- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient Gene-Editing in Human Pluripotent Stem Cells Through Simplified Assembly of Adeno-Associated Viral (AAV) Donor Templates
Published: Vol 14, Iss 21, Nov 5, 2024 DOI: 10.21769/BioProtoc.5097 Views: 3176
Reviewed by: Rajesh RanjanGuanghui YangAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2024
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Gene-edited human pluripotent stem cells provide attractive model systems to functionally interrogate the role of specific genetic variants in relevant cell types. However, the need to isolate and screen edited clones often remains a bottleneck, in particular when recombination rates are sub-optimal. Here, we present a protocol for flexible gene editing combining Cas9 ribonucleoprotein with donor templates delivered by adeno-associated virus (AAV) vectors to yield high rates of homologous recombination. To streamline the workflow, we designed a modular system for one-step assembly of targeting vectors based on Golden Gate cloning and developed a rapid protocol for small-scale isolation of AAV virions of serotype DJ. High homology-directed repair (HDR) rates in human pluripotent stem cells (hPSCs), ~70% in ACTB and ~30% in LMNB1, were achieved using this approach, also with short (300 bp) homology arms. The modular design of donor templates is flexible and allows for the generation of conditional and/or complex alleles. This protocol thus provides a flexible and efficient strategy workflow to rapidly generate gene-edited hPSC lines.
Key features
• Versatile approach combining AAV-DJ donors and CRISPR ribonucleoproteins, providing an efficient method for long and short edits, insertions, and deletions in human pluripotent stem cells
• One-step cloning method for rapid generation of customized AAV donor plasmids
• Simplified AAV purification pipeline for ready-to-infect virion preparations
Keywords: AAV purificationGraphical overview
Background
The increasing availability of human genetic data offers unprecedented opportunities to understand, diagnose, and treat human diseases, but challenges remain in our ability to interpret the functional significance of specific genetic variants. At the same time, CRISPR/Cas-based gene-editing technologies have vastly simplified the means to introduce specific variants for functional interrogations [1–3]. Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), are attractive model systems for such functional studies as they allow experimental manipulations in relevant human cell types [4,5]. Of particular importance, hPSCs allow disease modeling in cell types of the central nervous system, including neurons, astrocytes, and microglia [6], which are generally inaccessible from patients yet are highly relevant to study in the context of psychiatric and neuropsychiatric diseases such as autism and schizophrenia, which are both largely attributable to genetic variants that demand functional studies.
In a typical gene-editing experiment, Cas9 nuclease is introduced to a bulk of cells together with a gene-specific guide RNA (gRNA) and a repair template for homology-directed repair (HDR). This strategy provides a means for flexible and precise edits ranging from single-base-pair substitutions to large insertions. However, sub-optimal editing rates may require tedious screening of clones to obtain cells with the desired edit(s), in particular when bi-allelic edits are desired. Moreover, clonal isolation involves the inevitable risk of acquiring off-target differences between selected cells that may confound the interpretation of subsequent experiments [7], which necessitates validation in multiple clones or programmable strategies in which the same clone can give rise to both experimental and control conditions [8,9].
Recombinant adeno-associated viruses (AAVs) provide superior donor templates for HDR, especially when double-strand breaks (DSBs) are simultaneously introduced at targeted loci [10–12]. Previous studies have demonstrated high HDR rates in hPSCs when Cas9-induced DSBs were combined with AAV donors, reaching ~40%–90% without prior selection [13]. Multiple factors likely contribute to the superiority of AAV virions as vectors of HDR, including its single-stranded DNA genome and the nature of its flanking inverted terminal repeats (ITRs). Several AAV serotypes effectively transduce hPSCs, of which serotype 6 is commonly used [13,14]. AAV-DJ is a synthetic hybrid serotype with expanded tropism that provides comparable rates in hPSCs [13,15]. The use of AAV donor templates may thus overcome challenges related to limited targeting efficiencies. However, contrasting the off-the-shelf availability of Cas9 protein and synthetic sgRNAs, the multi-step cloning and production of AAV virions is time-consuming, which provides a barrier to this approach.
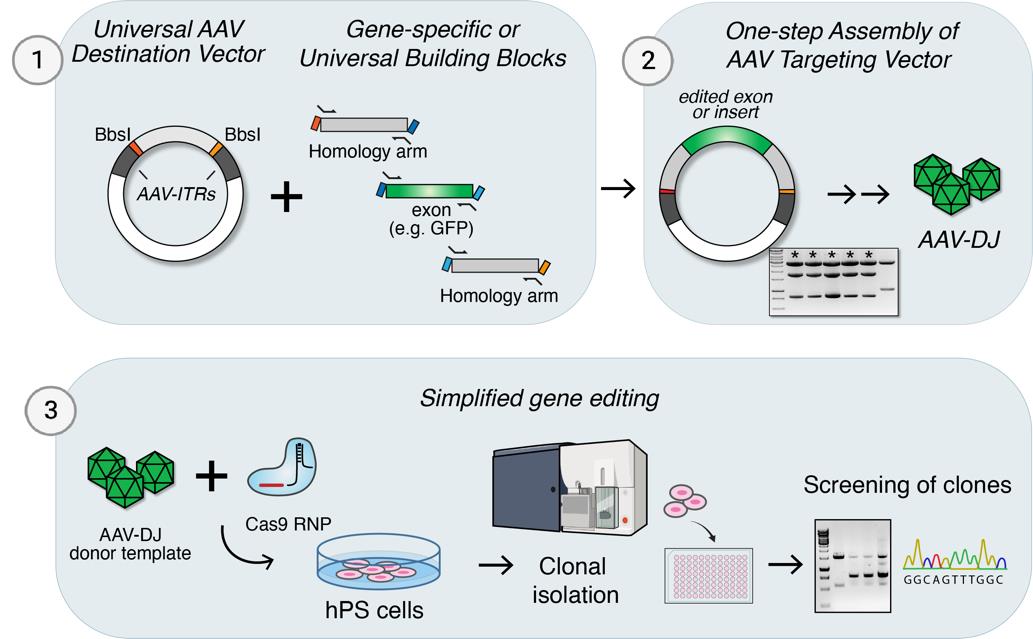
Here, we present a protocol for gene-editing of hPSCs that includes a system for one-step cloning of AAV targeting vectors by Golden Gate [16] assembly as well as a simplified protocol for rapid small-scale isolation of AAV virions. Combined with Cas9 ribonucleoprotein (RNP) complexes, this approach provides a highly efficient yet adaptable approach for gene editing in hPSCs, exemplified by large tag insertions, the generation of conditional alleles, as well as single base edits.
Materials and reagents
Biological materials
AAV-293 cells (Agilent, catalog number: 240073)
hESC H9, WA09 (WiCell, catalog number: WAe009-A)
One shot Stbl3 chemically competent E. coli (Thermo Fisher Scientific, catalog number: C737303)
AAV-GG plasmid vector (Addgene, # 213039)
AAV-packaging plasmids pHelper, pRC-DJ [15]
mTeSR Plus (STEMCELL, catalog number: 100-0276)
CloneR2 (STEMCELL, catalog number: 100-0691)
ReLeSR (STEMCELL, catalog number: 100-0483)
P3 Primary Cell 4D-Nucleofector Kit S (20 μL) (Lonza, catalog number: V4XP-3032)
DMEM, high glucose/GlutaMAX/pyruvate (Thermo Fisher Scientific, catalog number: 31966047)
MEM, no glutamine, no phenol red (Thermo Fisher Scientific, catalog number: 51200046)
Fetal bovine serum (FBS) (Merck, catalog number: F7524)
TrypLE Select enzyme, no phenol red (Thermo Fisher Scientific, catalog number: 12563011)
DPBS without calcium and magnesium (Thermo Fisher Scientific, catalog number: 14190250)
Bambanker freezing media (Techtum, catalog number: 23-BB01)
Matrigel hESC-qualified matrix (Corning, catalog number: 354277)
Accutase (STEMCELL, catalog number: 07920)
Thiazovivin (Cayman chemical, catalog number: 14245-5)
PrimeSTAR GXL DNA polymerase (Takara Clontech, catalog number: R050A)
Nuclease-free DEPC water, PCR grade (Thermo Fisher Scientific, catalog number: AM9937)
Alt-R S.p. HiFi Cas9 Nuclease V3, 500 μg (IDT, catalog number: 1081061)
Alt-R CRISPR-Cas9 crRNA, 2 nmol (IDT, custom design)
Alt-R CRISPR-Cas9 tracrRNA, 20 nmol (IDT, catalog number: 1072533)
BpiI FastDigest enzyme (Thermo Fisher Scientific, catalog number: FD1014)
T4 DNA ligase 2000 U/μL (NEB, catalog number: M0202T)
10× T4 DNA ligase buffer (NEB, catalog number: B0202S)
QIAEX II gel extraction kit (Qiagen, catalog number: 20021)
Plasmid Plus Midi Kit (Qiagen, catalog number: 12945)
DNEasy Blood and Tissue kit (50 st) (Qiagen, catalog number: 69504)
CaCl2 (Sigma-Aldrich, catalog number: S9888)
NaCl (Sigma-Aldrich, catalog number: 223506)
KCl (Merck, catalog number: 4936.1000)
Na2HPO4 (Merck, catalog number: 1.06586.0500)
D-(+)-Glucose (Sigma-Aldrich, catalog number: G7021)
HEPES (Sigma-Aldrich, catalog number: H3375)
MgCl2, (Sigma-Aldrich, catalog number: M9272)
6-well culture plates (Thermo Fisher Scientific, catalog number: 140675)
96-well culture plates (Corning, Falcon, catalog number: 353072)
1.5 mL Eppendorf tubes (VWR, catalog number: 89000-028)
15 mL centrifuge tubes (VWR, catalog number: 21008-216)
10 μL racked tips, low-retention sterile (VWR, catalog number: 10017-062)
200 μL racked tips, low-retention sterile (VWR, catalog number: 76322-150)
1000 μL low-retention tips (VWR, catalog number: 10017-090)
Nunc cell culture cryogenic tubes, 1 mL (500) (Thermo Fisher Scientific, catalog number: 377224PK)
PCR tubes and domed caps, strips of 8. (Thermo Fisher Scientific, catalog number: AB0266)
T25 EasYFlask, TC surface, filter cap (Thermo Fisher Scientific, catalog number: 156367T25)
5 mL round-bottom polystyrene tubes (VWR, catalog number: 734-0445)
Amicon Ultra-4 centrifugal filter unit (Merck, catalog number: UFC810024)
35 μm cell strainer cap polystyrene tubes (Falcon, catalog number: 08-771-23)
Equipment
T100 Thermal Cycler (Bio-Rad, model: 1861096)
Microcentrifuge 5418R (Eppendorf, catalog number: 5401000010)
Benchtop refrigerated centrifuge 5430R (Eppendorf, catalog number: 022620601)
4D-Nucleofector X Unit (Lonza, catalog number: AAF-1003X)
NanoDrop 1000 UV/VIS Spectrophotometer (Thermo Fisher Scientific, catalog number: ND-1000)
FACS Aria™ Fusion Flow Cytometer (BD Biosciences, serial number: R656700J40005)
Software and datasets
CHOPCHOP [17] (version 3, http://chopchop.cbu.uib.no)
TIDE [18] (version 3.3.0; http://tide.nki.nl)
Sequence analysis software, e.g., Geneious Prime (version 2024.0.2, BioMatters, 2024-02-14)
Procedure
Design strategy and selection of target gRNA
We design gRNAs for the desired target site aided by the CHOPCHOP design tool (http://chopchop.cbu.uib.no) [17]. Select a gRNA target site that will be disrupted by recombination with the targeting vector, preferably by disrupting its protospacer adjacent motif (PAM).
With this constraint in mind, choose 1–2 gRNAs per site based on their mismatch number and efficiency score from [19], preferably selecting guides with efficiency scores >45 and no off-target sites with <3 mismatches. The gRNAs should ideally cut no more than 30 bp away from the desired recombination site for optimal efficiency [20], and the target sequence should be edited by recombination to prevent recutting.
Design of targeting vector and generation of fragments by PCR or custom synthesis
Design the targeting vector by generating a desired target sequence flanked by ~300–500 bp homology arms (Figure 1A). Each fragment should contain Golden Gate–compatible overhangs listed in Table 1 (ensure that there are no BbsI sites in target sequences). The homology arms can be amplified by PCR with BbsI-compatible primer extensions shown in Table 2, using a high-fidelity polymerase (e.g., PrimeSTAR, Takara). It is preferable to use genomic DNA from the cell line to be targeted as the template for the PCR reaction. Alternatively, the homology arms and/or the edited sequence can be ordered as synthetic fragments (e.g., IDT gBlocks).
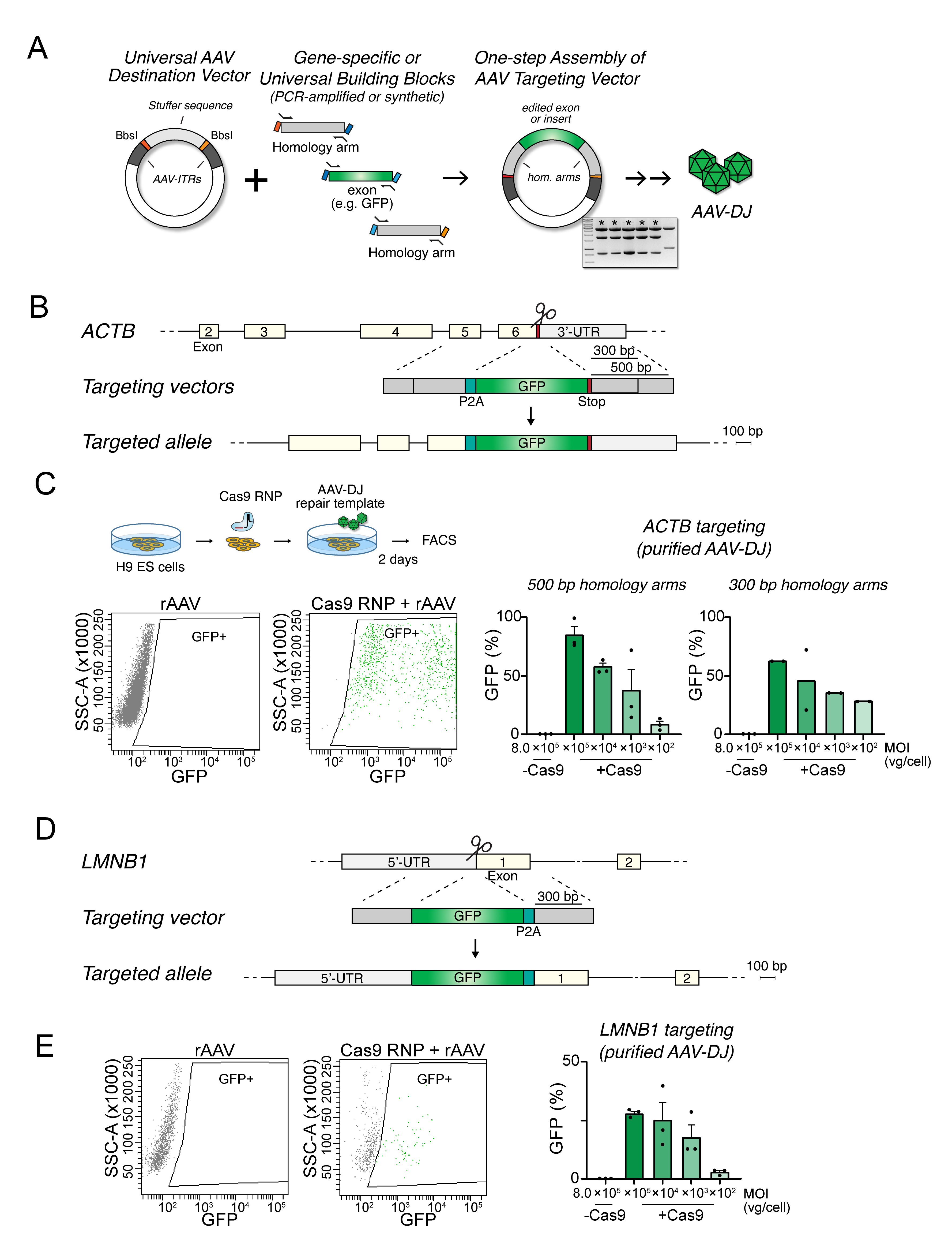
Figure 1. Efficient homologous recombination in human pluripotent stem cells (hPSCs) by combining Cas9 ribonucleoproteins (RNPs) and purified adeno-associated virus (AAV)-DJ donor templates. A. One-step assembly of AAV targeting vectors by Golden Gate assembly. The acceptor vector contains unique overhangs flanked by BbsI-sites and interspaced by a “stuffer sequence” to separate the inverted terminal repeats (ITRs). Transgenes, exons, and/or homology arms generated by PCR or as synthetic fragments can be assembled in a single step. The fragments are assembled in the AAV donor vector and used to package the donor template in AAV particles. B. Schematic of the human ACTB locus and strategy to insert 2A-GFP in the 3’-UTR. Target site for the gRNA is marked by a pair of scissors. C. Outline of the experiment (top) to measure homologous recombination efficiencies by FACS analysis. Targeting efficiencies were measured as the percentage of GFP-positive cells following transfection with AAV plasmid or transduction with different amounts of donor templates with ~500 bp or ~300 bp homology arm lengths, respectively (right). Kruskal-Wallis statistical comparison of 500 bp and 300 bp arms targeting for each dilution was not significant (ns, p > 0.999). D. Schematic of the human LMNB1 locus and strategy to insert GFP-2A in the 5’-UTR. Homology arms were of ~350 bp lengths. The target site for the gRNA is marked by a pair of scissors. E. Representative plots (left) and summary statistics on the proportion of GFP-positive cells. Multiplicity of infection (MOI) refers to the ratio of viral genomes (vg), as determined by quantitative PCR, per transduced cell. Data represented as mean ± standard error of the mean (SEM).Table 1. Example of Golden Gate fragment overhangs used for the modular assembly of AAV vectors (Figure 1A)
Fragment Left junction Right junction AAV-GG vector AGCG GCAA Left homology arm (LHA) GCAA AGTG Edited sequence AGTG TCCA Right homology arm (RHA) TCCA AGCG Table 2. PCR primer extensions for amplifying BbsI-compatible fragments.
The underlined sequence corresponds to ligated overhangs (Table 1).
Fragment Forward primer Reverse primer LHA 5’-CACCACAGAAGACGAGCAA…[primer] 5’-CACCACAGAAGACTCCACT…[primer] Edited 5’-CACCACAGAAGACGAAGTG…[primer] 5’-CACCACAGAAGACTCTGGA…[primer] RHA 5’-CACCACAGAAGACGATCCA…[primer] 5’-CACCACAGAAGACTCCGCT…[primer] Fragments amplified by PCR should be gel-purified, e.g., using the QIAEX II Gel Extraction Kit, resuspended in Milli-Q water. Assess quality and concentration using UV spectroscopy, e.g., using a NanoDrop instrument.
One-step assembly of targeting vector by Golden Gate assembly into universal AAV vector
Set up the Golden Gate assembly reaction in a PCR tube following the recipe in Table 3. Adjust volumes according to the concentrations and lengths of the fragments (total amount of each should be 40 fmol).
Table 3. Typical reaction mix for Golden Gate assembly of three fragments into the AAV-GG destination vector
Fragment Concentration (ng/μL) Volume (μL) Left homology arm (ca. 500 bp) 50 0.25 Right homology arm (ca. 500 bp) 50 0.25 Edited sequence (ca. 500 bp) 50 0.25 AAV-GG vector (ca. 4,000 bp) 100 1 10X T4 DNA ligase buffer - 1.25 BpiI (isochisomer to BbsI) - 0.75 T4 DNA ligase 2000U/μL - 0.25 Water - 8 Total 12.5 Perform the Golden Gate reaction in a thermal cycler using the following program (Table 4):
Table 4. Golden Gate cycling program
37 °C 1 min 30× 16 °C 1 min 60 °C 5 min Transform competent bacteria, according to standard procedures. We use chemically competent Stbl3 cells, with 40–50 μL of cells transformed with 1/10 vol of the Golden Gate reaction.
Plate transformed bacterial culture into LB-agar plates with ampicillin and incubate overnight at 37 °C.
Pick 4–6 colonies and grow in LB with ampicillin overnight; expect at least one positive colony. The amount of positive clones obtained differs depending on the purity and size of the fragments but, typically, efficiencies range from 30% to 100%.
Critical: Include a negative control Golden Gate reaction without one of the homology arm fragments to assess for background formed by re-ligation of the backbone plasmid.
Miniprep cultures and test for correct assembly, e.g., using diagnostic restriction enzyme digests.
Amplify a correct clone by growing a 35 mL LB culture and purify the plasmid using the Plasmid Plus Midi Kit, according to instructions.
Rapid isolation of AAV virions from AAV-293 cell conditioned media
Culture AAV-293 cells in DMEM supplemented with 10% (vol/vol) FBS.
One day prior to transfection, split near-confluent cells using TrypLE to a T25 flask (surface area 25 cm2) at 1:3 dilution.
One hour prior to transfection, replace to fresh DMEM-FBS media.
Co-transfect the AAV-293 cells with the AAV targeting vector along with pHelper and RC-DJ plasmids using the Cal-Phos method: Per T25 flask, prepare in a 1.5 mL Eppendorf tube 37 μL of 2 M CaCl2 solution, 3 μg of each plasmid, and water to a total volume of 300 μL. Add this dropwise to a 5 mL round-bottom polystyrene tube containing an equal volume (300 μL) of 2× HBS (0.4 M NaCl, 10 mM KCl, 1.5 mM Na2HPO4, 0.2% glucose, 38.4 mM HEPES pH 7.05) and immediately disperse on top of the cells.
Four hours after transfection, replace media with 5 mL of mTeSR Plus medium.
Collect the media (approximately 5 mL) 72 h post-transfection and clear dead cells and debris by centrifugation at 1,500× g for 10 min at 4 °C.
Equilibrate an Amicon Ultra-4 centrifugal filter (100 kDa) 15 mL spin column with DPBS + 2 mM MgCl2, followed by a brief centrifugation (1 min, until most of the PBS has passed the filter) at 1,500× g at 4 °C.
Load the cleared viral supernatant on the spin filter and centrifuge at 1,500× g for 5–15 min at 4 °C to concentrate the virus solution to ~500 μL, followed by a brief wash with mTeSR Plus medium and further concentration until a final volume of 250 μL of cleaned mTeSR Plus media containing virion particles. Caution: Do not allow the membrane to dry out, as viral titer may decrease due to particles becoming trapped in the filter membrane. Check the volume every 2–3 min during centrifugation to ensure volume reduction without complete drying.Pause point. AAV-DJ particles can be snap-frozen in small aliquots at liquid nitrogen. In contrast to AAV purified over iodixanol gradients, this rapid preparation of AAV particles will contain a significantly higher amount of non-infectious AAV genomes, making the determination of viral titers by quantitative PCR inaccurate. Hence, we empirically determine suitable viral amounts to use (see below).
Nucleofection of hPSCs with CRISPR-RNPs and subsequent transduction of cells with AAV carrying the targeting vector
Culture hPSCs for one week prior to performing nucleofection. Maintain cells in mTeSR Plus medium on Matrigel-coated plastic (0.5 mg/mL dilution in MEM) and keep in a humidified incubator with 5% CO2 at 37 °C. Replace media every other day and passage cells when required using ReLeSR according to the manufacturer’s instructions.
Anneal Alt-R crRNA with Alt-R tracr-RNA (both from IDT) in a 1:1 ratio at 50 μM final concentration of each gRNA by heating to 95 °C for 5 min in a PCR cycler followed by slow cool down to room temperature on the bench top for 3 min; then, place on ice. Annealed guides can be stored at -20 °C for 2–3 weeks and at -80 °C for 6 months.
Prepare Cas9 RNPs by mixing recombinant Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT) with the annealed crRNA and tracr-RNA for a total volume of 4 μL per reaction, as in Table 5. Incubate the mixed gRNA and Cas9 protein at room temperature for 10–15 min before transfection to ensure proper RNP complex formation. Pause point. RNPs can be stored at -20 °C for up to 2 weeks.
Table 5. CRISPR-Cas9 RNP reagents for a single nucleofection reaction
Reagent Final concentration (μM) Volume (μL) Annealed crRNA and tracr-RNA 50 2,3 Alt-R™ S.p. HiFi Cas9 Nuclease V3 (IDT) 61 1,7 Total 4 Nucleofection of hPSCs:
Treat cells with ROCK inhibitor (we use 2 μM thiazovivin) for ~2 h prior to transfection.
Detach cells with Accutase and count cells using an automated cell counter or Burker chamber. In a 15 mL Falcon tube, spin down (200× g for 5 min) 400,000 cells per transfection. Carefully remove all supernatant and resuspend cells in 20 μL of P3 solution (Lonza). Transfer the cell suspension to an Eppendorf tube and mix with 4 μL of RNP complex. Immediately transfer to a 16-Nucelocuvete strip using a 4D-Nucleofector system (Lonza). Tap strips to avoid the presence of bubbles and bring all material to the bottom of the cuvette.Caution: Work fast and avoid creating bubbles during the resuspension of cells with the P3 solution and transfer to the nucleocuvette.Critical: It is recommended to include transfection with a GFP plasmid (1.5 μg) as a control for nucleofection efficiency. A condition nucleofecting gRNA without Cas9 protein may be included as an additional negative control for downstream applications.
Electroporate in 4D nucleofector (Lonza) using the program CA-137.
Immediately after pulse completion, resuspend cells in 100 μL of mTeSR Plus with ROCK inhibitor (e.g., 2 μM thiazovivin) and plate onto Matrigel-coated 12-well plates previously filled with 1 mL of warm (37 °C) mTeSR Plus medium with ROCK inhibitor. Seed at a density of 200,000 cells per well.
Add AAV particles to the media within 10 min after nucleofection. We normally add 5 μL per well and a no-virus control.Recommended: As viral toxicity can be noticed in some experiments, we recommend empirically testing the optimal amount of viral particles by adding (e.g., 1:5) serial dilutions of the AAV preparation to 3–4 replicate wells of nucleofected cells and subsequently select the condition with the highest titer not showing apparent toxicity.
Place cells back in the incubator and culture for 24–72 h prior to analysis. Change media to mTeSR Plus without ROCK inhibitor 24 h after transfection. Monitor cell viability as high amounts of AAV particles may affect cell viability. Continue with the highest titer that does not show excessive cytotoxicity.
Optional: Isolation of single clones by FACS for expansion and PCR screening.
For isolation and analysis of single clones, we recommend sorting single cells by FACS 24–48 h after Cas9 RNP nucleofection and AAV transduction.
Prepare a 96-well plate coated with Matrigel for 1 h at 37 °C. Fill each well with 100 μL of mTeSR Plus media supplemented with CloneR2 (to promote cell survival).
Detach cells with Accutase, spin down (200× g for 5 min), and resuspend in mTeSR Plus supplemented with CloneR2. Pass cells through a 35 μm cell strainer cap of a polystyrene tube.
Configure gates and flow of an automated cell sorter such as a BD FACSAria III to sort single viable cells into 96-well plates.
Following sorting, place the plate in the incubator. Replace media with mTeSR Plus for clonal expansion 48 h post-sorting and change media every other day. Colonies will appear within one week.
For analysis and banking, expand clones into two duplicate 24-well plates such that one well is used to collect DNA (e.g., using QIAGEN DNAeasy kit) and the other to freeze cells. For freezing, detach cells with Accutase, spin down (200× g for 5 min), and resuspend with 300 μL of Bambanker freezing media.
Validation of protocol
Efficient gene editing in hPSCs by AAV-DJ donors obtained by one-step assembly
A previous study demonstrated high rates of HDR in hPSCs when Cas9-induced DSBs were combined with repair templates delivered by AAVs [13]. However, a drawback of this approach is the requirement to design and generate allele-specific AAV vectors with sometimes complex cloning pipelines. We sought to simplify this workflow by first designing a simplified strategy to assemble donor templates in an AAV backbone by one-step Golden Gate assembly (Figure 1A) [16,21].
To test the utility of our approach, we assembled a targeting vector to insert a sequence encoding a self-cleaving 2A peptide and a green fluorescent protein (P2A-GFP) in the 3’-untranslated region (UTR) of the ACTB locus. GFP expression is thus driven by endogenous ACTB expression in correctly targeted cells only. This approach is analogous to one previously used in mice [22] but employs shorter homology arms of only 300 or 500 bp (Figure 1B) to further simplify vector build. A gRNA sequence (5’-CGTCCACCGCAAATGCTTCT) to cut near the stop codon in exon 6 of ACTB was selected. The AAV targeting vectors were generated by PCR using primer overhangs shown in Table 2. The P2A sequence was added by a long primer, amplifying the GFP fragment of the plasmid pEGFP_N1 (Clontech). Flanking homology arms (300 or 500 bp) were similarly prepared by PCR from synthetic DNA fragments (gBlock; IDT) corresponding to the following genomic coordinates (hg38): LHA300 (chr7:5,527,751-5,528,055); LHA500 (chr7:5,527,751-5,528,304); RHA300 (chr7:5,527,441-5,527,749); RHA500 (chr7:5,527,184-5,527,748); see Supplementary information for full targeting sequence and primers used for cloning. PCR products were gel-purified using the QIAEX II Gel Extraction Kit and subjected to the single-tube reaction assembly into the AAV destination vector (Table 3), using the following program: 30× (37 °C, 5 min, 16 °C, 5 min) 60 °C, 5 min. After completion of the reaction, another 0.5 μL of Bpil was added, followed by a final incubation at 37 °C for 60 min to remove background. Colonies were screened by restriction digests and verified by Sanger sequencing.
Human H9 ES cells were electroporated with the Cas9 RNP and AAV particles, here purified over an iodixanol gradient [23]. Two days later, the proportion of GFP-expressing cells was quantitatively measured by flow cytometry (Figure 1C). We observed a dose-dependent response, with editing rates of up to ~80% editing in conditions using a high titer of AAV-DJ virions (8 × 105 viral genomes per cell). As expected, recombination rates were lower albeit not significantly different when homology arms were shortened to 300 bp, but in the same order of magnitude. The combination of Cas9 RNP and AAV-DJ donor templates thus yields high HDR rates also with relatively short homology arms.
To test the universality of this approach, we took a similar promoterless approach by instead targeting GFP upstream of the coding sequence of LMNB1 (Figure 1D). We similarly designed 300 bp homology arms from synthetic DNA fragments (gBlocks; IDT) corresponding to the genomic coordinates (hg38) LHA (chr5:126,776,986-126,777,467) and RHA (chr5:126,777,597-126,778,005), and assembled with a GFP-2A sequence for one-step assembly in our universal AAV targeting vector, here using a synthetic fragment (IDT gBlock) because a high GC content made genomic PCR challenging (see Supplementary information for full targeting sequences and primers used for cloning). AAV-DJ particles purified by density gradient centrifugation were delivered to human H9 ES cells following electroporation with Cas9 RNPs with a gRNA targeting exon 1 of LMN1B (5’-GGGGTCGCAGTCGCCATGG). The proportion of GFP-positive cells two days later was about 30% (Figure 1E). The lower efficiency compared with the targeting of ACTB may reflect allele-specific differences in targeting efficiencies. The apparent lack of dose-dependent increase with higher AAV concentrations may also reflect toxicity from viral particles, as described in the hematopoietic stem and progenitor cells (HSPCs) [24], and/or allele-specific effects related to manipulation of the LMNB1 locus at the 5’UTR region [22].
Rapid small-scale AAV purification simplifies editing pipeline
In our first experiments, we used dilutions of concentrated AAV particles highly purified through gradient density centrifugation. As this procedure is laborious, here we compared this to simply collecting, briefly washing and concentrating viral particles from the media of AAV-producing cells on spin filter columns (Figure 2A). Using the same assays to test for insertion by HDR as shown in Figure 1, we found high degrees of GFP-positive cells in both ACTB and LMNB1 loci. The rates were almost as high as with the purified vectors, in a dose-dependent manner (Figure 2B-2C). This simplified protocol can reduce the time and cost needed to generate AAV virions for in vitro applications. We have also used this protocol to efficiently introduce a tag in the NRXN1 locus [25].
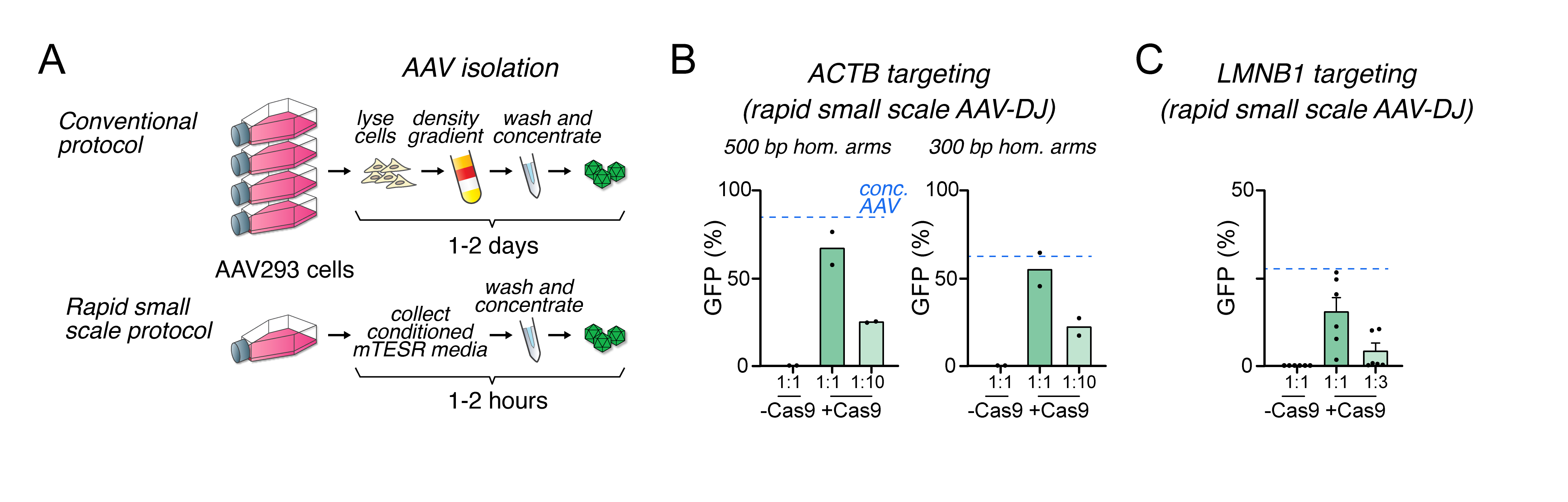
Figure 2. Rapid small-scale production of adeno-associated virus (AAV)-DJ targeting vectors permits efficient homologous recombination.
A. Schematic comparing conventional purification of AAV particles by density gradient (top) with our simplified protocol for faster, small-scale production for in vitro use (bottom). B. Summary statistics of homology recombination rates in the ACTB locus obtained by small-scale purification of AAV-DJ. Dashed lines indicate the rates obtained with the highest titer of purified viral particles, used in Figure 1C. The 1:1 dilution is estimated to correspond to 8 × 103 viral genomes/cell. C. Summary statistics of homology recombination rates in the LMN1B locus obtained by small-scale purification of AAV-DJ. Dashed lines indicate the rates obtained with the highest titer of purified viral particles, used in Figure 1E. Data represented as mean ± standard error of the mean (SEM).
General notes and troubleshooting
| Step | Problem | Possible reason | Suggested solution |
| Cloning AAV-GG backbone | Low yield of bacterial colonies after Golden Gate cloning. | Low purity of HDR-template fragments; overhangs incompatibilities due to design inaccuracies; presence of additional BbsI/Bpil recognition sites in fragments; low transformation efficiency. | Check design of primers and/or synthetic fragments; ensure purity of fragments by gel electrophoresis and UV spectroscopy; use fresh lots recommended enzymes (Bpil Invitrogen, Cat# FD1014) and high-concentration T4 DNA ligase (NEB, Cat# M0202T) with fresh aliquots of T4 ligase buffer (NEB); include positive and negative controls to test for competency of bacteria and selection on plates. |
| No bands, multiple bands, or low yield following genomic PCR to generate cloning fragments. | Primer or template sequences incompatible with PCR; suboptimal quality of template DNA. | Check primers, template GC-content, annealing temperature, and optimize PCR accordingly; use DNA polymerase optimized for challenging templates (e.g., PrimeSTAR GXL, Takara, Cat# R050A); order target sequences as synthetic fragments. | |
| High number of background colonies in the Golden Gate reaction. | Incomplete digestion of the AAV-GG backbone plasmid. | Perform an extra step of BbsI/Bpil digestion after the Golden Gate reaction to linearize unligated backbone vector and reduce the number of background colonies. | |
| hPSC transfection | Insufficient cell survival after transfection and AAV infection. | Cell confluency before transfection < 60%; Nucleofection-related toxicity; AAV-related toxicity; gene-editing affects cell survival. | Ensure a cell density of at least 80% before transfection and perform gentle pipetting when detaching cells with Accutase. Work swiftly after resuspension of cells with the P3 transfection solution to avoid toxicity-derived cell death and add ROCK inhibitor to media before and after nucleofection; dilute AAV particles even further; consider the potential cell-lethality of the intended gene edit. |
| Low-efficiency transfection observed as low yield of GFP positive upon nucleofection with control plasmid. | Suboptimal cell health prior to nucleofection; suboptimal handling of cells during nucleofection; expired P3 transfection solution. | See above; also ensure the quality of reagents including the GFP plasmid and that the P3 solution is not expired. | |
| AAV generation | Low efficiency of gene-editing events. | Low yield of AAV donor templates; low-efficiency sgRNA targeting or sgRNA degradation; low nucleofection rates; inefficient gRNA. | Use AAV-293T cells that produce higher AAV titers than HEK293 cells. Ensure high density (around 70% confluency) of HEK cell culture before transfection; compare targeting efficiencies with or without Cas9 RNP or AAV donors and assess efficiency by a functional readout (e.g., GFP fluorescence) or analysis of bulk DNA (e.g., T7 assay); try alternative gRNAs. |
Acknowledgments
We thank Susann Li and Stefanie Fruhwürth for assistance with FACS analysis and Swaroop Achuta for the initial cloning of the AUTS2 targeting vector. Funding: S.M. is supported by a grant from the Assar Gabrielsson Foundation. F.H.S. is supported by a grant from the Swedish Research Council (dnr 2017-03331). F.H.S. and ES are supported by the Knut and Alice Wallenberg Foundation via the Wallenberg Centre for Molecular and Translational Medicine at the University of Gothenburg, Sweden.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A. and Charpentier, E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337(6096): 816–821. https://doi.org/10.1126/science.1225829.
- Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science. 339(6121): 819–823. https://doi.org/10.1126/science.1231143.
- Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E. and Church, G. M. (2013). RNA-guided human genome engineering via Cas9. Science. 339(6121): 823–826. https://doi.org/10.1126/science.1232033.
- Sabitha, K. R., Shetty, A. K. and Upadhya, D. (2021). Patient-derived iPSC modeling of rare neurodevelopmental disorders: Molecular pathophysiology and prospective therapies. Neurosci Biobehav Rev. 121: 201–219. https://doi.org/10.1016/j.neubiorev.2020.12.025.
- Shi, Y., Inoue, H., Wu, J. C. and Yamanaka, S. (2017). Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 16(2): 115–130. https://doi.org/10.1038/nrd.2016.245.
- Wang, H., Yang, Y., Liu, J. and Qian, L. (2021). Direct cell reprogramming: approaches, mechanisms and progress. Nat Rev Mol Cell Biol. 22(6): 410–424. https://doi.org/10.1038/s41580-021-00335-z.
- Panda, A., Suvakov, M., Mariani, J., Drucker, K. L., Park, Y., Jang, Y., Kollmeyer, T. M., Sarkar, G., Bae, T., Kim, J. J., et al. (2022). Clonally selected lines after CRISPR/Cas editing are not isogenic. bioRxiv: 2022.2005.2017.492193. https://doi.org/10.1101/2022.05.17.492193.
- Pak, C., Danko, T., Zhang, Y., Aoto, J., Anderson, G., Maxeiner, S., Yi, F., Wernig, M. and Südhof, T. C. (2015). Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused by Heterozygous Mutations in NRXN1. Cell Stem Cell. 17(3): 316–328. https://doi.org/10.1016/j.stem.2015.07.017.
- Zhou, B., Lu, J. G., Siddu, A., Wernig, M. and Sudhof, T. C. (2022). Synaptogenic effect of APP-Swedish mutation in familial Alzheimer's disease. Sci Transl Med. 14(667): eabn9380. https://doi.org/10.1126/scitranslmed.abn9380.
- Miller, D. G., Petek, L. M. and Russell, D. W. (2003). Human gene targeting by adeno-associated virus vectors is enhanced by DNA double-strand breaks. Mol Cell Biol. 23(10): 3550–3557. https://doi.org/10.1128/MCB.23.10.3550-3557.2003.
- Porteus, M. H., Cathomen, T., Weitzman, M. D. and Baltimore, D. (2003). Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol Cell Biol. 23(10): 3558–3565. https://doi.org/10.1128/MCB.23.10.3558-3565.2003.
- Hanlon, K. S., Meltzer, J. C., Buzhdygan, T., Cheng, M. J., Sena-Esteves, M., Bennett, R. E., Sullivan, T. P., Razmpour, R., Gong, Y., Ng, C., et al. (2019). Selection of an Efficient AAV Vector for Robust CNS Transgene Expression. Mol Ther Methods Clin Dev. 15: 320–332. https://doi.org/10.1016/j.omtm.2019.10.007.
- Martin, R. M., Ikeda, K., Cromer, M. K., Uchida, N., Nishimura, T., Romano, R., Tong, A. J., Lemgart, V. T., Camarena, J., Pavel-Dinu, M., et al. (2019). Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell. 24(5): 821–828.e825. https://doi.org/10.1016/j.stem.2019.04.001.
- Fu, Y. W., Dai, X. Y., Wang, W. T., Yang, Z. X., Zhao, J. J., Zhang, J. P., Wen, W., Zhang, F., Oberg, K. C., Zhang, L., et al. (2021). Dynamics and competition of CRISPR-Cas9 ribonucleoproteins and AAV donor-mediated NHEJ, MMEJ and HDR editing. Nucleic Acids Res. 49(2): 969–985. https://doi.org/10.1093/nar/gkaa1251.
- Grimm, D., Lee, J. S., Wang, L., Desai, T., Akache, B., Storm, T. A. and Kay, M. A. (2008). In Vitro and In Vivo Gene Therapy Vector Evolution via Multispecies Interbreeding and Retargeting of Adeno-Associated Viruses. J Virol. 82(12): 5887–5911. https://doi.org/10.1128/JVI.00254-08.
- Engler, C., Gruetzner, R., Kandzia, R. and Marillonnet, S. (2009). Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 4(5): e5553. https://doi.org/10.1371/journal.pone.0005553.
- Labun, K., Montague, T. G., Krause, M., Torres Cleuren, Y. N., Tjeldnes, H. and Valen, E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47(W1): W171-W174. https://doi.org/10.1093/nar/gkz365.
- Brinkman, E. K., Kousholt, A. N., Harmsen, T., Leemans, C., Chen, T., Jonkers, J. and van Steensel, B. (2018). Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 46(10): e58-e58. https://doi.org/10.1093/nar/gky164.
- Doench, J. G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E. W., Donovan, K. F., Smith, I., Tothova, Z., Wilen, C., Orchard, R., et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 34(2): 184–191. https://doi.org/10.1038/nbt.3437.
- Paquet, D., Kwart, D., Chen, A., Sproul, A., Jacob, S., Teo, S., Olsen, K. M., Gregg, A., Noggle, S. and Tessier-Lavigne, M. (2016). Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533(7601): 125–129. https://doi.org/10.1038/nature17664.
- Engler, C., Kandzia, R. and Marillonnet, S. (2008). A one pot, one step, precision cloning method with high throughput capability. PLoS One 3(11): e3647. https://doi.org/10.1371/journal.pone.0003647.
- Tran, N. T., Sommermann, T., Graf, R., Trombke, J., Pempe, J., Petsch, K., Kuhn, R., Rajewsky, K. and Chu, V. T. (2019). Efficient CRISPR/Cas9-Mediated Gene Knockin in Mouse Hematopoietic Stem and Progenitor Cells. Cell Rep. 28(13): 3510–3522 e3515. https://doi.org/10.1016/j.celrep.2019.08.065.
- Xu, W., Morishita, W., Buckmaster, P. S., Pang, Z. P., Malenka, R. C. and Sudhof, T. C. (2012). Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron 73(5): 990–1001. https://doi.org/10.1016/j.neuron.2011.12.036.
- Bak, R. O. and Porteus, M. H. (2017). CRISPR-Mediated Integration of Large Gene Cassettes Using AAV Donor Vectors. Cell Rep. 20(3): 750–756. https://doi.org/10.1016/j.celrep.2017.06.064.
- Marco de la Cruz, B., Campos, J., Molinaro, A., Xie, X., Jin, G., Wei, Z., Acuna, C. and Sterky, F. H. (2024). Liprin-alpha proteins are master regulators of human presynapse assembly. Nat Neurosci. https://doi.org/10.1038/s41593-024-01592-9
Article Information
Publication history
Received: Jun 29, 2024
Accepted: Sep 6, 2024
Available online: Sep 25, 2024
Published: Nov 5, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
de La Cruz, B. M., Mitra, S., He, B., Çelik, M., Kaminski, D., Smedler, E. and Sterky, F. H. (2024). Efficient Gene-Editing in Human Pluripotent Stem Cells Through Simplified Assembly of Adeno-Associated Viral (AAV) Donor Templates. Bio-protocol 14(21): e5097. DOI: 10.21769/BioProtoc.5097.
Category
Stem Cell > Pluripotent stem cell > Cell-based analysis
Molecular Biology > DNA > Gene expression
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.