1% 10 μL PBS n/a 9,990 μL Total n/a 10 mL You can downscale the volume. It will be fast to dissolve Triton X-100 by using a magnetic stirrer bar in a small beaker. Alternatively, making a 10% Triton X-100 stock solution helps this process. If a 10% stock solution is used, the volume of Triton X-100 should be 100 μL.
Laboratory supplies
1.5 mL or 2.0 mL microcentrifuge tubes
Micropipette (P200 and P1000)
1.5 mL tube rack
15 mL tubes
100 mm plastic Petri dishes or square dishes (e.g., Carolina Biological Supply, catalog number: 741470)
Gloves
Flat-tip forceps
35 mm glass bottom dishes (Mattek, catalog number: P35G-1.5-7-C)
Note: As long as the thickness is #1.5, any glass-bottom dishes for cellular imaging would be suitable.
1.5 mm spacer glass plates (Bio-Rad, catalog number: 1653312)
Short glass plates (Bio-Rad, catalog number: 1653308)
Mini-PROTEAN tetra cell casting stand & clamps (Bio-Rad, catalog number: 1658050)
Mini-PROTEAN casting stand gasket (Bio-Rad, catalog number: 1653305)
(alternative for 10/11) glass tube (the bottom has to be tightly sealed with a removable plug) or plastic disposable syringes (no needle required; the top has to be cut off)
Equipment
LSM 880 (Carl Zeiss)
20× objective lens; a high numerical aperture, longer working distance objective lens is strongly recommended, such as Plan-Apochromat 20×/N.A. = 0.8 M27 (Carl Zeiss, catalog number: 420650-9901-000)
-20 °C freezer (Thermo Fisher Scientific)
UV crosslinker (such as Spectronics Corporation, catalog number: XL-1000) or a cell culture hood equipped with a UV lamp
Software and datasets
Fiji/Image J (2.15.1), available without any cost
Microsoft Excel (no specific version as long as Office 2003 or newer versions are used)
Fluorescence Spectraviewer (Bio-Rad; https://www.bio-rad-antibodies.com/spectraviewer.html), available without any costs. Only required if you want to confirm the compatibility of your microscopic illumination settings.
Procedure
Preparation of gelatin cryogels
Set up the gel casting apparatus
Use either a protein gel apparatus (to prepare a sheet-type cryogel), glass tubes, or small syringes (to prepare a tube-type cryogel).
Make sure to measure the volume of liquid that your casting can hold (depending on this volume, you may scale up/down the total volume of the cryogel) (Recipe 1). Mix water, gelatin, and glutaraldehyde in an appropriate tube, such as a 15 mL plastic tube.
Critical: Add glutaraldehyde solution after gently mixing water and gelatin, because chemical crosslinking will start soon after adding glutaraldehyde. After the addition of glutaraldehyde, gently mix the solution again.
Pour the mixed solution into an apparatus. Leave on the bench (room temperature) for 30 min.
Carefully place an apparatus in a -20 °C freezer. Leave in a freezer overnight (~16 h).
Critical: Freeze-thawing is the critical step for the formation of porous structure. Make sure that other people do not open the freezer door (especially during the initial freezing process).
Taking out cryogels
Take out a gel apparatus from the -20 °C freezer and leave it on a bench until completely thawed.
Critical: If a cryogel is not completely thawed, gels or pore structures may be damaged during the process of retrieving a cryogel from an apparatus. See General note 1.
Set up a dish (such as a 100 mm Petri dish or a rectangular plastic dish) filled with 0.1 M sodium carbonate buffer (Recipe 2).
Carefully separate glass plates or slowly push the gel (if a glass tube or a syringe is used).
Rinse cryogels with deionized water. Then, transfer cryogels to a dish filled with 0.1 M sodium carbonate buffer.
Add a small pinch of sodium borohydride powder to the solution. One previous study indicates 0.1 M freshly prepared sodium borohydride solution [4]. This is equivalent to the concentration of 3.78 mg/mL. You will start seeing bubbles (sodium borohydride reacts with remaining glutaraldehyde and producing hydrogen). To quench enough, you may leave cryogels in the sodium borohydride solution overnight on a rocking shaker (Pause point). Cryogels can be kept in sodium carbonate buffer with or without sodium borohydride. However, we recommend starting the quenching with sodium borohydride soon after taking out gels from the casting apparatus, as it will take at least overnight to quench residual sodium borohydride well. Remaining sodium borohydride can cause cell death if you grow cells.
You can keep cryogels in 0.1 M sodium carbonate buffer. Keep it in a refrigerator (Pause point). Rinse well with deionized water before use.
Staining of cryogels
Cutting cryogels
Use a razor blade to slice a piece of cryogel. Make a small rectangular piece (approximately 5–10 mm is sufficient)
Transfer a piece of cryogel onto the glass surface of a glass-bottom dish.
(Not necessary: We conducted this to compare pyronin Y staining in 2D and 3D) To stain ECM coating, the surface of a glass bottom dish should be coated with 1 mg/mL of ECM solution first. Then, the same staining process (step B2) needs to be done.
Pyronin Y staining
Critical: If MSCs are included, go to section E, then come back here after Alexa Fluor 647-phalloidin staining of MSCs.
Place a piece of cryogel from the previous step onto a glass bottom dish (#1.5 thickness).
Critical: The thickness of coverslips is critical to acquiring microscopic images with the optimum conditions.
Add the pyronin Y solution (Recipe 3) to cover the entire gel. Although the volume will depend on the size of a piece of cryogel, typically 50–200 μL of the pyronin Y solution is sufficient.
Leave at room temperature for 5 min.
Rinse the stained cryogel with deionized water three times (5 min each). If over-staining happens, try again with a shorter staining time (see step C1h and General note 5).
Imaging of cryogels with a laser-scanning confocal microscope*
*Many people may use their core imaging facility. It may be easy for you to consult with your facility manager/director.
Critical: Please let them know that the settings described in steps C1c–d are critical, as pyronin Y’s imaging probably requires less laser power and relatively low digital gain; see General note 2.
Determine the acquisition settings
Place a 35 mm glass-bottom dish with a piece of the stained cryogel onto the stage of a confocal microscope.
Use an appropriate objective lens (see General note 2).
Set the lowest laser power that is necessary to obtain sufficient signals. With a Zeiss 880 equipped with a highly sensitive Gallium-Arsenide-Phosphide (GaAsP) detector, ~0.1% was sufficient for the excitation of pyronin Y (with a 561 nm laser). When MSCs (stained with Alexa Fluor 647-phalloidin) are also included for dual-color imaging, a slightly higher laser power for a 633 nm laser may be needed (see General note 3).
Select the laser for pyronin Y excitation (such as 561 nm).
Set the scanning area, digital zoom, and scanning speed (important; see General note 4). We used the highest scanning speed (8; resulting in a resolution of 0.415 μm). The scanning area is up to your choice. Because the magnification is low in this case (20×), we set 1,024 × 1,024. 512 × 512 may be too small.
Set the master gain. Probably ~500 is appropriate. (Critical: Too high master gain can damage the detector.) Because pyronin Y staining of gelatin cryogels gives fairly high signals, we strongly suggest keeping the digital gain at 1.0 and digital offset at 0.
Set the pinhole diameter. In Zeiss confocal operated with Zen, you simply need to hit 1 AU, which will set the optimum pinhole diameter (with a 561 nm excitation wavelength, it is 36 μm).
Hit Live to check that the setting is suitable for imaging. Keep your eye on the histogram of the signal to make sure that the signal is not saturated (see General note 5).
If you conduct dual-color imaging with MSCs stained with Alexa Fluor 647-phalloidin, repeat steps C1f–h with the laser for the excitation of Alexa Fluor 647-phalloidin. [In our case, we used a 633 nm laser; an excitation wavelength closer to 647 nm is fine (better) if available.] Also, carefully check what MSCs look like (see Troubleshooting).
Acquire images
In the control panel, first set live to determine the start and the end plane.
When you set the z-axis step, a 1.0 μm interval may be sufficient, because a relatively low magnification of an objective lens is used.
Then, hit the start experiment button. The following YouTube video may be helpful: https://www.youtube.com/watch?v=QhFb6vU1Iyg.
Save z-stack images. Keep the original data files without modification (important).
Quantification of pores using Image J/Fiji [13]
Export files from the original z-stack data files
Open the original data files with the software to control the microscope and export them to create TIFF files (see General note 6). If Zeiss confocal is used, the original files can be opened with Fiji.
Adjust the contrast if necessary (see General note 7).
Quantification of pores (see General note 8)
Copy the selected z-plane image from the stack (Image → Duplicate). When a small window pops up, type the desired single-plane range (for example, 8-8).
Set the threshold (Image → Adjust → Threshold).
The following YouTube video may be very useful: https://www.youtube.com/watch?v=4gm9Q_TdRvY.
MSC culture
Rinsing cryogels
Rinse a piece of cryogel with deionized water extensively.
After rinsing, dip a piece of gel in 70% ethanol, then place it on a 35 mm dish (or a 12-well plate for more convenient handling). Then, place a dish/plate in a UV crosslinker for approximately 2 min. If a UV crosslinker is not available, placing it under a cell culture hood for approximately 5–10 min (then turning on the UV lamp) can be an alternative.
Caution: Do not overdo this process, especially with a UV crosslinker. It can dry the gel too much and cause shrinking. Also, cryogels should be handled aseptically after this process. Contamination of microorganisms has to be avoided. We used a UV crosslinker only to sterilize cryogels. Chemical crosslinking is not the purpose of this procedure; therefore, sterilization under cell culture hoods with UV lamps will also work fine.
MSC culture and staining
Slowly seed 10 μL of trypsinized MSCs suspension (~400,000 cells/mL) onto a piece of cryogel in a 12-well tissue culture plate to absorb the cell suspension.
Put the plate back in a CO2 incubator and leave it for 30 min to allow MSCs to adhere to a cryogel piece.
Caution: If your gel is too dry, you may want to add a little drop of cell culture medium after the previous step to avoid cells dying due to drying.
Add 1 mL of culture medium.
Aspirate the medium a few hours later and fix cells with 4% paraformaldehyde in PBS for 20 min.
Permeabilize the cell membrane with 0.1% Triton X-100 in PBS (5 min).
Rinse with PBS three times (5 min each).
Stain F-actin with Alexa Fluor 647-phalloidin (1/100) in PBS for 15–30 min (see General note 9).
Rinse with PBS three times (5 min each).
Conduct the Pyronin Y staining (as described in B2; also see General note 10).
Data analysis
Although no special computational skills are needed, some try-and-error type patience is required if you try a semi-automated quantification of pores. The video mentioned in step C2c is useful.
Validation of protocol
In order to test the specificity of pyronin Y dye to different ECMs, pyronin Y was first applied to stain the various ECMs in 2D. Glass-bottom dishes were coated with each ECM protein and then incubated with pyronin Y solution. The comparison of the average maximum fluorescence intensity showed minimal staining of the ECMs coating of the glass surface (Figure S1). The fluorescence intensity, however, yielded a slightly higher signal level in gelatin- and collagen-coated dishes. This result implied that pyronin Y may potentially recognize a specific structure present in those proteins, such as glycine-proline-hydroxyproline triplets. Remarkably, gelatin cryogels showed approximately 8-fold higher average signals than gelatin or collagen coating (Figure S1). Indeed, pyronin Y staining of a gelatin cryogel resulted in exceptionally clear visualization of porous structures (Figure 1).
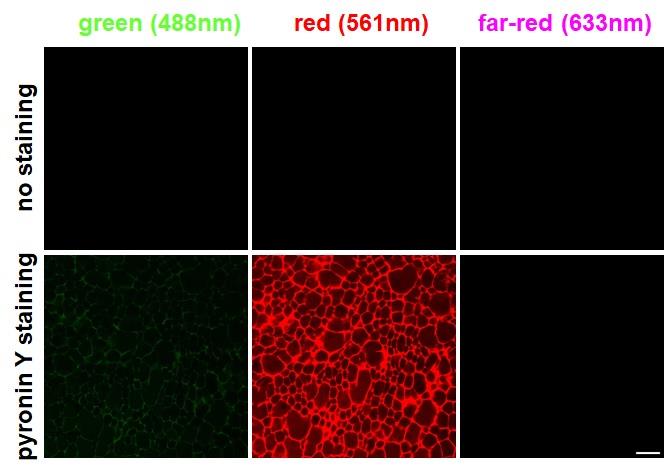
Figure 1. Examination of pyronin Y staining in three channels. No staining (top) and pyronin Y staining (bottom) of cryogels imaged with three different laser lines (488, 561, and 633 nm). Scale bar = 50 μm.
Through threshold adjustment, followed by conversion to a black-and-white format and inversion of the binary image, the production of a clear image of the mesh-like pores (Figure 2A) was possible. Using the object counting function in Fiji software allowed the semi-automatic outlining and quantification of pore sizes (section C).
Employing a 561 nm laser resulted in more than 50% of the peak emission of pyronin Y, which has an optimum excitation peak of around 544 nm (Figure S2). In contrast, a 488 nm laser, though less efficient, still presented approximately 15% of the maximum emission. Consequently, some degree of bleed-through was expected by combining pyronin Y with green fluorescent dyes for dual-color imaging. However, at 633 nm, pyronin Y is not expected to emit any fluorescence. This theoretical bleed-through was experimentally validated by imaging a pyronin Y-stained gelatin cryogel with three laser lines (488, 561, and 633 nm). As presented in Figure 1, excitation of a pyronin Y–stained cryogel with a 488 nm laser yielded a low, yet recognizable level of bleed-through, whereas excitation with a 633 nm laser showed no detectable emission.
Therefore, we hypothesized that combining pyronin Y with a far-red fluorescence dye, such as Alexa Fluor 647, may be more effective for dual-color imaging. Based on these insights, we conducted dual-color imaging of MSCs and a piece of cryogel. Figure 2B demonstrates that the combination of pyronin Y and Alexa Fluor 647 phalloidin staining enabled successful dual-color confocal imaging of cryogels and MSCs. Although there was a subtle staining inside of MSCs observed in the red channel (Figure 2B), it was believed to be due to the binding of pyronin Y to RNA inside of MSCs. The far-red channel clearly shows distinct actin staining (center, Figure 2B). This compelling evidence reinforces the versatility of pyronin Y as a powerful tool in fluorescence microscopy, particularly for dual-color imaging applications. The successful combination of pyronin Y with Alexa Fluor 647 phalloidin in our experiments not only validates our hypothesis but also sets a new precedent for advanced imaging techniques in cellular and molecular biology.
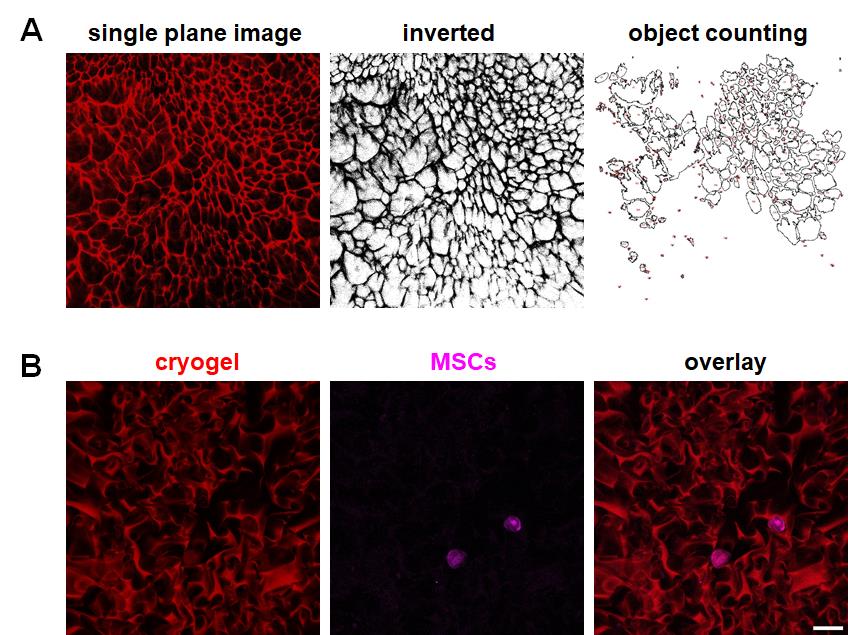
Figure 2. Examples of semi-automated pore analysis and dual-color imaging of a pyronin Y–stained cryogel with MSCs. (A) An example of a single-plane staining image of a gelatin cryogel processed for quantification. (B) Dual-color imaging of a cryogel and MSCs. red: pyronin Y-stained cryogel; magenta: MSCs stained with Alexa Fluor 647. Scale bar = 50 μm.
General notes and troubleshooting
General notes
Pyronin Y solution is stable at room temperature for at least a few weeks. The solution shows a slightly dark yet vivid color. However, if this color does not appear to be vivid, preparing a fresh pyronin Y solution is necessary. If you have not used the stock solution for a while, we strongly recommend conducting a quick staining test with a piece of gelatin cryogel. You can quickly see if fluorescence is present by checking with a conventional fluorescent microscope equipped with a Texas Red/X-Rhodamine filer. However, a conventional epi-fluorescence microscope gives significantly higher background signals and images will not be suitable to semi-automatically quantify with Fiji/Image J. Therefore, a combination of a confocal microscope and the indicated laser(s) wavelength would give you the best result. If you just want a routine quality check of cryogels, the use of a conventional epi-fluorescence microscope is sufficient. We have not tried imaging pyronin Y–stained cryogels with a spinning disk confocal microscope, although it should not be a problem. Note that the detector(s) used for a spinning disk confocal microscope could be a charge-coupled device (CCD), a complementary metal oxide semiconductor (cMOS), or an electron-multiplying charge-coupled device (EM-CCD) camera. We do not think EM-CCD is necessary for the imaging of pyronin Y staining; however, please note that the sensitivity of CCD and cMOS will be lower than that of the GaAsP detector. You need to adjust (increase) the laser power. To minimize photobleaching, increased exposure time is efficient for acquiring high-contrast images when those cameras with a spinning disk confocal are used. Because magnification is relatively low, binning (such as 2 × 2) is also effective, depending on the pixel size of CCD and cMOS cameras. However, those acquisition settings are beyond the scope of this protocol, and we will omit the discussion.
Please note that the protocol using Fiji/Image J shown here does not allow 3D measurements―such tasks may be done with Imaris or other software (not free).
Despite some possible limitations, we would like to emphasize that pyronin Y staining is a quite reasonably performed, very simple, easy, and yet reproducible fluorescent labeling method for gelatin cryogels.
When cryogels are successfully made, it appears as a uniform, very slightly yellow/tan-colored material. If something is wrong during the freeze-thawing, you may see a half-transparent appearance (Maybe ice crystals, but we do not know exactly.) In such cases, it would be better to start all over again to prepare a new cryogel. After the successful quenching with sodium borohydride, a subtle yellow/tan color of cryogels will disappear and cryogels’ color will change to white (Figure 3).

Figure 3. Cryogels before (left arrow) and after quenching (right arrow) with sodium borohydrideIf higher magnifications, such as 60× (63× in Zeiss) and 100×, are used, please keep in mind that the working distance of microscopic objective lenses becomes shorter and thus may not be suitable for pore imaging of cryogels. Because the purpose of this protocol is to stain and evaluate the overall architectures of cryogels, we think that lower magnifications are more appropriate. 10–20× objective lenses with relatively high numerical aperture (N.A.) values work best and, therefore, we used 20× (N.A. = 0.8) objective in this example (N.A. higher than 0.8 is great but such lenses are optimized for multi-photon imaging and thus very expensive). If conventional 10–20× objective lenses are used, Plan Apochromat or Plan Fluor quality lenses are highly preferred. Please note that the use of lower N.A. lenses will result in less signal; thus, acquisition settings would need to be adjusted accordingly (a little more laser power and slightly higher master gain of a GaAsP detector). Also, if you use a laser scanning confocal (either earlier models from Zeiss or other manufacturers’ laser scanning confocal microscopes) without high-sensitive detectors such as a GaAsP detector, you need to increase the laser power (and probably digital gain as well). Traditionally, photomultipliers (PMT) have been used as detectors in laser scanning confocal microscopy. However, its sensitivity is approximately half for pyronin Y’s emission wavelength and ~30% for Alexa Fluor 647’s emission wavelength.
We confirmed that the combination of 488 and 561 nm cannot avoid breed-through because of the excitation spectrum of pyronin Y (see Figure S2). A 633 nm laser was used to excite Alexa Fluor 647-phalloidin because a 633 nm laser is the standard configuration for Zeiss 880. If your microscopic settings allow the use of a laser closer to 647 nm, even better.
Please note that the scan line speed was set at the highest in our case. Although higher scan line speed can be disadvantageous, such as reduced signal-to-noise ratios and spatial resolution, please note that too low scan line speed (= more laser scanning) is not necessary with 20× objective lenses. If you use the formula d = 0.61 λ/N.A., the spatial resolution in our case (using a 20× objective lens with a 561 nm laser) should be 428 nm (0.428 μm). Thus, the maximum scan line speed (again, our case is Zeiss 880 with Zen) gives a sufficient spatial resolution.
Overstaining of cryogels must be avoided because it does not allow quantitative imaging. Too much pyronin Y signals might also bother the dual-color imaging. Overstaining of cryogels can easily occur when pyronin Y staining takes longer than 5 min. It may be necessary to optimize the staining time (may be a little shorter). Saturation of digital signals also occurs by the detector’s settings (the image would be 16-bit, so the maximum signal value in grayscale should be 65,535).
When you export files from the original data, make sure to split the channels if you conduct dual-color imaging (i.e., pyronin Y–stained cryogels and Alexa Fluor 647-phalloidin-labeled MSCs). This can be done with microscopic software (Zen) when you export; alternatively, you can also split the channels after opening the original stack files with Fiji.
Changing the Gamma value is not necessary and not recommended. Although the later procedure using Fiji/Image J will create binary images for semi-automated quantification, note that modifying the Gamma value will introduce the nonlinear fluorescence intensity adjustment in an image [14]. With pyronin Y, gelatin cryogels should be able to give relatively high signal-to-noise ratio images. If not, either staining or acquisition conditions may need to be checked.
Please note that successful quantification relies on image quality. If the signal-to-noise ratio of the original images is not great, semi-automated quantification will become quite difficult.
We did not attempt blocking (of non-specific binding of Alexa Fluor 647-phalloidin to cryogels) with bovine serum albumin, serum, or skim milk. Prolonged incubation of this step may cause significant Alexa Fluor 647-phalloidin binding to cryogels, but we have not tested it. One fear of doing enough blocking is that it might also bother the staining of MSCs. At least, a relatively short incubation time was sufficient for dual-color imaging of pyronin Y–stained cryogels and Alexa Fluor 647-phalloidin-labeled MSCs.
We conducted Alexa Fluor 647-phalloiding staining (MSCs) first, then moved on to the pyronin Y staining of a cryogel. The order could be reversed. However, it is important to keep in mind that 1) the cell membrane needs to be permeabilized with a detergent such as Triton X-100, as shown in this protocol, to stain F-actin, and 2) pyronin Y is not covalently bound to gelatin. So, if pyronin Y staining is done first, you may lose significant pyronin Y staining during Triton X-100 treatment.
Troubleshooting
To culture MSCs, you should rinse cryogels very well before setting the cell culture. Residual buffer or glutaraldehyde from the manufacturing process of cryogels can cause unhappy MSCs (or, in the worst case, cell death). After the quenching of glutaraldehyde with sodium borohydride, cryogels are submerged in 0.1 M sodium carbonate buffer, whose pH is basic. It may be necessary to rinse cryogels with deionized water for more than a day.
Acknowledgments
We would like to thank Dr. Karen Kasza and Christian Cupo (Columbia University) for allowing us to use the Zeiss 880 confocal microscope. The authors also appreciate Dr. Björn Kafsack (Weill Cornell Medicine) for sharing pyronin Y. This study was partially supported by a St. Francis College faculty research grant (K.K.). St. Francis College STEM Resource Center (funded by the Department of Education, STEM Success & Articulation grant P031C210140; for B.R. and E.B.) and Collegiate Science and Technology Entry Program (CSTEP) from the New York State Education Department (CSTEP grant #0537-22-2016; for E.B.) provided support for faculty-mentored research of students. Lastly, we thank Dr. Allen Burdowski (St. Francis College) and Noemi Rivera (St. Francis College STEM Resource Center) for their tireless effort to support student engagement in research. This is an original work and has not been published in any other papers.
Competing interests
There are no conflicts of interest or competing interests.
References
- Masson, P. (1929). Some histological methods. Trichrome staining and their preliminary technique. J Tech Methods. 12: 75–90.
- Junqueira, L. C. U., Bignolas, G. and Brentani, R. R. (1979). Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 11(4): 447–455.
- Lanir, Y., Walsh, J. and Soutas-Little, R. W. (1984). Histological staining as a measure of stress in collagen fibers. J Biomech Eng. 106(2): 174–176.
- Dainiak, M. B., Allan, I. U., Savina, I. N., Cornelio, L., James, E. S., James, S. L., Mikhalovsky, S. V., Jungvid, H. and Galaev, I. Y. (2010). Gelatin-fibrinogen cryogel dermal matrices for wound repair: Preparation, optimisation and in vitro study. Biomaterials. 31(1): 67–76.
- Shapiro, H. M. (1981). Flow cytometric estimation of DNA and RNA content in intact cells stained with Hoechst 333442 and pyronin Y. Cytometry. 2(3): 143–150.
- Li, B., Wu, Y. and Gao, X. -M. (2002). Pyronin Y as a fluorescent stain for paraffin sections. Histochem J. 34(6–7): 299–303.
- Andrews, L. M., Jones, M. R., Digman, M. A. and Gratton, E. (2013). Detecting Pyronin Y labeled RNA transcripts in live cell microenvironments by phasor-FLIM analysis. Methods Appl Fluoresc. 1(1): 015001.
- Jones, L. O., Williams, L., Boam, T., Kalmet, M., Oguike, C. and Hatton, F. L. (2021). Cryogels: recent applications in 3D-bioprinting, injectable cryogels, drug delivery, and wound healing. Beilstein J Org Chem. 17: 2553–2569.
- Hixon, K. R., Lu, T., Sell and S. A. (2017). A comprehensive review of cryogels and their roles in tissue engineering applications. Acta Biomater. 62: 29–41.
- Liu, D., Nikoo, M., Boran, G., Zhou, P. and Regenstein J. M. (2015). Collagen and gelatin. Annu Rev Food Sci Technol. 6: 527–557.
- Yoshii, A., Koji, T., Ohsawa, N. and Nakane, P. K. (1995). In situ localization of ribosomal RNAs is a reliable reference for hybridizable RNA in tissue sections. J Histochem Cytochem. 43(3): 321–327.
- Kita, K, Gauglitz, G. G., Phan, T. T., Herndon, D. N. and Jeschke, M. G. (2010). Isolation and characterization of mesenchymal stem cells from the sub-amniotic human umbilical cord lining membrane. Stem Cells Dev. 19(4): 491–502.
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods. 9(7): 676–682.
- Rossner, M. and Yamada, K. M. (2004). What’s in a picture? The temptation of image manipulation. J Cell Biol. 166(1): 11–15.
Supplementary information
The following supporting information can be downloaded here:
- Figure S1. A graph summarizing the average of maximum fluorescence intensity in a field (n = 3–5 pictures)
- Figure S2. The comparison of pyronin Y (A) excitation (dotted line) and emission spectra (solid line) data with three laser lines (488, 561, and 633 nm; B–D)
Article Information
Publication history
Received: Jun 17, 2024
Accepted: Sep 23, 2024
Available online: Oct 22, 2024
Published: Nov 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Reece, B., Bahar, E. V., Pereira, A. C., Witek, L. and Kita, K. (2024). A Simple Staining Method Using Pyronin Y for Laser Scanning Confocal Microscopy to Evaluate Gelatin Cryogels. Bio-protocol 14(22): e5115. DOI: 10.21769/BioProtoc.5115.
Category
Biological Engineering > Biomedical engineering
Cell Biology > Cell isolation and culture > 3D cell culture
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.