- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

An In Vitro Model of Murine Osteoclast-Mediated Bone Resorption
Published: Vol 14, Iss 21, Nov 5, 2024 DOI: 10.21769/BioProtoc.5100 Views: 2244
Reviewed by: Valérian DORMOYJaira Ferreira de VasconcellosNona Farbehi
Original research article
The authors used this protocol in:

Apr 2023

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Osteoclasts are terminally differentiated multinucleated giant cells that mediate bone resorption and regulate skeletal homeostasis under physiological and pathological states. Excessive osteoclast activity will give rise to enhanced bone resorption, being responsible for a wide range of metabolic skeletal diseases, ranging from osteoporosis and rheumatoid arthritis to tumor-induced osteolysis. Therefore, the construction of in vitro models of osteoclast-mediated bone resorption is helpful to better understand the functional status of osteoclasts under (patho)physiological conditions. Notably, it is essential to provide an in vivo–relevant bone substrate that induces osteoclasts to generate authentic resorption lacunae and excavate bone. Here, we summarize the experimental design of a reproducible and cost-effective method, which is suitable for evaluating the regulatory mechanisms and influence of molecular agonists and antagonists as well as therapeutics on osteoclast-mediated bone-resorbing activity.
Key features
• Experiments are performed using bovine cortical bone slices to simulate bone substrate resorption by murine osteoclasts in vivo.
• The method allows for quantification of bone resorption in vitro.
• The method is suitable for evaluating the regulatory mechanisms that control osteoclast-mediated bone-resorbing activity.
Keywords: OsteoclastGraphical overview
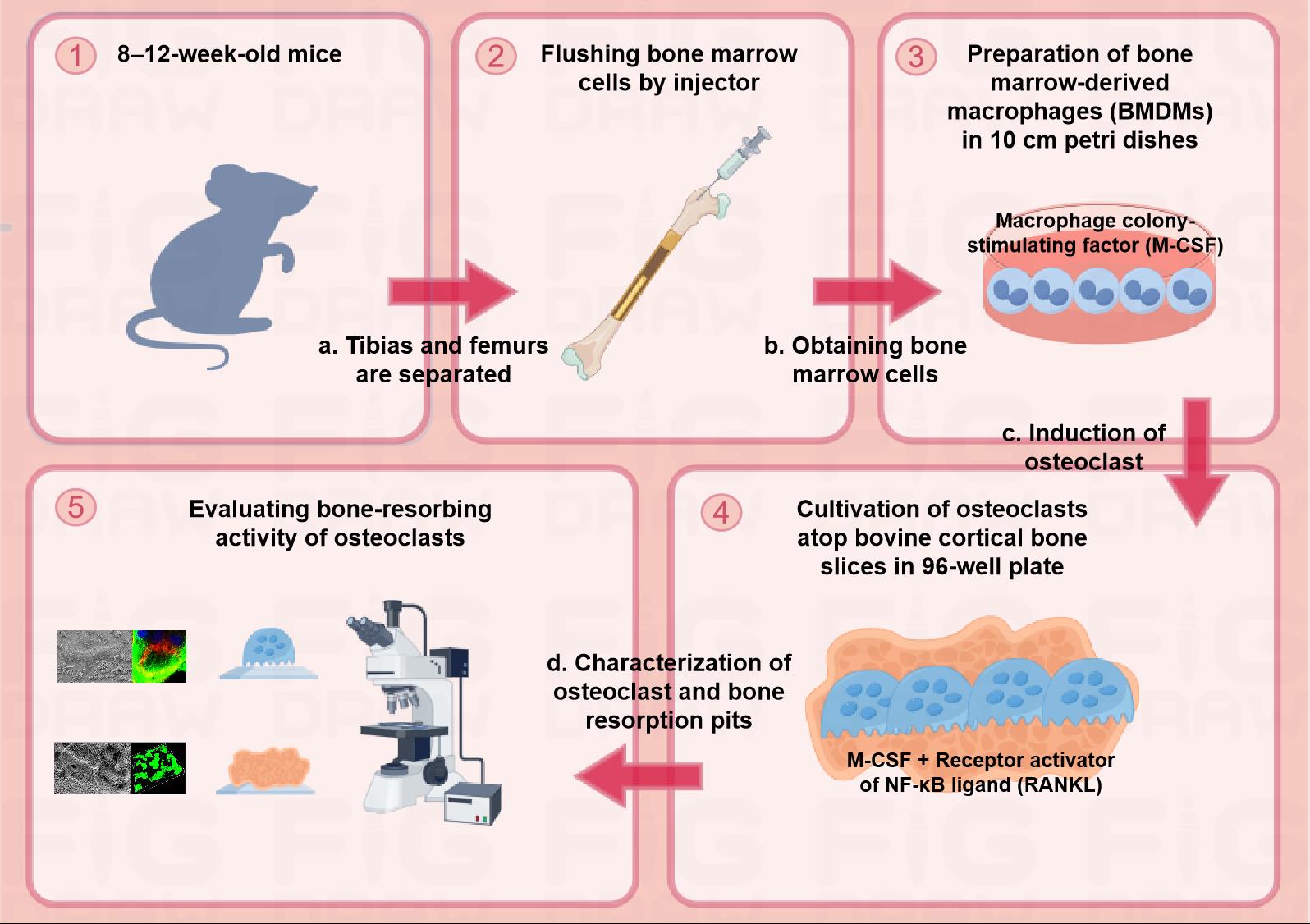
Schematic diagram of the protocol for assessing osteoclast-mediated bone-resorbing activity in vitro. Step 1: Euthanizing 8–12-week-old male mice to separate tibias and femurs. Step 2: Flushing bone marrow cells by injector and obtaining the cells. Step 3: Preparation of BMDMs with complete medium containing 20 ng/mL M-CSF in 10 cm Petri dishes. Step 4: Cultivation of osteoclasts with complete medium containing 20 ng/mL M-CSF and 30 ng/mL RANKL atop bovine cortical bone slices in a 96-well plate. Step 5: Evaluating bone-resorbing activity of osteoclasts.
Background
Osteoclasts are exclusive and specialized multinucleated bone-resorbing cells derived from both embryonic and hematopoietic stem cell precursors of erythromyeloid and myeloid lineages [1]. An imbalance of skeletal remodeling as a consequence of increased osteoclast-mediated bone resorption is responsible for a wide range of metabolic skeletal diseases, ranging from osteoporosis and rheumatoid arthritis to tumor-induced osteolysis. Therefore, the construction of in vitro osteoclast-mediated bone resorption models is helpful to better characterize the functional status of osteoclasts under both physiological and pathological conditions.
The transition of cells of the monocyte/macrophage lineage into osteoclasts in vitro requires two essential cytokines—macrophage colony-stimulating factor (M-CSF) and receptor activator of NF-kB ligand (RANKL). The presence of M-CSF promotes the proliferation of osteoclast precursor cells, and RANKL further induces precursor cells to express an osteoclast phenotype [2]. Mature osteoclasts at the bone surfaces undergo a polarization process marked by extensive morphologic changes, including the formation of an actin ring at their basal surface, termed the sealing zone. The sealing zone surrounds a differentiated and intricately folded region of the plasma membrane called the ruffled border, where protons, chloride ions, and proteases are secreted into the underlying resorption lacunae where bone demineralization and extracellular matrix degradation proceed [3]. As the organic phase of bone is dominated by type I collagen, the proteolytic release of C-terminal telopeptide fragments of type collagen (CTX-1) is a characteristic biochemical marker of osteoclast-mediated bone resorption [4]. When cultured on bone or dentin substrate, osteoclasts assemble resorption lacunae, thereby generating resorption pit structures similar to those formed during bone resorption in vivo [5]. Notably, bovine cortical bone slices are superior to synthetic materials or dentine-derived matrix as they better mimic bone substrates encountered in vivo [6]. When combined with an appropriate detection assay, osteoclast-mediated bone-resorbing activity can be quantified under a variety of experimental conditions [7,8].
Here, we describe a step-by-step protocol for in vitro bone-resorbing activity using an osteoclast-cortical bone slice co-culture system. This protocol consists of three main steps: mouse bone marrow–derived macrophage (BMDM) preparation, osteoclast induction and characterization, and bone resorption pit quantification. This method can be universally utilized to evaluate the bone-resorbing activity of osteoclasts in response to both agonists and antagonists.
Materials and reagents
Biological materials
8–12-week-old male C57BL/6J mice (The Jackson Laboratory, catalog number: 000664)
Bovine cortical bone slices (Immunodiagnostic Systems, catalog number: DT-1BON1000-96)
Reagents
Alpha minimum essential medium (α-MEM) (Hyclone, Cytiva, catalog number: SH30265.01)
Fetal bovine serum (FBS) (Sigma-Aldrich, catalog number: F0193)
Penicillin-streptomycin solution (Hyclone, Cytiva, catalog number: SV30010)
Red blood cell lysing buffer (Biosharp, catalog number: BL504A)
Phosphate buffered saline (PBS) (Servicebio, catalog number: G0002)
Ethylenediaminetetraacetic acid (EDTA) solution, 0.5 M (Sigma-Aldrich, catalog number: 03690)
Recombinant murine M-CSF (R&D Systems, catalog number: 416-ML)
Recombinant murine RANKL (R&D Systems, catalog number: 462-TEC)
Bovine serum albumin (Sigma-Aldrich, catalog number: A1933)
Paraformaldehyde powder (Sigma-Aldrich, catalog number: 158127)
Glutaraldehyde solution, 50% (Sigma-Aldrich, catalog number: 49629)
Tartrate-resistant acid phosphatase (TRAP) stain kit (Sigma-Aldrich, catalog number: 387A)
Triton X-100 solution, 10% (Sigma-Aldrich, catalog number: 49629)
Phalloidin-tetramethylrhodamine B isothiocyanate (Sigma-Aldrich, catalog number: P1951)
α-Tubulin mouse mAb (Cell Signaling Technology, catalog number: 3873)
Donkey anti-rat Alexa 488-conjugated secondary antibodies (Invitrogen Molecular Probes, catalog number: A-21202)
Sodium hydroxide (NaOH) powder (Sigma-Aldrich, catalog number: 655104)
Peroxidase-conjugated wheat germ agglutinin (WGA) (Sigma-Aldrich, catalog number: L3892)
Diaminobenzidine (DAB) tablets (Sigma-Aldrich, catalog number: D4418)
FITC conjugate WGA (Sigma-Aldrich, catalog number: L4895)
CrossLaps ELISA kit for quantifying CTX-1 release (Immunodiagnostic Systems, catalog number: AC-07F1)
Solutions
5 mM EDTA solution (see Recipes)
0.1% Bovine serum albumin solution (see Recipes)
Murine recombinant M-CSF solution (see Recipes)
Murine recombinant RANKL solution (see Recipes)
4% Paraformaldehyde solution (see Recipes)
2.5% Glutaraldehyde solution (see Recipes)
NaOH solution (see Recipes)
0.1% Triton X-100 solution (see Recipes)
Peroxidase-conjugated WGA solution (see Recipes)
FITC-conjugated WGA solution (see Recipes)
Recipes
5 mM EDTA solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time 0.5 M EDTA solution 5 mM 0.5 mL Room temperature Sterile PBS NA 49.5 mL Room temperature Final volume 50 mL 4 1 week 0.1% Bovine serum albumin solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time Bovine serum albumin 0.1% 0.02 g 4 Double-distilled water NA 20 mL Room temperature Final volume 20 mL -20 1 month Murine rM-CSF solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time Murine rM-CSF 50 μg/mL 50 μg -80 Sterile PBS containing 0.1% bovine serum albumin NA 1 mL -20 Final volume 1 mL -80 1 year Murine rRANKL solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time Murine rRANKL 50 μg/mL 50 μg -80 Sterile PBS containing 0.1% bovine serum albumin NA 1 mL -20 Final volume 1 mL -80 1 year 4% Paraformaldehyde solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time Paraformaldehyde powder 4% (w/v) 0.4 g -4 Sterile PBS NA 10 mL Room temperature Final volume 10 mL -4 1 week 2.5% Glutaraldehyde solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time 50% glutaraldehyde solution 2.5% (v/v) 0.5 mL Room temperature Sterile PBS NA 9.5 mL Room temperature Final volume 10 mL -4 1 week NaOH solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time NaOH powder 0.5 N 1 g Room temperature Double-distilled water NA 50 mL Room temperature Final volume 50 mL -4 1 month 0.1% Triton X-100 solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time 10% Triton X-100 solution 0.1% 0.01 mL Room temperature Double-distilled water NA 9.99 mL Room temperature Final volume 10 mL -4 1 month Peroxidase-conjugated WGA solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time Peroxidase-conjugated WGA 2 mg/mL 1 mg -20 Sterile PBS NA 0.5 mL Room temperature Final volume 0.5 mL -20 1 year FITC-conjugated WGA solution
Reagent Final concentration Quantity or Volume Recommended storage condition Recommended storage time FITC-conjugated WGA 2 mg/mL 1 mg -20 Sterile PBS NA 0.5 mL Room temperature Final volume 0.5 mL -20 1 year
Laboratory supplies
10 mL syringe with 25 G needle
Sterile absorbent gauze
70 µm cell strainer (Corning, catalog number: 431751)
Pipette
15 mL conical tube (Nest, catalog number: 601001)
50 mL conical tube (Nest, catalog number: 601002)
10 × 10 cm Petri dish with cover (Nest, catalog number: 752001)
Cell lifter (Corning, catalog number: CLS3008)
96-well cell culture plates (Nest, catalog number: 713011)
Equipment
Dissecting tweezers and scissors (Autoclaved)
Cell counter (Thermo Fisher Scientific Inc, model: Countess 3)
Cell culture incubator (37 °C, 5% CO2)
Centrifuge (Eppendorf)
Inverted microscope (Olympus)
Confocal laser scanning microscope (CLSM) (Nikon A1)
Critical point drying (Balzers Union)
Au/Pg sputtered (Polaron Equipment Ltd, model: E-5100)
Field emission scanning electron microscopy (FE-SEM) (AMRAY 1910)
Software and datasets
ImageJ is a Java-based (runs on Mac OS X, Linux, and Windows) freeware available for download at: http://rsb.info.nih.gov/ij/
GraphPad Prism 7
Procedure
Preparation of bone marrow-derived macrophages (BMDMs)
Euthanize 8–12-week-old male C57BL/6J mice by carbon dioxide inhalation and soak in 50 mL of 75% ethanol for 5 min.
Remove the mouse hind limbs intact and bilaterally along the greater trochanter of the femur using sterile scissors and forceps. Remove excess skin and muscle tissues from the femur and tibia with sterile gauze and then place the bones in 10 mL of pre-prepared PBS solution at 4 containing 2% penicillin-streptomycin.
Separate tibiae and femurs using sterile scissors and then wash twice with 2 mL of PBS solution containing 2% penicillin-streptomycin for 5 min. Figure 1B shows the bones and both ends of each bone type cut to expose the bone marrow cavity.
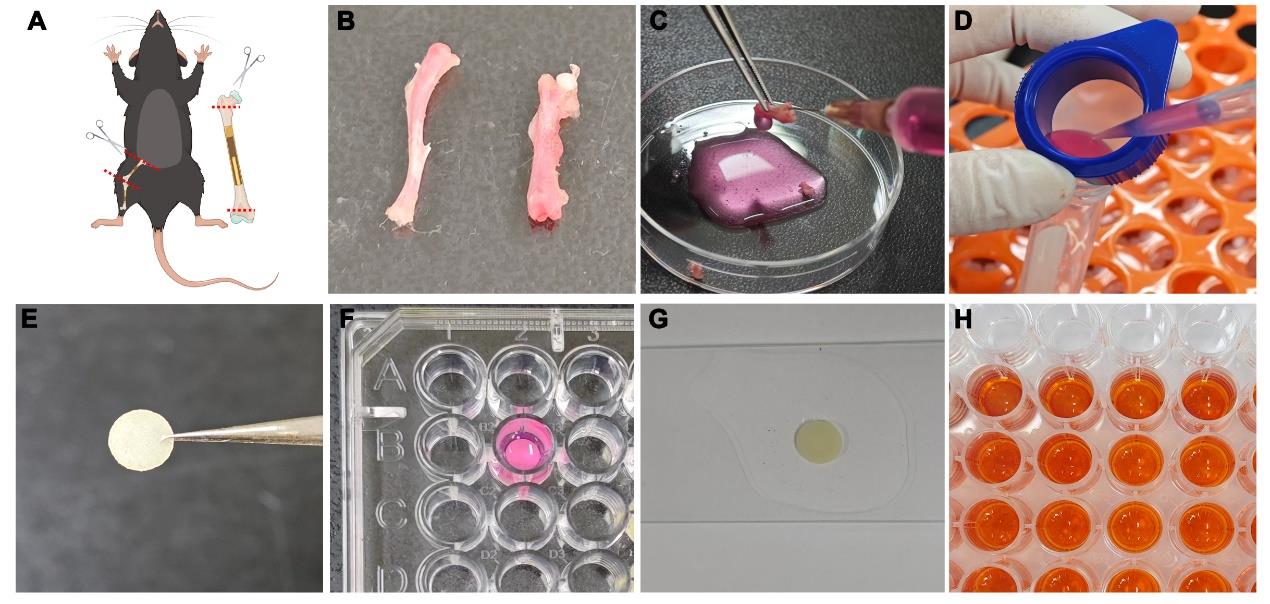
Figure 1. Overview of osteoclastogenesis and bone-resorbing assay. (A) Schematic diagram of separating tibiae and femurs and removing the ends of the bones. (B) Separating the tibia and femur. (C) Irrigating bone marrow cavity. (D) Filtering through a cell strainer. (E) Preparing bovine cortical bone slices. (F) Culturing osteoclasts atop bone slice in a 96-well plate. (G) Preparing for observation of bone resorption. (H) ELISA analysis of the cultured supernatants.Flush bone marrow cells from both ends of the bone with α-MEM using a 10 mL syringe with a 25 G needle and then pass them through a 70 μm cell strainer into a 50 mL conical tube (Figure 1C, D). Rinse repeatedly until there is no visible redness remaining in the bone marrow cavity. Use approximately 8 mL of α-MEM per bone to flush cells and temporarily store all bone marrow cells at room temperature in 50 mL conical tubes.
Pellet bone marrow cells by centrifuging at 400× g for 5 min at room temperature and discard the supernatant. Resuspend cells thoroughly with 3 mL of red blood cell lysing buffer and then let them stand for 2–3 min at room temperature to lyse red blood cells.
Add 5 mL of room-temperature α-MEM medium supplemented with 10% FBS to terminate lysis, pellet cells by centrifuging at 300× g for 3 min at room temperature, and discard the supernatant.
Resuspend cells in the 20 mL prepared complete medium containing 20 ng/mL rM-CSF and then transfer cells into two 10 cm Petri dishes for overnight incubation in 5% CO2/95% air atmosphere at 37 °C.
After 12 h, collect non-adherent cells and transfer them to fresh α-MEM medium containing 10% FBS and 20 ng/mL rM-CSF in six to eight 10 cm Petri dishes for further expansion of the cell population (Figure 2A). The expected yield of non-adherent cells is approximately 5 × 106 cells/mL.
Change medium every other day for 4–5 days until the adherent cells reach 80%–90% confluence (Figure 2B).
After rinsing twice with PBS, add 5 mL of a 5 mM EDTA solution to each Petri dish. After incubation in 5% CO2/95% air atmosphere at 37 °C for 5 min, detach adherent cells using a cell lifter.
When most cells are observed to be suspended under the microscope, cell detachment is terminated by adding 5 mL per dish of α-MEM medium containing 10% FBS. After centrifugation of cells in 15 mL conical tubes at 400× g for 5 min, resuspend the cells with α-MEM medium with 10% FBS containing 20 ng/mL rM-CSF.
Note: Cells should be used immediately for culture and not prepared for cryopreservation.
Induction and characterization of osteoclast
Disinfect bovine cortical bone slices with 75% ethanol for 24 h and then wash twice with PBS solution for 5 min (Figure 1E). Place bone slices in a 96-well plate (one slice per well) with 200 μL of α-MEM medium containing 10% FBS.
Harvest bone marrow cells and culture atop individual bone slices in the presence of 20 ng/mL rM-CSF at a final cell concentration of 6 × 104–8 × 104 cells/mL in a final volume of 200 μL/well (Figure 1F).
After 12 h, change the medium to α-MEM medium with 10% FBS containing 20 ng/ml rM-CSF and 30 ng/ml rRANKL. Then, change full medium every other day for 6 days to induce osteoclast formation (days 4–5) and bone resorption pits (days 5–6).
Note: If the specific factors or compounds are of interest, add them to the cell culture medium as well. Due to the opacity of the bone slices, it is difficult to observe the state of the cells during this incubation period (Figure 2C); the reference times we have provided are suitable for strict adherence to our protocols.
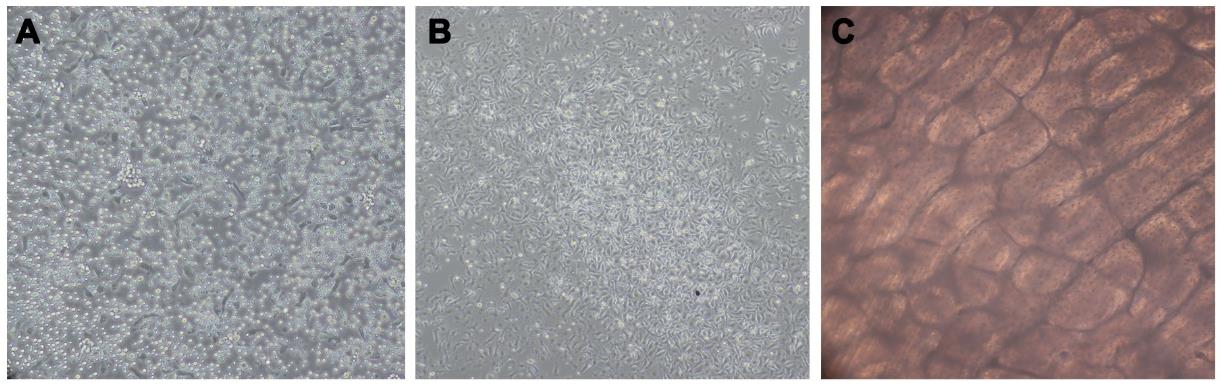
Figure 2. State of cells at different stages under the light microscope. (A) Non-adherent cells (step A8). (B) Adherent cells reaching 80%–90% confluence (step A9). (C) Culturing osteoclasts atop bone slice (step B3).Fix bone slices with 4% paraformaldehyde or 2.5% glutaraldehyde in PBS solution for 30 min at room temperature. Transfer the cultured medium to a centrifuge tube, centrifuge at 400× g for 10 min at 4 °C, and then collect the supernatant.
Use the TRAP stain kit to label cells on a 96-well plate according to the manufacturer’s instructions. Then, examine microscopically to observe TRAP-positive osteoclasts. TRAP-positive cells, which are dark red to purple, are identified as osteoclasts if they have three or more nuclei (Figure 3A).
For F-actin staining, fix osteoclasts on bone slices with 4% paraformaldehyde, permeabilize with 200 μL of 0.1% Triton X-100 for 10 min, and block with 2.5% bovine serum albumin for 1 h prior to an overnight incubation at 4 °C with primary antibodies direct against α-Tubulin (1:1,000). Following primary antibody incubations, incubate osteoclasts with donkey anti-mouse Alexa 488-conjugated secondary antibodies for 1 h and subsequently with phalloidin-tetramethylrhodamine B isothiocyanate for 30 min at 37 °C (Figure 3B).
For scanning electron microscopy (SEM) analysis, process osteoclasts atop bone slices for critical point drying and Au/Pg sputter; then, image them on a FE-SEM (Figure 3B).
Quantification of bone resorption pits
After incubating bone slices in 0.5 N NaOH for 30 s, gently scrape off cells using a medical cotton swab.
Label bone slices on the opposite side without cells to serve as a marker for the cell-containing side to observe. Dehydrate samples in ascending ethanol series (70%, 80%, 90%, 95%, 100%) at room temperature.
For SEM analysis, process bone slices for critical point drying and Au/Pg sputter. The resorption pits are imaged on a FE-SEM to clearly observe their morphology (Figure 3C).
For WGA-DAB analysis, soak bone samples in PBS for 5 min, stain with 100 μL of 20 μg/mL Peroxidase-WGA for 45 min, and then incubate with DAB tablets for 15 min. Picture bone resorption pits, which are stained in brown using a light microscope (Figure 3C), and determine the resorbed area in three random sites in one bone slice using ImageJ software. Access the irregular graphics box to outline resorption pits (stained brown) and then use the Analyze and Measure function to determine the resorption area.
For FITC-WGA analysis, sonicate bone samples in PBS and stain with 100 μL of 20 μg/mL FITC-WGA for 45 min in darkness (Figure 1G). Use a confocal laser scanning microscope to characterize three-dimensional profiles of resorption pits (Figure 3C) and process to obtain images of the sagittal angle. Quantitative analysis of resorption pit depth is performed in three random sites per bone slice using ImageJ software. Use ImageJ software to open the image of the sagittal angle, mark the depth of the bone resorption pits with the straight-line tool, and then click on Analyse and Measure to get the straight-line length.
Detect the supernatant from the cultured medium by CrossLaps ELISA kit for CTX-1 according to the manufacturer’s instructions to measure the concentration of collagen fragments (Figure 1H).
Data analysis
Figure 3 shows the characterization of osteoclast morphology and bone resorption pits.

Figure 3. Osteoclasts were induced on bone slices and formed bone resorption lacunae. (A) Bone marrow–derived macrophages (BMDMs) were then harvested and cultured for 4–5 days with rM-CSF (20 ng/mL) and rRANKL (30 ng/mL) on plastic substrata. Following TRAP staining, large multinucleated cells (MNCs) were formed (scale bar = 50 μm). Black arrows indicate large multinucleated cells. (B) BMDMs were cultured atop bovine bone slices in the presence of rM-CSF and rRANKL for 6 days. SEM analysis shows bone adherent osteoclasts. Sealing zone formation is assessed by phalloidin staining to visualize F-actin rings, while microtubules and nuclei are identified by staining with alpha-tubulin and DAPI, respectively. Scale bar = 10 μm, 10 μm, and 2 μm, from left to right panels). (C) Resorption pits were visualized by wheat germ agglutinin–diaminobenzidine (WGA-DAB) staining, scanning electron microscopy (SEM), and 3D imaging of WGA-FITC staining by confocal microscopy (scale bar = 100 μm, 10 μm, and 100 μm, from left to right panels). Black and white arrows indicate bone resorption pits.
Validation of protocol
This protocol has been used and validated in the following research article:
Zhu et al. [9]. Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci Transl Med. (Figures 2 and 3)
Zhu et al. [10]. A Zeb1/MtCK1 metabolic axis controls osteoclast activation and skeletal remodeling. EMBO J. (Figure 3)
Zhu et al. [11]. Proteolytic regulation of a galectin-3/Lrp1 axis controls osteoclast-mediated bone resorption. J Cell Biol. (Figures 2 and 5)
Ng et al. [12]. Sugar transporter Slc37a2 regulates bone metabolism in mice via a tubular lysosomal network in osteoclasts. Nat Commun. (Figure 8)
Li et al. [13]. Dynamic changes in O-GlcNAcylation regulate osteoclast differentiation and bone loss via nucleoporin 153. Bone Res. (Figures 2 and 3)
General notes and troubleshooting
Problem 1: BMDMs were unable to attach and culture atop bone slices.
Possible cause: Bone slices were not activated by the cultured medium.
Solution: Before BMDMs are inoculated, bone slices need to be soaked in the medium for 2 h.
Problem 2: BMDMs were unable to fuse to form osteoclasts.
Possible cause: Our recommended cell density is 6 × 104–8 × 104 cells/mL, 200 μL each well. The cell density is much higher than conventional RANKL-induced osteoclast in 96-well plates.
Solution: Use our recommended cell densities of BMDMs when inoculating cells on bone slices, since an appropriate density of BMDMs contributes to their osteoclastogenesis and fusion.
Problem 3: There was no F-actin ring formation of osteoclasts.
Possible cause: Osteoclast bone resorption function was not activated.
Solution: Before final fixation of osteoclasts, fresh rRANKL should be stimulated for 12–14 h.
Problem 4: There were few resorption pits on the bone slices despite sufficient osteoclast numbers observed.
Possible cause: Osteoclast and cell debris were not successfully removed.
Solution: Incubate bone slices in 0.5 N NaOH for 30 s and gently scrape off the cells using a medical cotton swab.
Problem 5: No cells or resorption pits were observed on the bone slices.
Possible cause: The cells were cultured on the other side of the bone surface.
Solution: We recommend that the bone slices be marked with a pencil on the surface devoid of cells after fixation.
Acknowledgments
This work was supported by the NSFC 82370914 and 81970919, the Fundamental Research Funds for the Central Universities 2042024YXA010 (L.Z.), the NSFC 82201042 and the Natural Science Foundation of Hubei Province 2022CFB658 (X.S.), the R01-AR075168 from the NIH (S.J.W.)
Competing interests
The authors declare no conflict of interest.
Ethical considerations
All animal experiments were approved by the Animal Research Ethics Committee of Wuhan University, China.
References
- Jacome-Galarza, C. E., Percin, G. I., Muller, J. T., Mass, E., Lazarov, T., Eitler, J., Rauner, M., Yadav, V. K., Crozet, L., Bohm, M., et al. (2019). Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature. 568(7753): 541–545.
- Udagawa, N., Takahashi, N., Akatsu, T., Tanaka, H., Sasaki, T., Nishihara, T., Koga, T., Martin, T. J. and Suda, T. (1990). Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci USA. 87(18): 7260–7264.
- Uehara, S., Udagawa, N., Mukai, H., Ishihara, A., Maeda, K., Yamashita, T., Murakami, K., Nishita, M., Nakamura, T., Kato, S., et al. (2017). Protein kinase N3 promotes bone resorption by osteoclasts in response to Wnt5a-Ror2 signaling. Sci Signaling. 10(494): eaan0023.
- Zhang, Y., Rohatgi, N., Veis, D. J., Schilling, J., Teitelbaum, S. L. and Zou, W. (2018). PGC1β Organizes the Osteoclast Cytoskeleton by Mitochondrial Biogenesis and Activation. J Bone Miner Res. 33(6): 1114–1125.
- Inoue, K., Deng, Z., Chen, Y., Giannopoulou, E., Xu, R., Gong, S., Greenblatt, M. B., Mangala, L. S., Lopez-Berestein, G., Kirsch, D. G., et al. (2018). Bone protection by inhibition of microRNA-182. Nat Commun. 9(1): 1114–1125.
- Nishi, Y., Atley, L., Eyre, D. E., Edelson, J. G., Superti-Furga, A., Yasuda, T., Desnick, R. J. and Gelb, B. D. (1999). Determination of Bone Markers in Pycnodysostosis: Effects of Cathepsin K Deficiency on Bone Matrix Degradation. J Bone Miner Res. 14(11): 1902–1908.
- Rumpler, M., Würger, T., Roschger, P., Zwettler, E., Sturmlechner, I., Altmann, P., Fratzl, P., Rogers, M. J. and Klaushofer, K. (2013). Osteoclasts on Bone and Dentin In Vitro: Mechanism of Trail Formation and Comparison of Resorption Behavior. Calcif Tissue Int. 93(6): 526–539.
- Shemesh, M., Addadi, S., Milstein, Y., Geiger, B. and Addadi, L. (2015). Study of Osteoclast Adhesion to Cortical Bone Surfaces: A Correlative Microscopy Approach for Concomitant Imaging of Cellular Dynamics and Surface Modifications. ACS Appl Mater Interfaces. 8(24): 14932–14943.
- Zhu, L., Tang, Y., Li, X. Y., Keller, E. T., Yang, J., Cho, J. S., Feinberg, T. Y. and Weiss, S. J. (2020). Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci Transl Med. 12(529): eaaw6143.
- Zhu, L., Tang, Y., Li, X., Kerk, S. A., Lyssiotis, C. A., Feng, W., Sun, X., Hespe, G. E., Wang, Z., Stemmler, M. P., et al. (2023). A Zeb1/MtCK1 metabolic axis controls osteoclast activation and skeletal remodeling. EMBO J. 42(7): e2022111148.
- Zhu, L., Tang, Y., Li, X. Y., Kerk, S. A., Lyssiotis, C. A., Sun, X., Wang, Z., Cho, J. S., Ma, J., Weiss, S. J., et al. (2023). Proteolytic regulation of a galectin-3/Lrp1 axis controls osteoclast-mediated bone resorption. J Cell Biol. 222(4): e202206121.
- Ng, P. Y., Ribet, A. B. P., Guo, Q., Mullin, B. H., Tan, J. W. Y., Landao-Bassonga, E., Stephens, S., Chen, K., Yuan, J., Abudulai, L., et al. (2023). Sugar transporter Slc37a2 regulates bone metabolism in mice via a tubular lysosomal network in osteoclasts. Nat Commun. 14(1): 906.
- Li, Y. N., Chen, C. W., Trinh-Minh, T., Zhu, H., Matei, A. E., Györfi, A. H., Kuwert, F., Hubel, P., Ding, X., Manh, C. T., et al. (2022). Dynamic changes in O-GlcNAcylation regulate osteoclast differentiation and bone loss via nucleoporin 153. Bone Res. 10(1): 51.
Article Information
Publication history
Received: Jun 20, 2024
Accepted: Sep 3, 2024
Available online: Oct 13, 2024
Published: Nov 5, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Sun, X., Wang, Z., Tang, Y., Weiss, S. J. and Zhu, L. (2024). An In Vitro Model of Murine Osteoclast-Mediated Bone Resorption. Bio-protocol 14(21): e5100. DOI: 10.21769/BioProtoc.5100.
- Zhu, L., Tang, Y., Li, X. Y., Kerk, S. A., Lyssiotis, C. A., Sun, X., Wang, Z., Cho, J. S., Ma, J., Weiss, S. J., et al. (2023). Proteolytic regulation of a galectin-3/Lrp1 axis controls osteoclast-mediated bone resorption. J Cell Biol. 222(4): e202206121.
Category
Developmental Biology > Cell growth and fate > Differentiation
Cell Biology > Cell isolation and culture > Cell differentiation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.