- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In Vitro GT-array (i-GT-ray), a Platform for Screening of Glycosyltransferase Activities and Protein–Protein Interactions

Published: Vol 14, Iss 18, Sep 20, 2024 DOI: 10.21769/BioProtoc.5066 Views: 2644
Reviewed by: David PaulWeidong AnAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Mar 2024

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Progress in bioinformatics has facilitated the identification of a large number of putative glycosyltransferases (GTs) associated with many physiological processes. However, many of these GTs remain with unknown biochemical function due to numerous technical limitations. One of these limitations is the lack of innovative tools for large-scale screening of enzyme activity in vitro and testing protein–protein interactions (PPIs) between GT partners. Currently, testing the enzyme activity of a protein requires its production in a heterologous expression system and purification before enzyme assays, a process that is time-consuming and not amenable to high-throughput screening. To overcome this, we developed a platform called in vitro GT-array (i-GT-ray). In this platform, 96-well microplates are coated with plasmid DNA encoding for tagged GTs and a capture antibody. Tagged GTs are produced from plasmid DNA via a cell-free in vitro transcription/translation (IVTT) system and captured through the anti-tag capture antibody directly on microplates. After washing to remove IVTT components, the captured enzymes can be considered purified, and their activity can be tested directly on microplates. The whole process can be performed in less than two days, compared to several weeks for currently available screening methods. The i-GT-ray platform has also been adapted to investigate PPIs between GTs. Here, we provide a practical user guide for the preparation of GT-arrays coated with plasmid DNA and a capture antibody that can be used for monitoring enzyme activity and PPIs of GTs in a high-throughput manner.
Key features
• Synthesis of tagged proteins directly from plasmid DNA, which are captured by anti-tag antibody attached to microplates.
• Captured tagged proteins can be considered as purified proteins ready for enzyme assays.
• Our platform can be used for high-throughput screening of enzyme activity and protein–protein interactions in vitro in a short time.
• Our platform can be used for biochemical characterization of difficult proteins such as membrane-integrated glycosyltransferases.
• Our platform can be adapted to downstream analytical methods such as mass spectrometry (i.e., DPS-MS).
Keywords: Protein synthesisGraphical overview
Background
Advancements in high-throughput genomics technologies, including DNA sequencing, DNA microarrays, RNA-Seq, and proteomics, have greatly facilitated the identification of numerous genes across various species. However, methods for high-throughput biochemical analyses of proteins encoded by these genes are lacking [1]. Determining the enzyme activity of a protein can be achieved through direct testing in vitro or indirectly via genetic complementation of mutants, which are poorly adapted for high throughput. Currently, characterizing the biochemical function of an enzyme involves expression in heterologous organisms that typically lack that protein, followed by measurement of enzyme activity of solubilized and partially purified protein [2–4]. This approach has several drawbacks, including high cost, low yield, time consumption, and limited applicability to high-throughput analyses.
The development of protein microarrays requires the production of substantial quantities of purified proteins for binding onto a solid support and necessitates specialized instrumentation, such as microarray printers and scanners [5–7]. A recent advancement in protein microarray technology, called nucleic acid programmable protein array (NAPPA), has facilitated the efficient generation of protein microarrays through in vitro cell-free transcription/translation (IVTT) systems directly from plasmid DNA [8]. Currently, NAPPA is primarily employed in screening for protein–protein interactions (PPIs) [5,9–12]. We have recently developed and validated a NAPPA-based platform for enzyme assays of glycosyltransferases (GTs) named in vitro GT-array (i-GT-ray) [13,14]. Additionally, it enables the study of PPIs of GTs [15] in a high-throughput manner. Most eukaryotic GTs are difficult to characterize biochemically through conventional methods because they are integral membrane proteins [16] and are present (along with their corresponding mRNAs) in very low quantities within the cell. These characteristics pose challenges in production and purification [16,17]. Interestingly, in this platform, all GTs tested during validation work could be produced in a soluble and active form even though the cell-free coupled IVTT systems do not contain any lipids or detergent (membrane mimic). The i-GT-ray platform utilizes 96-well microplates pre-coated with plasmid DNA-encoding tagged proteins and anti-tag capture antibody (CAb). This enables simultaneous protein production using IVTT systems and protein capturing via the CAb on microplates (Figure 1A and B). Following washing, the captured GTs can be screened for enzyme activity directly on microplates (Figure 1C). The products resulting from transferase reactions can be detected using mass spectrometry or the GLO system directly on microplates (Figure 1D). This unique platform facilitates the rapid and effective creation of GT arrays, significantly reducing the time required for enzyme activity screening. In this manuscript, we provide a step-by-step protocol for the preparation of GT arrays, which can be utilized for in vitro enzyme activity screening and PPIs of GTs. The potential pitfalls of using full-length membrane integral proteins are discussed.
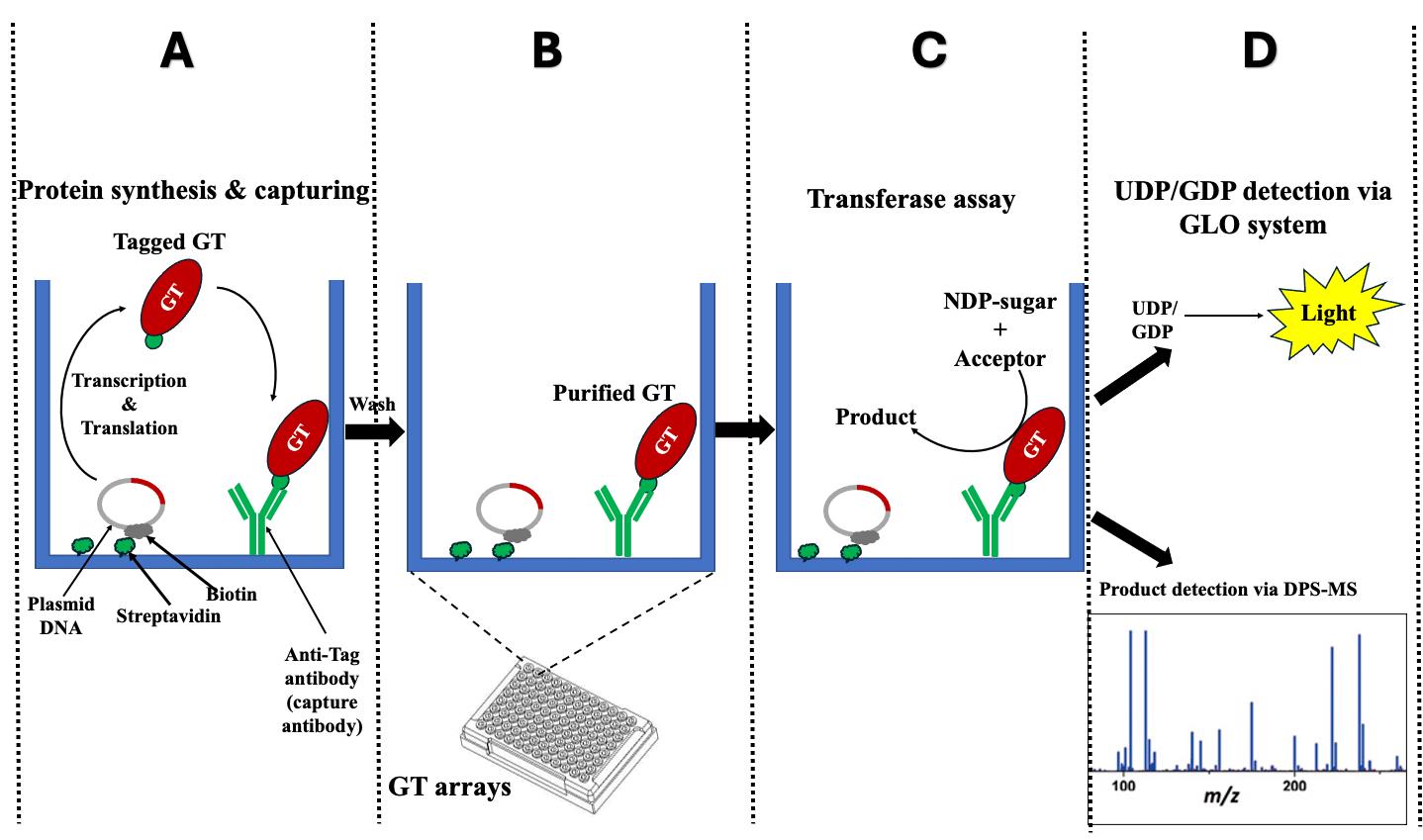
Figure 1. Schematic representation illustrates the fundamental principle of the i-GT-ray platform. (A) The tagged glycosyltransferases (GTs) are simultaneously synthesized and attached on microplate wells using plasmid DNA and anti-tag capture antibody. (B) After washing, the captured tagged GTs are considered purified. (C) Transferase reactions can be carried out by adding nucleotide diphosphate (NDP) sugars and acceptors. (D) The monitoring of transferase reactions is achieved through various methods, including the UDP/GDP GLO system, radioactive assays, or DPS-MS.
Materials and reagents
Biological materials
Chemically competent E. coli cloning strain (e.g., One ShotTM TOP10 Chemically Competent E. coli, Invitrogen, catalog number: C404010 or equivalent)
Expression vector pJFT7_nHALO_DC (Ampicillin resistant, DNASU Plasmid Repository, Clone ID: EvNO000424503) and pANT7_cGST (Ampicillin resistant, DNASU Plasmid Repository, Clone ID: EvNO00023103)
GatewayTM donor vector (non-ampicillin resistant, e.g., pCRTM8/GW/TOPOTM TA, Fisher Scientific, catalog number: K250020)
LR ClonaseTM II enzyme mix (Invitrogen, catalog number: 11791-020)
High-fidelity polymerase (e.g., KAPA HiFi DNA Polymerase, Roche Sequencing and Life Science, catalog number: KK2102)
Taq DNA polymerase (New England Biolabs, catalog number: M0273L)
Template for full-length protein-coding sequences (CDS) of interest or a CDS clone
1-Step Human Coupled IVT Kit, DNA (Thermo Scientific, catalog number: 88882)
TnT® Quick Coupled Transcription/Translation System (Promega, catalog number: L1170)
Anti-GST antibody (Cytiva, catalog number: 27457701)
Anti-HaloTag® pAb (Promega, catalog number: G9281)
Secondary mouse anti-goat IgG HRP conjugated (Santa Cruz Biotechnology, catalog number: sc-2354)
Secondary anti-rabbit IgG HRP conjugated (Promega, catalog number: W4011)
PfoI restriction enzyme (Thermo Scientific, catalog number: ER1751)
Streptavidin (Thermo Scientific, catalog number: 434302)
Reagents
Gel-purification kit (Zymo Research, catalog number: D4007)
DNA Clean and concentratorTM (Zymo Research, catalog number: D4004)
LB broth (Fisher Scientific, catalog number: BP1426-2)
Agar (Research Products International, catalog number: A20020-100.0)
Agarose ME (Research Products International, catalog number: A20085-500.0)
Ampicillin (Sigma-Aldrich, CAS number: 69-52-3)
Spectinomycin (Research Products International Corp., catalog number: 22189-32-8)
NucleoBond Xtra Midi kit (MACHEREY-NAGEL, catalog number: 740420.10)
2× Laemmli sample buffer (Bio-Rad, catalog number: 1610737EDU)
SuperSignalTM West Femto maximum sensitivity substrate (Thermo Scientific, catalog number: 34096)
Tween-20 (Sigma-Aldrich, CAS number: 9005-64-5)
EDC [1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride] (Thermo Scientific, catalog number: 22980)
EZ-LinkTM Hydrazide-Biotin (Thermo Scientific, catalog number: 21339)
Non-fat dried milk bovine (Sigma-Aldrich, catalog number: M7409-1BTL)
Acrylamide/bis 37.5:1 (Bio-Rad, catalog number: 1610158)
Ammonium persulfate (APS) (Sigma-Aldrich, catalog number: 7727-54-0)
TEMED (Bio-Rad, catalog number: 1610801)
Imidazole (Sigma-Aldrich, CAS number: 288-32-4)
ZebaTM Micro Spin desalting columns, 7 K MWCO (Thermo Scientific, catalog number: 89877)
AccuBlue Next Gen dsDNA Quantitation kit (Biotium, catalog number: 31060-T)
UDP-GLO Kit (UDP-GloTM Glycosyltransferase Assay) (Promega, catalog number: V6961)
GDP-GLO Kit (GDP-GloTM Glycosyltransferase Assay) (Promega, catalog number: VA1090)
DOWEX 1X8-100 resin (Cl) (Fisher Scientific, catalog number: AAL142570B)
Ethanol (Fisher Chemical, catalog number: A962-4)
ScintiVerseTM BD Cocktail (ScintanalyzedTM) (Fisher Chemical, catalog number: SX18-4)
Methanol (Fisher Scientific, catalog number: A434-20)
Tris-HCL (Sigma-Aldrich, catalog number: 1185-53-1)
Tris-Base (Sigma-Aldrich, catalog number: 77-86-1)
EDTA (Sigma-Aldrich, catalog number: 60-00-4)
Glycine (Bio-Rad, catalog number: 1610718)
HCl (Fisher Scientific, catalog number: A144-50)
SDS (Vivantis, catalog number: 151-21-3)
NaCl (Sigma-Aldrich, catalog number: 7647-14-5)
KCl (Sigma-Aldrich, catalog number: 7447-40-7)
NaOH pellets (Fisher Scientific, catalog number: S318-100)
Na2HPO4 (Fisher Scientific, catalog number: P285-500)
NaH2PO4 (Fisher Scientific, catalog number: S381-500)
KH2PO4 (Fisher Scientific, catalog number: S375-212)
Na2CO3 (Sigma-Aldrich, catalog number: 497-19-8)
NaHCO3 (Sigma-Aldrich, catalog number: 144-55-8)
DMSO (Sigma-Aldrich, catalog number: 67-68-5)
MgCl2 (Sigma-Aldrich, catalog number: 7786-30-3)
MnCl2 (Sigma-Aldrich, catalog number:7773-01-5)
Solutions
LB medium (1 L) (see Recipes)
100 mg/mL antibiotic stock (Ampicillin or Spectinomycin) (10 mL) (see Recipes)
LB agar plate with antibiotics (1 L) (see Recipes)
SDS-PAGE gel working solutions (see Recipes)
SDS-PAGE running buffer (1 L) (see Recipes)
SDS-PAGE transfer buffer (1 L) (see Recipes)
100 mM phosphate buffer pH 7.2 (1 L) (see Recipes)
10 mM phosphate buffer pH 7.2 (1× PBS) (1 L) (see Recipes)
10 mM phosphate buffer pH 7.2 (1× PBS) containing 5% (w/v) fat-free milk (100 mL) (see Recipes)
10 mM phosphate buffer pH 7.2 (1× PBS) containing 0.05% (v/v) Tween 20 (1 L) (see Recipes)
10 mM phosphate buffer pH 7.2 (1× PBS) containing 5% (w/v) fat-free milk and 0.05% (v/v) Tween 20 (100 mL) (see Recipes)
10 mM phosphate buffered saline with EDTA (PBS with EDTA) (1 L) (see Recipes)
Sodium bicarbonate buffer (pH 9.6) (see Recipes)
50 mM biotin hydrazide (1 mL) stock (see Recipes)
2.5 mM biotin hydrazide (1 mL) working solution (see Recipes)
100 mM Tris-HCl buffer (pH 7.2) (see Recipes)
Recipes
LB medium (1 L)
Reagent Final concentration Quantity LB broth 25 g/L 25 g ddH2O n/a ~980 mL Total n/a 1,000 mL Adjust the pH to 7.4 with NaOH. Autoclave at 121 °C for 15 min. Store at RT.
100 mg/mL antibiotic stock (ampicillin or spectinomycin)
Reagent Final concentration Quantity Ampicillin or Spectinomycin 100 mg/mL 1 g ddH2O n/a 10 mL Total n/a 10 mL Sterilize the solution with a 0.2 μm syringe filter. Store at -20 °C.
LB agar plate with antibiotics (1 L)
Reagent Final concentration Quantity LB broth 25 g/L 25 g Agar 15 g/L 15 g Antibiotic stock (100 mg/mL) 100 μg/mL 1 mL ddH2O n/a ~960 mL Total n/a 1,000 mL Adjust the pH to 7.4 with NaOH. Add agar after adjusting pH. Autoclave at 121 °C for 15 min. Allow to cool to 35–45 °C. Add 1 mL of 100 mg/mL antibiotic solution and mix well. Transfer 25 mL of the solution to petri dishes. Allow the agar plates to cool and dry in a laminar flow hood. Store the plates at 4 °C.
SDS-PAGE gel working solutions (for three 1.0 mm gels)
Separation gel (10%)
Reagent Quantity ddH2O 6.7 mL 30% acrylamide/bis 37.5:1 5.3 mL Lower solution (1.5 M Tris-HCl pH 8.8 and 0.2% SDS) 4 mL Persulfate (10% Sol) 150 μL TEMED 10 μL Stacking gel
Reagent Quantity ddH2O 6 mL 30% acrylamide/bis 37.5:1 1.5 mL Upper solution (1 M Tris-HCl pH 6.8 and 0.2% SDS) 2.5 mL Persulfate (10% Sol) 40 μL TEMED 10 μL SDS-PAGE running buffer (1 L)
Reagent Final concentration Quantity Tris 30 mM 3.03 g Glycine 200 mM 14.5 g SDS 0.1% (w/v) 1 g ddH2O n/a ~980 mL Total n/a 1,000 mL Store at room temperature (RT).
SDS-PAGE transfer buffer (1 L)
Reagent Final concentration Quantity Tris 30 mM 2.525 g Glycine 200 mM 13.824 g ddH2O n/a ~780 mL Methanol 20% (w/v) 200 mL Total n/a 1,000 mL Adjust the pH to approximately 8.3 after adding Tris and glycine in ~780 mL of ddH2O. Then, add 200 mL of 100% methanol and add ddH2O to 1 L. Store buffer at RT.
100 mM phosphate buffer pH 7.2 (1 L)
Reagent Final concentration Quantity NaCl 1.36 M 80 g KCl 26.83 mM 2 g Na2HPO4 141.96 mM 14.4 g KH2PO4 17.64 mM 2.4 g ddH2O n/a ~900 mL Total n/a 1,000 mL Adjust pH to approximately 7.2. Autoclave at 121 °C for 35 min. Store at 4 °C.
10 mM phosphate buffer pH 7.2 (1× PBS) (1 L)
Reagent Final concentration Quantity 100 mM phosphate buffer pH 7.2 10 mM 100 mL ddH2O n/a ~900 mL Total n/a 1,000 mL Adjust pH to 7.2. Store buffer at 4 °C.
10 mM phosphate buffer pH 7.2 (1× PBS) containing 5% (w/v) fat-free milk (100 mL)
Reagent Final concentration Quantity 100 mM phosphate buffer pH 7.2 10 mM 10 mL Fat-free milk 5% 5 g ddH2O n/a ~90 mL Total n/a 1,00 mL Adjust pH to 7.2 for 1× PBS, add fat-free milk, and mix for 1 h. Store buffer at 4 °C.
10 mM phosphate buffer pH 7.2 (1× PBS) containing 0.05% (v/v) Tween 20 (1 L)
Reagent Final concentration Quantity 100 mM phosphate buffer pH 7.2 10 mM 100 mL Tween 20 0.05% (v/v) 0.5 mL ddH2O n/a ~900 mL Total n/a 1,000 mL Adjust pH to 7.2 for 1× PBS, add Tween 20, and mix for 1 h. Store buffer at 4 °C.
10 mM phosphate buffer pH 7.2 (1× PBS) containing 5% (w/v) fat-free milk and 0.05% (v/v) Tween 20 (100 mL)
Reagent Final concentration Quantity 100 mM phosphate buffer pH 7.2 10 mM 10 mL Tween 20 0.05% (v/v) 0.05 mL Fat-free milk 5% 5 g ddH2O n/a ~99 mL Total n/a 100 mL Adjust pH to 7.2 for 1× PBS, add Tween 20 and fat-free milk, and mix for 1 h. Store buffer at 4 °C.
10 mM phosphate buffered saline with EDTA (PBS with EDTA) (1 L)
Reagent Final concentration Quantity NaCl 150 mM 8.8 g EDTA (0.5 M, pH 8.0) 10 mM 20 mL Na2HPO4 6.7 mM 0.95 g NaH2PO4 3.34 mM 0.4 g ddH2O n/a ~950 mL Total n/a 1,000 mL Adjust the pH to approximately 7.2. Autoclave at 121 °C for 35 min. Store at 4 °C.
Sodium bicarbonate buffer (pH 9.6)
Reagent Final concentration Quantity Na2CO3 15 mM 1.59 g NaHCO3 34.87 mM 2.93 g ddH2O n/a ~900 mL Total n/a 1,000 mL Adjust the pH to approximately 9.6. Autoclave at 121 °C for 35 min. Store at 4 °C.
50 mM biotin hydrazide (1 mL) stock
Reagent Final concentration Quantity Hydrazide-Biotin 50 mM 12.917 mg DMSO n/a 1 mL Total n/a 1 mL Store the solution at -20 °C.
2.5 mM biotin hydrazide (1 mL) working solution
Reagent Final concentration Quantity Hydrazide-biotin (50 mM) 2.5 mM 5 mL 0.1 M imidazole, pH 6 n/a 995 mL Total n/a 1 mL 100 mM Tris-HCl buffer (pH 7.2)
Reagent Final concentration Quantity Tris-HCl 100 mM 12.14 g MgCl2 2 mM 0.19 g MnCl2 2 mM 0.252g ddH2O n/a ~900 mL Total n/a 1,000 mL Adjust the pH to approximately 7.2. Sterilize the solution by filtration with a 0.2 μm syringe filter. Store at 4 °C.
Laboratory supplies
Pipette tips 0.5–10 µL, 20–200 µL, 100–1,000 µL (Fisher Scientific, FisherbrandTM, catalog numbers: 02-707-438, 02-707-417, 02-707-403)
1.5 mL Eppendorf tubes (Fisher Scientific, FisherbrandTM, catalog number: 02-681-321)
2 mL Eppendorf tubes (Fisher Scientific, FisherbrandTM, catalog number: 02-681-320)
Liquid scintillation vials (DWK Life Sciences, catalog number: 986730)
0.2 mL PCR tubes (Fisher Scientific, Thermo ScientificTM, catalog number: AB-0620)
100 mm × 15 mm Petri dishes (Fisher Scientific, FisherbrandTM, catalog number: FB0875712)
14 mL culture tubes (Globe Scientific, catalog number: 110178)
0.2 μm sterile syringe filter (e.g., Corning, catalog number: 431227)
Immobilon membrane (Millipore Sigma, catalog number: IPFL85R)
96-well microplate (clear, flat bottom, half area, high binding, polystyrene) (Corning, catalog number: 3690)
96-well microplate (flat bottom, half area, non-binding, white polystyrene) (Corning, catalog number: 3642)
Whatman® cellulose chromatography papers (Whatman, catalog number: 3017-820)
Equipment
250 mL Erlenmeyer flask
Micropipettes 0.1–2.5 µL, 2–20 µL, 20–200 µL (Eppendorf, models: 3123000217, 3123000233, 3123000250)
Multichannel pipettes (8 channels) 0.1–10 µL, 10–100 µL (Eppendorf, models: 2231300043, 2231300045)
Benchtop microcentrifuge (Beckman Coulter, model: Microfuge 22R centrifuge, catalog number: 8043-30-1145)
PCR thermocycler (Applied biosystems, Veriti 96-well Thermal Cycler 2990215201, catalog number: 4375305)
Incubation shaker (New Brunswick Scientific, Classic series, model: M1247-0004, catalog number: 19525)
Incubator (Fisher Scientific, Thermo ScientificTM, model: HerathermTM IGS60, catalog number: 51028063)
Mini-PROTEAN Tetra vertical electrophoresis system (Bio-Rad, catalog number: 1658002EDU)
Gel electrophoresis tank (Bio-Rad Laboratories, model: Wide Mini-Sub®, catalog number: 1704468)
Gel electrophoresis power supply (Bio-Rad Laboratories, model: PowerPacTM Basic, catalog number: 1645050)
Gel imager (Bio-Rad Laboratories, model: Gel DocTM XR+, catalog number: 1708195)
Plate reader (BioTek Synergy/HTX multi-mode reader, model: S1LFA, catalog number: BTS1LFA)
ChemiDocTM imaging system (Bio-Rad Laboratories, Gel Doc EZ, catalog number: 1708270)
Multi-purpose scintillation counter (Beckman, model: LS6500, SKU: 8043-30-1194)
Freeze dry system (Labconco, model: 4.5 FreeZone, catalog number: 7750020)
Vortex
Water bath
Software and datasets
SnapGene Viewer v.7.1.1
Gen5 v.2.09.2
Microsoft® Excel V 16.86
Procedure
Part 1: i-GT-ray platform for efficient screening of enzyme activity of glycosyltransferases (Figure 2)
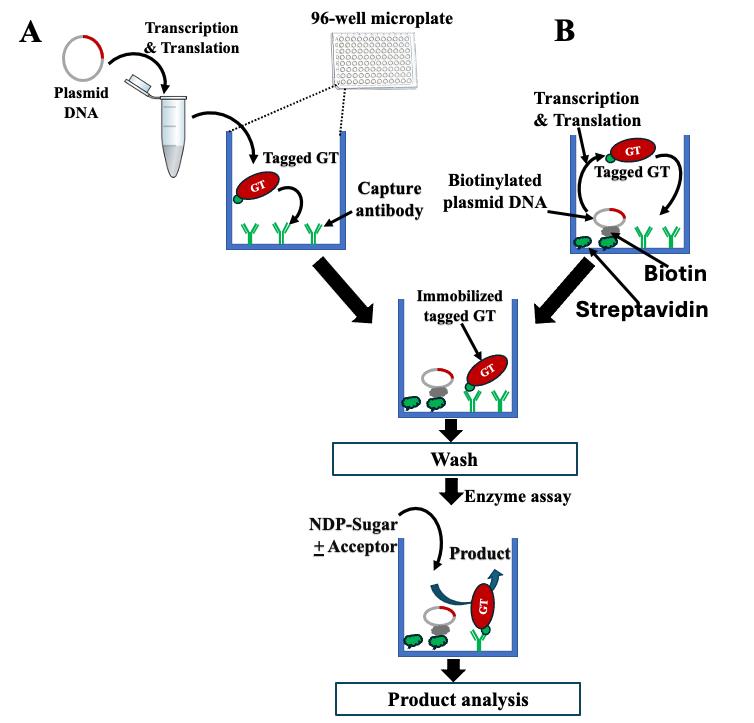
Figure 2. Schematic overview of enzyme assays performed on i-GT-ray platform. (A) Microplates can be pre-coated with anti-tag capture antibody (CAb) only to capture tagged glycosyltransferases (GTs) synthesized from plasmid DNA in an Eppendorf tube using in vitro transcription/translation (IVT) system. (B) Microplates can be pre-coated with both CAb and streptavidin to which the biotinylated plasmid DNA is attached. The IVT system is then added to wells for synthesis of tagged GTs that are captured by CAb. After washing, the captured tagged GTs are considered purified and transferase reactions are carried out by adding NDP-sugars and acceptors.
I. Cloning of protein coding sequences (CDSs)
Cloning of CDSs into a gateway donor vector
Note: After the PCR, two forms of CDS are created: one with a stop codon, and one without a stop codon. Therefore, for the PCR process, the primers are designed accordingly. The reverse primers are designed with and without a stop codon to ensure the generation of both forms of the CDSs.
The CDSs are PCR-amplified using a high-fidelity polymerase (e.g., KAPA HiFi DNA Polymerase or Phusion polymerase).
Purify the fragment from an agarose gel using a standard kit (e.g., Zymoclean Gel DNA Recovery Kits).
Add extra As to the CDSs using a reaction made of reagents listed in Table 1.
Table 1. Conditions for addition of As to the CDSs.
Component Final concentration Quantity 10× ThermoPol reaction buffer 1× 1 µL 10 mM dNTPs 500 mM 0.5 µL Purified CDSs PCR DNA 25 ng/mL Variable (250 ng) Taq DNA polymerase 2.5 units 0.5 µL ddH2O n/a ~10 µL Total n/a 10 µL Incubate at 72 °C for 25 min in a PCR machine.
Create an entry clone by transferring the CDS to the GatewayTM donor vector (e.g., pCRTM8/GW/TOPOTM TA) through TA cloning following the manufacturer’s protocol.
Introduce the constructs into the chemically competent E. coli cloning strain (e.g., DH5a) using the heat-shock method.
Spread the transformed competent bacteria cells onto LB plates containing 100 mg/L spectinomycin and incubate overnight at 37 °C to form colonies.
Screen the colonies and confirm the reading frame, correct orientation, and full sequence of the inserted CDS through Sanger sequencing.
Cloning of CDSs into an expression vector
Perform Gateway cloning from pCRTM8/GW/TOPOTM entry vector to pJFT7_nHALO_DC or pANT7_cGST destination vectors using LR ClonaseTM II enzyme mix following the manufacturer’s protocols.
Note: For the creation of the N-terminal Halo-Tagged GT proteins, an entry vector containing the full-length GT gene with a stop codon is required. However, to generate the C-terminal GST-tagged GT proteins, an entry vector containing the full-length GT gene without a stop codon is necessary.
Transfer the constructs into the chemically competent E. coli cloning strain by the heat-shock method.
Spread the transformed competent cells onto LB plates containing 100 mg/L ampicillin and incubate overnight at 37 °C to form colonies.
Screen the colonies by PCR and confirm the reading frame, correct orientation, and full sequence of the insert through Sanger sequencing.
The correct colony is cultured in 200 mL of LB liquid medium supplemented with 100 mg/L ampicillin. Grow at 37 °C with shaking at 180 rpm until the OD600 value reaches 0.6–1.0. Then, extract and purify large amounts of plasmid using the NucleoBond Xtra Midi kit following the manufacturer’s protocol.
II. Recombinant protein production and analysis
Produce the tagged GT fusion proteins either in 1.5 mL Eppendorf tubes or on 96-well microplates using 1 µg of GT plasmid DNA and an expression kit (e.g., 1-Step Human Coupled IVT Kit, Thermo Scientific) according to the manufacturer’s recommendations at 30 °C for 6 h.
Confirm the production of Halo-GT and GT-GST fusion proteins by running western blotting as follows:
Transfer 4 µL of expressed protein to Eppendorf tubes, add 4 µL of 2× Laemmli sample buffer, and denature the proteins at 100 °C for 10 min.
Prepare an SDS-PAGE gel using separating gel and stacking gel solutions.
Run the proteins on SDS-PAGE gel using running buffer. After running is completed, transfer the proteins onto Immobilon membranes using the transfer buffer in the Mini-Protein Tetra cell system (Bio-Rad) for 80–90 min, maintaining the cold environment using the ice pack (or place in a cold room).
After transfer, rinse the membrane with 1× PBS and proceed to blocking the membrane with 5% (w/v) fat-free dry milk in 1× PBS at 4 °C overnight (or at least 1 h at room temperature).
Wash the membrane with 1× PBS containing 0.05% (v/v) Tween 20 three times for 10 min each with gentle shaking (approximately 50 rpm).
Apply the primary antibody (anti-GST or anti-Halo) at 1:10,000 dilution in 1× PBS containing 5% (w/v) fat-free milk with 0.05% (v/v) Tween 20 for 2 h with gentle shaking (approximately 50 rpm) at room temperature.
Wash the membrane with 1× PBS containing 0.05% (v/v) Tween 20, three times for 10 min each with gentle shaking (approximately 50 rpm).
Apply the corresponding secondary antibody (anti-goat or anti-rabbit) at 1:15,000 dilution in 1× PBS containing 5% (w/v) fat-free milk with 0.05% (v/v) Tween 20 for 1 h with gentle shaking (approximately 50 rpm) at room temperature.
Wash the membrane with 1× PBS containing 0.05% (v/v) Tween 20, three times for 10 min each with gentle shaking (approximately 50 rpm).
Wash the membrane with 1× PBS (without Tween 20), two times for 10 min each with gentle shaking (approximately 50 rpm).
Detect the presence of the fusion protein using SuperSignal West Femto Maximum Sensitivity Substrate according to the manufacturer’s protocols and observing the membrane in the ChemiDocTM Imaging system (Bio-Rad).
III. Coating of microplates with capture antibody (CAb) with or without plasmid DNA
Coating of microplate with CAb alone
Apply 50 µL of CAb (anti-GST or anti-Halo antibody) at a 1/200 dilution in 50 mM sodium bicarbonate buffer, pH 9.6, to the 96-well plate and incubate at 4 °C overnight (or at least 1 h at room temperature).
Wash the wells with 1× PBS three times, each time for 10 min.
Block the wells with 5% fat-free dry milk in 1× PBS at 4 °C for 4–6 h.
Wash the wells three times with 1× PBS, each time for 10 min. These coated microplates can be used immediately or stored at 4 °C for up to 60 days (without liquid).
Coating of microplates with both CAb and plasmid DNA
DNA linearization and biotinylation
For linearization of plasmid DNA:
• Use approximately 60 µg of plasmid DNA (containing GT with tag) using PfoI restriction enzyme according to the manufacturer’s recommendations.
Note: During linearization of plasmid DNA, choose the restriction site far from the gene of interest (at least 1,000 bp).
• Freeze-dry the linearized plasmid DNA and resuspend in 10 μL of water to achieve a concentration of 6 mg/μL.
Note: For freeze-drying of plasmid DNA, the linearized plasmid DNA solution in water is frozen rapidly in a -80 °C freezer or by immersing in liquid nitrogen. Then, the frozen sample is placed in the freeze-dryer after it is pre-cooled and the vacuum system is functioning properly. The freeze-drying process is carried out according to the manufacturer's instructions. The sample is placed in the freeze-dryer until the samples are thoroughly dried. They should appear as a dry, powdery material.
For biotinylation of plasmid DNA:
• Mix 7.5 µL (40–50 µg) of the linearized plasmid DNA with ~1.25 mg (6.52 μmol) of the linker EDC (1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride) and then immediately add 5 μL of biotin hydrazide working solution (2.5 mM in 100 mM imidazole).
• Vortex the mixture until the contents are dissolved completely and add 20 μL of 100 mM imidazole pH 6 after a short spin (to collect the solution to the bottom of the tube); then, incubate overnight at 37 °C in a water bath.
• Remove the non-reacted EDC and its by-products using spin desalting column (ZebaTM Spin Desalting Column) with PBS with EDTA (Recipe 12), following the manufacturer’s recommendations.
• Store the biotinylated DNA plasmid at -20 °C until use.
Coating of microplate with plasmid DNA followed by CAb
First, coat microplates with streptavidin by applying 50 µL of 50 mM sodium bicarbonate buffer, pH 9.6 containing 2 µg of streptavidin and incubating overnight at 4 °C.
Wash the microplate wells with 1× PBS three times, each for 10 min.
Apply 50 µL of 1× PBS buffer containing CAb (anti-GST or anti-Halo antibody) at 1/200 dilution to the well and incubate at 4 °C for 4 h.
Wash the microplate wells with 1× PBS three times, each for 10 min.
Block the wells with 5% fat-free dry milk in 1× PBS for 2–4 h at 4 °C.
Wash the microplate wells with 1× PBS three times, each for 10 min.
Apply 1 µg of linearized and biotinylated DNA plasmid (prepared in step III.B.1.a beforehand) dissolved in 50 µL of 5% (w/v) fat-free dry milk in 1× PBS to the wells and incubate overnight at 4 °C.
Wash the wells with 1× PBS three times, each for 10 min. These microplates can be used immediately or stored at 4 °C for up to 30 days (without liquid).
Note: To estimate the amount of plasmid DNA attached to each well, use the AccuBlue Next Gen dsDNA quantitation kit (Biotium) according to the manufacturer’s protocols.
IV. Binding of tagged GT fusion proteins
For microplates pre-coated with CAb only (Figure 2A)
Note: Proteins are synthesized in 1.5 mL Eppendorf tubes using in vitro transcription/translation kit according to the manufacturer’s recommendations at 30 °C for 6 h and then transferred to the microplates pre-coated with CAb. For synthesis of fusion protein, 25 µL of in vitro transcription/translation mixture is carried out containing 1 µg of plasmid DNA according to the manufacturer’s recommendations.
Add 50 µL of protein mixture [12.5 µL of tagged proteins + 37.5 µL of 1× PBS containing 5% (w/v) fat-free milk] to microplates pre-coated with CAb.
Incubate at 14 °C for 4–6 h with shaking at 250 rpm.
Wash the microplates three times with 1× PBS, each time for 10 min.
Perform transferase assays according to the procedure outlined below (section V).
For microplates pre-coated with CAb and plasmid DNA (Figure 2B)
Note: Proteins are synthesized directly on microplates pre-coated with CAb and plasmid DNA using in vitro transcription/translation kit according to the manufacturer’s recommendations. The procedure involves simultaneous protein synthesis and binding.
Directly load 50 µL of in vitro transcription/translation mixture onto the wells of microplates pre-coated with CAb and plasmid DNA, following the manufacturer’s recommendations.
Incubate the microplates at 30 °C for 4 h.
Incubate the microplates at 14 °C for 4–6 h with shaking at 250 rpm.
Wash the microplates three times with 1× PBS, each for 10 min.
Perform the transferase assays according to the procedure below (section V).
V. Transferase assays on microplates
For testing transferase activity using non-radioactive NDP-sugars
Note: The standard transferase assays are conducted in 50 µL volume utilizing NDP-sugars and acceptors (oligosaccharides and polysaccharides) in microplates pre-coated with fusion proteins.
In a 1.5 mL Eppendorf tube, combine the assemble transferase reactions as indicated in Table 2 and mix well before transferring to wells.
Table 2. Transferase reaction mixture using non-radioactive NDP-sugars
Reagents Final concentration Volume Tris-HCl buffer (100 mM, pH 7.2) 50 mM 25 µL NDP-sugar (50 mM) 3 mM 3 µL Acceptor (10 mg/mL) 2 mg/mL 10 µL ddH2O n/a To achieve 50 µL Total n/a 50 µL Transfer 50 µL of transferase reaction mix to each well containing captured tagged GTs.
Incubate the reactions at 25 °C for 90 min.
Prepare the NDP-detection reagent (GDP or UDP following manufacturer’s protocol) 10 min before transferase reactions are completed.
Transfer 25 µL of reactions from each well to a new white polystyrene 96-well microplate.
Note: If the donor used is UDP-sugar, the luminescence activity is measured using UDP-GLO kit; if the donor is GDP-sugar, then GDP-GLO kit must be used.
Add 25 µL of the pre-prepared NDP-detection reagent and mix properly (with a pipette) without creating air bubbles in the well.
Incubate in the dark for 1 h at room temperature.
Observe the luminescence activity in the microplate reader using Gen5 software with a gain setting of 100. (Figure 5 represents an example of the results obtained.)
For testing the transferase activity using the radioactive NDP-sugars
Note: The standard assays are similar to non-radioactive assays, except that the appropriate NDP-[14C] sugar is used in addition to non-radioactive NDP-sugars.
In a 1.5 mL Eppendorf tube, combine the assemble transferase reactions as indicated in Table 3 and mix well before transfer to wells.
Table 3. Transferase reaction mixture using radioactive NDP sugars
Reagents Final concentration Volume Tris-HCl buffer (100 mM, pH 7.2) 50 mM 25 µL NDP-Sugar (50 mM) 1 mM 1 µL NDP-[14C]sugars (90,000–100,000) cpm/50 µL 1–5 µL Acceptor (10 mg/mL) 2 mg /mL 10 µL ddH2O n/a To 50 µL Total n/a 50 µL Transfer 50 µL of the transferase reaction mix to each well containing the captured tagged GTs.
Incubate the microplates for 90 min at 25 °C.
The detection method of the incorporated radioactive differs according to the charge of the oligosaccharide products formed. For neutral oligosaccharide products (i.e., for testing of AtXXT1), excess unused NDP-[14C] sugars are removed by chelation with DOWEX 1X8-100 resin (Cl) 1:1 (v/v). For charged oligosaccharide products (i.e., for testing of AtGUX1), excess unused NDP-[14C] sugars are removed through thin layer chromatography (TLC) according to published works [18–21]. When the acceptor used is a polysaccharide (i.e., such as xyloglucan for testing AtFUT1 and AtMUR3), the reactions are collected from wells into 2 mL Eppendorf tubes and treated as follows:
Add 1 mL of cold 70% ethanol and incubate overnight at 4 °C to precipitate the radioactive polysaccharides.
Centrifuge at 15,000× g for 15 min to collect the radioactive polysaccharides.
Rinse the pellets five times with 1 mL of cold 70% ethanol.
The incorporated radioactivity (in both oligosaccharides and polysaccharides) is measured as cpm using a LS 6500 multi-purpose scintillation counter (Beckman) by resuspension of the radioactive products in 0.3 mL of water and mixing with 3–5 mL of liquid scintillation solution in scintillation vials.
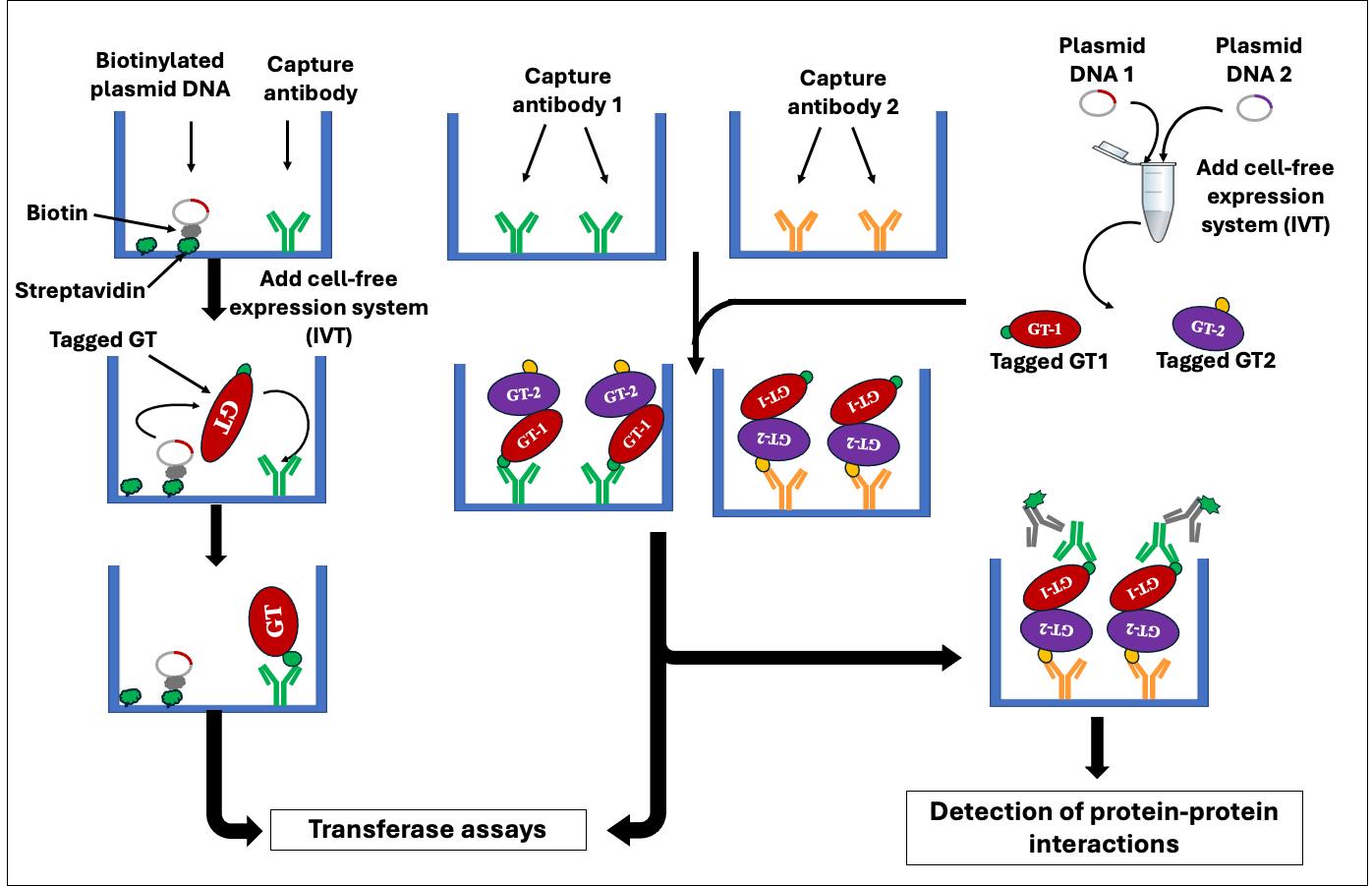
Part 2: i-GT-ray platform for screening of protein–protein interactions of glycosyltransferases (Figure 3 and Figure 4)
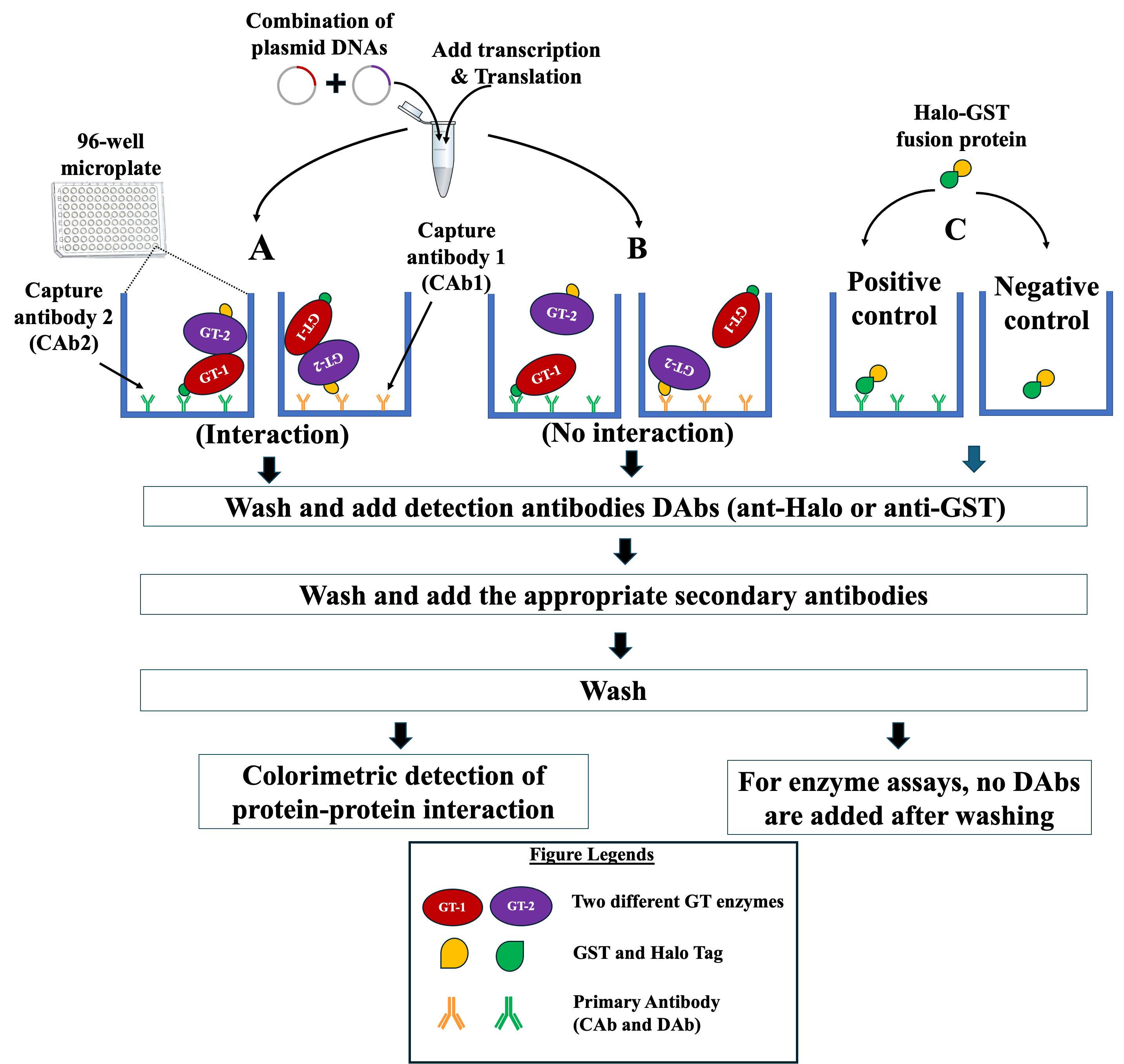
Figure 3. Schematic representation of the strategy used to investigate protein–protein interactions (PPIs) on microplates using NAPPA-based technology. Tagged proteins (GT1 and GT2) to be tested for PPIs are synthesized in an Eppendorf tube from their respective plasmid DNAs using in vitro IVT kit. The synthesized glycosyltransferases (GTs) are applied to microplates that are pre-coated with one of the two anti-tag capture antibodies (CAb1 or CAb2). (A) If the two GTs interact, the formed complex is detected by adding the appropriate detection antibody (DAb, anti-Halo, or anti-GST) and the appropriate HRP-linked secondary antibody. One GT acts as the prey and the second is the interacting partner (acts as a bait). (B) If there is no interaction between the two GTs, the addition of DAbs and HRP-linked secondary antibody will not result in a signal. (C) Halo-GST fusion protein is applied to the microplate pre-coated with CAb (positive control) and the microplate with no pre-coating (negative control) to assess the status of binding of CAb on the microplates. The detection of the formed complex (positive interaction) or the binding of Halo-GST fusion protein is performed via TMB colorimetric method using a microplate reader and absorbance at 450 nm.
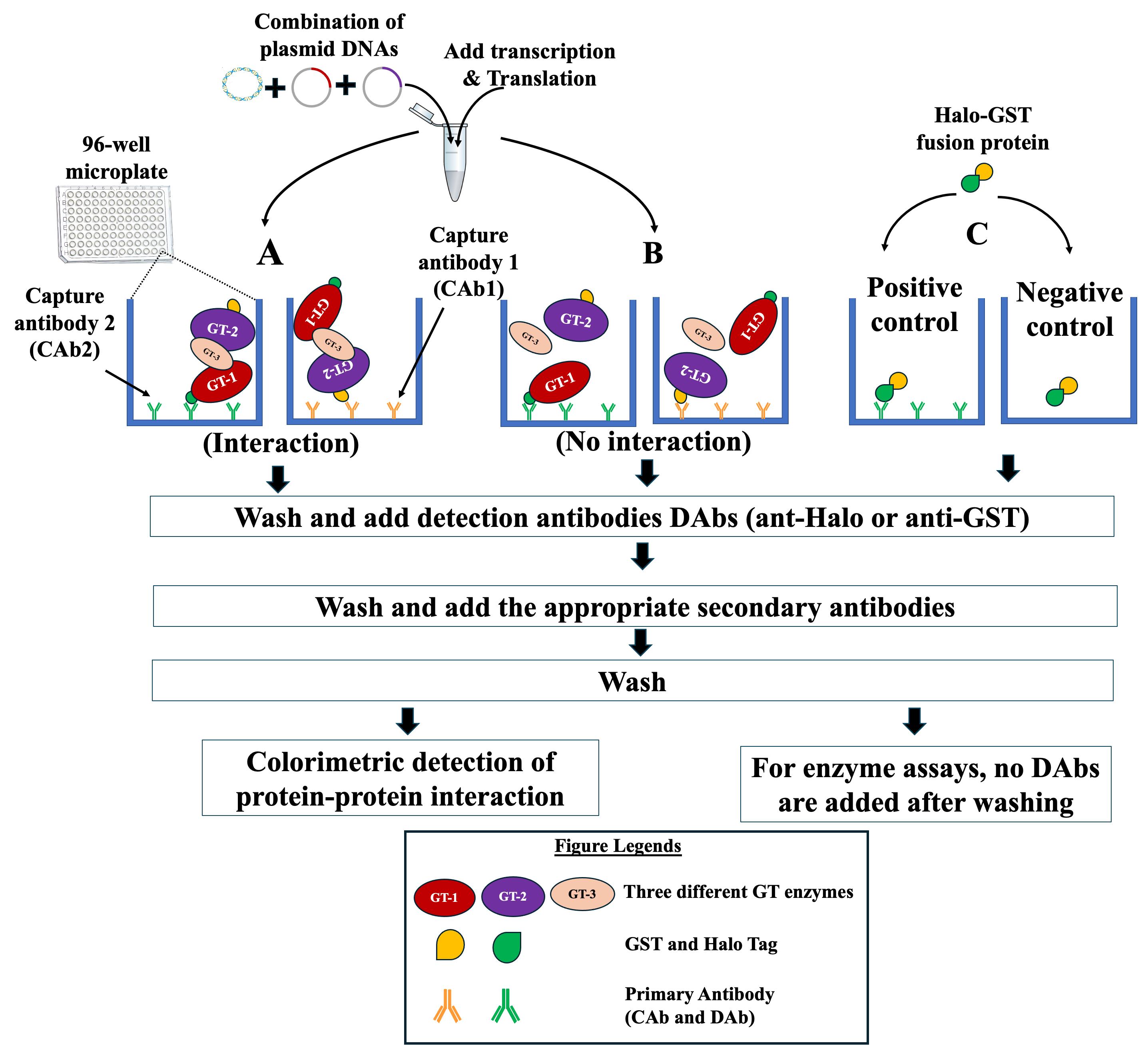
Figure 4. Schematic representation of the strategy used to investigate protein–protein interactions (PPIs) of three GTs on microplates using NAPPA-based technology. Tagged proteins (GT1 and GT2) and untagged protein (GT3) to be tested for PPIs are synthesized in an Eppendorf tube from their respective plasmid DNAs using in vitro IVT kit. The synthesized glycosyltransferases (GTs) are applied to microplates that are pre-coated with one of the two anti-tag capture antibodies (CAb1 or CAb2). (A) If the three GTs interact, the formed complex is detected by adding the appropriate detection antibody (DAb, anti-Halo, or anti-GST) and the appropriate HRP-linked secondary antibody. One GT acts as the prey, the second is the bait, and the third GT forms the bridge between GT1 and GT2. As a control, the testing of GT1 and GT2 does not result in a signal. (B) If there is no interaction between the three GTs, the addition of DAbs and HRP-linked secondary antibody will not result in a signal. (C) Halo-GST fusion protein is applied to microplate pre-coated with CAb (positive control) and microplate with no pre-coating (negative control) to assess the status of binding of CAb on the microplates. The detection of the formed complex (positive interaction) or the binding of Halo-GST fusion protein is performed via TMB colorimetric method using a microplate reader and absorbance at 450 nm.
I. Cloning of protein coding sequences (CDSs) into expression vector
CDSs in pCRTM8/GW/TOPOTM entry vector are generated as described in Part 1, Section I.A (see above).
Gateway cloning is performed using pCRTM8/GW/TOPOTM entry vector and pJFT7_nHALO_DC or pANT7_cGST destination vectors in presence of LR ClonaseTM II enzyme mix according to manufacturer’s protocols.
Note: For creation of the N-terminal Halo-Tagged GTs, the full-length GT genes are cloned into the entry vector with a stop codon. Similarly, to generate the C-terminal GST-tagged GT protein, the full-length GT genes without a stop codon are cloned into the entry vector. However, to generate the untagged GT protein, an entry vector containing the full-length GT gene with a stop codon is gateway cloned to pANT7_cGST destination vectors.
Transformation, screening, and plasmid extraction are performed as described in Part 1, section I.B (see above).
II. Screening of protein–protein interactions (PPIs)
Coating of microplates with CAb
Coating of microplates with CAb and blocking with 5% fat-free dry milk are performed as described above (Part 1, section III.A). For PPIs testing, the microplates are pre-coated with anti-GST or anti-Halo CAb depending on the tagged GTs.
PPI assays
Note: The two tagged GTs with different tags (GST or Halo) are co-produced in vitro (Figure 3). In case more than two GTs are co-produced, the third GT should be untagged (Figure 4).
The tagged GTs to be tested are co-produced in 1.5 mL Eppendorf tubes using equal amounts of plasmid DNA and the expression kit (e.g., 1-Step Human Coupled IVT Kit, Thermo Scientific) according to the manufacturer’s recommendations at 30 °C for 6 h.
Note: For co-production of two tagged GTs, ~500 ng of each GT is used, while for co-production of three GTs, 330 ng of each of GT plasmid DNA is used.
Apply 50 µL of produced protein mixture [12.5 µL of co-expressed proteins + 37.5 µL of 1× PBS containing 5%(w/v) fat-free milk] to each well of the microplate that was pre-coated with anti-Halo or anti-GST CAb.
Incubate the microplates for 4–6 h at 14 °C with shaking at 250 rpm.
Wash the microplates three times with 1× PBS, each time for 10 min.
Apply 50 µL of detecting antibody (DAb) [1/200 dilution in 1× PBS containing 5%(w/v) fat-free milk].
Note: If the microplate is coated with anti-GST, the detection antibody should be anti-Halo, and vice versa.
Incubate at 14 °C for 2–4 h with shaking at 250 rpm.
Wash the microplates three times with 1× PBS, each time for 10 minutes.
Apply 50 µL of the secondary antibody fused to Horseradish peroxidase (HRP) [anti-goat or anti-rabbit; 1/200 dilution in 1× PBS containing 5% (w/v) fat-free milk].
Incubate the microplates for 1 h at 14 °C with shaking at 250 rpm.
Wash the microplates three times with 1× PBS containing 0.05% (v/v) Tween 20, each time for 10 min.
Wash the microplates two times with 1× PBS only, each time for 10 min.
Apply 50 μL of TMB solution (substrate for HRP) to each well.
Incubate at room temperature until the appearance of color (approximately 5 min).
Stop the reaction by adding 50 μL of 2 M H2SO4.
Take the absorbance values at 450 nm using the microplate reader (Figure 6 represents an example of the results obtained).
Data analysis
For validation of the i-GT-ray platform, we have conducted enzyme activity screening of 22 non-processive putative fucosyltransferases (FUTs) from the GT37 family against five acceptor substrates and one donor substrate (GDP-fucose). The screening involved a total of 280 assays that were performed in high-throughput manner [14]. Furthermore, we validated the i-GT-ray platform using five synthases involved in cellulose, xyloglucan, (gluco)mannan, and b-(1,3)(1,4)-mixed-linkage glucan synthesis [13]. All data are expressed as the mean and standard deviation of the mean (Figure 5).
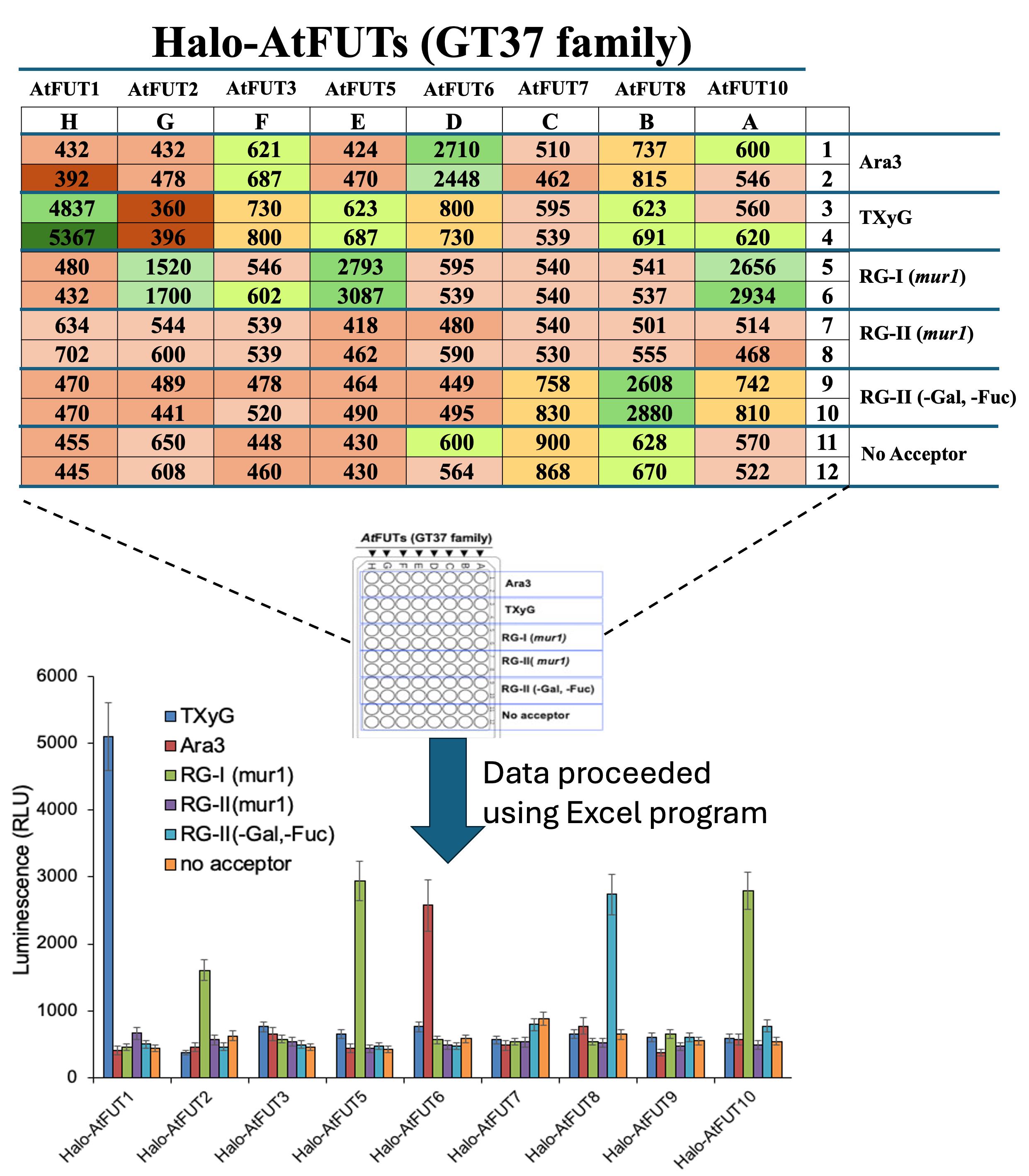
Figure 5. Example of data collected showing the fucosyltransferase activity of eight Arabidopsis AtFUTs from GT37 family on five acceptor substrates and one donor substrate (GDP-fucose). The layout of the assays in a 96-well microplate is indicated (center). Duplicate values were obtained using a second 96-well microplate. Data were obtained as luminescence (RLU, top panel) that measures the release GDP from GDP-fucose during the transferase reaction using GDP-GLO kit. Average data analysis showed that the activity of FUTs was more than three times higher than the control without the acceptor. Data are expressed as the mean ± SD using Excel software (bottom panel). Reprinted/adapted from Bhattarai et al. [14]. © 2024 The Authors. Published by Elsevier Inc on behalf of American Society for Biochemistry and Molecular Biology. Distributed under a Creative Commons Attribution NonDerivatives NonCommercial License 4.0 (CC BY-NC-ND license). https://creativecommons.org/licenses/by-nc-nd/4.0/
For protein–protein interaction (PPI), the i-GT-ray platform was validated by conducting PPI assays between eight rice GTs from OsGT43 and OsGT47 families [15]. Each OsGT43 protein was used as a prey to probe interactions with each of the OsGT47s (using both Halo- and GST-tagged proteins in an 8 × 8 protein interaction matrix in triplicates, for a total of 192 assays) (Figure 6).
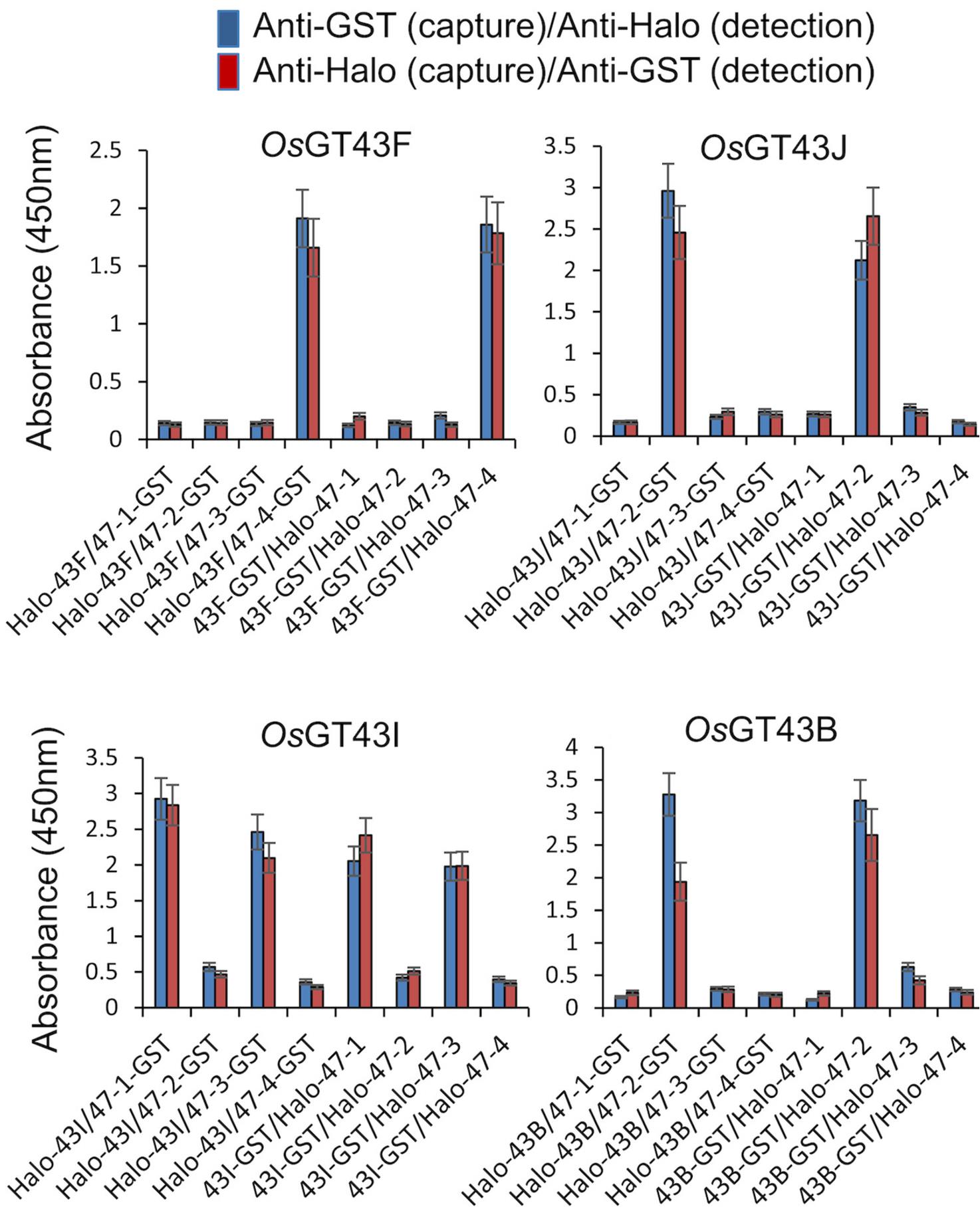
Figure 6. Example of data collected from PPI assays between eight members of OsGT43s and OsGT47s families using a 96-well microplate. Data are expressed as the mean ± SE of triplicate assays using Excel software. Reprinted/adapted from Javaid et al. [15]. © 2024 The Authors. The Plant Journal published by Society for Experimental Biology and John Wiley & Sons Ltd. Distributed under a Creative Commons Attribution NonDerivatives NonCommercial License 4.0 (CC BY-NC-ND license). https://creativecommons.org/licenses/by-nc-nd/4.0/
Validation of protocol
This protocol or parts of it has been used and validated in the following research articles:
Bhattarai et al. [14]. Streamlining assays of glycosyltransferases activity using in vitro GT-array (i-GT-ray) platform: Application to family GT37 fucosyltransferases. Journal of Biological Chemistry. 300:105734. (Figures 2–6)
Bhattarai et al. [13]. New Insights on Beta-Glycan Synthases Using in Vitro GT-Array (i-GT-Ray) Platform. SSRN 4753263. (Figures 1–5)
Javaid et al. [15]. Specific protein interactions between rice members of the GT43 and GT47 families form various central cores of putative xylan synthase complexes. The Plant Journal. 118: 856–878. (Figures 6 and 7)
General notes and troubleshooting
General notes
Despite this protocol being validated for GTs of plant cell wall, it could be applicable to other enzymes.
The in vitro biochemical characterization and PPI are technically challenging; therefore, all steps in this protocol must be carefully attended. Optimization of these steps contributes to the success of the assay.
Before starting the enzyme and PPI assays, it is very important to confirm the synthesis of fusion protein; thereafter, it is crucial for the validation of enzyme activity and PPIs.
Before starting the PPI assay, it is important to validate that the fusion proteins are synthesized at similar levels.
The detection of the final product of enzyme assays is also equally important, since they can synthesize products at very low amounts, and it depends on the sensitivity of the detection methods.
Limitations
Although the full-length GTs (which are type-II membrane proteins with transmembrane domains) were expressed in vitro and produced soluble tagged proteins with the expected enzyme activity, it is always possible that their folding is not optimal, which may impact their enzyme activity. Therefore, efforts should be taken to minimize issues with solubility and folding of membrane proteins. i-GT-ray platform will benefit from the use of soluble and active forms of GTs and below we propose some improvements to achieve this.
Proposed improvements
We propose the following improvements using practical approaches to enhance the solubility and functional assessment of the GTs (and other integral membrane proteins):
The easiest practical approach is to design GTs without the predicted TMDs (cloning of genes encoding for GTs without TMDs). There are several publicly available bioinformatics tools that can be used to efficiently predict the location of TMDs (e.g., TMHMM, TOPCONS). The in vitro biochemical function of many non-processive GTs (type-II membrane proteins) has been determined using this approach (standard method). However, this approach is less successful for processive GTs.
The second easy practical approach is the inclusion of detergents/membrane mimics (such as 0.1%–0.4% Triton-X, CHAPS, deoxycholic acid) during the in vitro synthesis of the GTs. This can help with the proper folding and solubilization of GTs. However, the detergent concentrations must be optimized to limit the impact on the IVTT systems.
The third quick practical approach is to determine the stability of the proteins after synthesis by implementing the ThermoFluor assay, which is a temperature-based assay. In this approach, a small quantity of protein (5 mM) is mixed with SYPRO Orange dye that binds to hydrophobic patches/denatured protein/molten globules and fluoresces. As the temperature increases and the protein unfolds, it is possible to determine a melting temperature, which provides information about the stability of the proteins. The temperature and fluorescence can be monitored using a real time PCR machine [22].
The fourth approach is more complex and consists of using nanodiscs (NDs) to mimic native-like membrane environment for GTs. Nanodiscs are soluble nanoscale phospholipid bilayers used to self-assemble integral membrane proteins for enzymatic or structural studies [23] that provide an excellent solution for full-length GTs in i-GT-ray platform. In this approach, GTs are produced in the presence of phospholipids along with an encircling amphipathic helical protein belt, termed a membrane scaffold protein (MSP), which is a derivative from the human apolipoprotein 1A [24]. The MSP would allow GTs to simultaneously assemble with phospholipids into a discoidal bilayer, bringing the assay closer to what might occur in a native cell membrane. Another advantage of ND technology is the possibility of forming small (~10 nm diameter) edge-stabilized lipid bilayer discs containing a single GT or a multi-protein complex with possible control of oligomerization of GTs, which might be necessary for proper activity of some GTs. This can be achieved by using MSP of various sizes. ND technology has been used to characterize the biochemical activity of a bacterial cellulose synthase complex [25].
The fifth approach is the use of a computational design to generate soluble and functional membrane protein analogues [26]. In this approach, a robust computational pipeline is used for de novo protein design, using inversion of the AF2 network [27] coupled with sequence design using ProteinMPNN [28]. The soluble analogues could be designed in soluble form while preserving native functional motifs. In this approach, inversion of the AF2 network (AF2seq) is used to generate sequences that adopt a desired target fold and then apply ProteinMPNN sequence optimization [28]. The structures of the resulting sequences are re-predicted with AF2 and filtered based on their structural similarity to the target topology.
Troubleshooting
Problem 1: Lack of expression of fusion proteins or expression of more truncated protein.
Possible cause: The expression of fusion proteins is influenced by several critical factors, including the quality and concentration of the plasmid DNA, the choice of in vitro expression kit, and the specific expression conditions employed. Furthermore, degradation of proteins during IVTT reactions can lead to the truncation of proteins, affecting its functionality and downstream applications.
Solution: Ensure high-quality plasmid DNA extraction by consistently utilizing an appropriate plasmid extraction kit to obtain purified DNA. Maintain the concentration of plasmid DNA within the range of 500–1,000 ng per reaction during protein expression. Employ only sterilized materials, such as pipette tips, tubes, and water, to minimize contamination risks during protein synthesis. Experiment with multiple in vitro expression kits initially to identify the most suitable kit for achieving minimal or no truncation of the protein of interest. It is advisable to assess the protein synthesis profile using western blotting when employing a new in vitro expression kit to verify the integrity and quality of the synthesized proteins.
Problem 2: Lack of binding of fusion protein to the well.
Possible cause: The binding of the fusion protein is influenced by several key factors, including the concentration of the capture antibody (CAb), the presence of truncated protein variants, and the duration of the binding process. Additionally, other factors such as the affinity of the CAb for the tag of fusion protein, the specificity of the interaction, and the purity of the CAb can also impact the binding efficiency.
Solution: It is essential to optimize the dilution of the CAb in the well to ensure the capture of the fusion proteins at the appropriate amounts. Excessive synthesis of truncated proteins can hinder the binding of functional proteins in the well. The ideal duration for effective binding of the fusion protein in the well typically falls within the range of 4–12 h. Additionally, using a new batch of CAb can enhance the binding capacity, resulting in increased capture of fusion proteins in the well.
Problem 3: Production of non-functional protein.
Possible cause: The expression of fusion proteins using an in vitro expression kit may yield a large quantity of protein, but it can often be in an inactive form. This can occur due to various factors, including improper protein folding, lack of post-translational modifications, or the absence of necessary co-factors. Additionally, issues such as incorrect buffer composition, inadequate incubation conditions, or suboptimal reaction pH can also contribute to protein inactivity.
Solution: Experiment with various in vitro expression kits to evaluate their ability to facilitate proper folding of the synthesized fusion protein. Select a kit that contains all the essential components necessary for correct protein folding, including chaperone proteins or other factors crucial for post-translational modifications. Furthermore, optimize enzyme activity by testing different co-factors that may enhance protein functionality. Consider factors such as temperature, pH, and incubation time during optimization to ensure maximal enzyme activity.
Problem 4: Lack of binding of plasmid DNA to the wells.
Possible cause: The binding of plasmid DNA in wells is influenced by several factors, including the biotinylation of the plasmid DNA, the quantity and quality of streptavidin present, and the amount of biotinylated plasmid DNA applied to the well.
Solution: The use of new batches of chemicals can enhance the efficiency of biotinylation of plasmid DNA. It is essential to optimize the amount of biotinylation of DNA to achieve maximum protein synthesis in the wells. Additionally, factors such as reaction time, temperature, and the ratio of biotin to DNA should be carefully considered and optimized to ensure optimal biotinylation efficiency.
Problem 5: Lack of enzyme activity detection using the GLO assay.
Possible cause: Enzyme activity is determined by various factors such as no or less capture antibody, high concentration of CAb, the loss of enzyme activity, or not suitable buffer system; a high amount of captured enzyme on the wells can lower accessibility of donor and acceptor to the enzyme. Therefore, it is critical to determine the optimal amount to attach to the wells. The lack of transferase activity can be due to the lack of metal ions in the buffer system. The production of truncated protein that binds to the capture antibody can prevent the capture of functional protein.
Solution: Include a positive control to make sure that CAb is attached to the microplates, use Halo-GST fusion protein (commercially available) to determine the capturing capacity of the CAb, and use AccuBlue Next Gen dsDNA quantitation kit (Biotium) to determine the amounts of plasmid DNA attached to microplates. For enzyme assays, potential issues can be identified through meticulous adjustments to antibody concentration, buffer composition, enzyme concentration, and metal ion presence to maximize enzyme activity detection.
Problem 6: Detection of enzyme activity using GLO assay, but inability to detect the oligosaccharide products.
Possible cause: Some GTs may have hydrolytic activity that effectively releases sugar from NDP-sugar substrates without sugar transfer to the acceptor. Low binding of GTs on the microplates (see Problem 1–5).
Solution: Consider including a control without the acceptor, stabilizing NDP-sugars with metal ions (Mn and/or Mg). If the amount of the products formed is low, increase the number of reactions that are subsequently combined, which can significantly enhance detection sensitivity and accuracy. If the amount of GT captured on the microplate surface is low, the potential reasons could include the lack of expression of fusion proteins, the expression of more truncated protein, the lack of binding of fusion protein to the well, or the lack of binding of plasmid DNA to the wells; see troubleshooting 1, 2, and 4 to solve the problem.
Problem 7: Detecting the PPI in negative controls.
Possible cause: Detection of PPI in negative controls may arise from inadequate washing of the secondary antibody and insufficient blocking of the wells to prevent non-specific binding.
Solution: Thorough washing to ensure that the secondary antibody is removed by incorporating 0.05% Tween 20 in the washing buffer and subsequent removal of Tween 20 residues through additional washing with buffer alone. Enhance blocking efficiency by incubating the wells with 5% fat-free milk for an extended period, such as overnight at 4 °C.
Acknowledgments
This work was supported by an USDA-NIFA award (#1019179). This protocol is adapted from Bhattarai et al. [14] and Javaid et al. [15] and has also been used in Bhattarai et al. [13].
Competing interests
The authors declare no competing interests.
References
- Gygi, S. P., Rochon, Y., Franza, B. R. and Aebersold, R. (1999). Correlation between Protein and mRNA Abundance in Yeast. Mol Cell Biol. 19(3): 1720–1730.
- Edwards, M. E., Dickson, C. A., Chengappa, S., Sidebottom, C., Gidley, M. J. and Reid, J. S. G. (1999). Molecular characterisation of a membrane-bound galactosyltransferase of plant cell wall matrix polysaccharide biosynthesis. Plant J. 19(6): 691–697.
- Perrin, R. M., DeRocher, A. E., Bar-Peled, M., Zeng, W., Norambuena, L., Orellana, A., Raikhel, N. V. and Keegstra, K. (1999). Xyloglucan Fucosyltransferase, an Enzyme Involved in Plant Cell Wall Biosynthesis. Science. 284(5422): 1976–1979.
- Yin, L., Verhertbruggen, Y., Oikawa, A., Manisseri, C., Knierim, B., Prak, L., Jensen, J. K., Knox, J. P., Auer, M., Willats, W. G., et al. (2011). The Cooperative Activities of CSLD2, CSLD3, and CSLD5 Are Required for Normal Arabidopsis Development. Mol Plant. 4(6): 1024–1037.
- LaBaer, J. and Ramachandran, N. (2005). Protein microarrays as tools for functional proteomics.Curr Opin Chem Biol. 9(1): 14–19.
- Ramachandran, N., Hainsworth, E., Demirkan, G. and LaBaer, J. (2006). On-Chip Protein Synthesis for Making Microarrays. In: New and Emerging Proteomic Techniques. Methods in Molecular BiologyTM, vol 328, 1–14. Humana Press.
- Hahm, J. I. (2011). Polymeric Surface-Mediated, High-Density Nano-Assembly of Functional Protein Arrays. J Biomed Nanotechnol. 7(6): 731–742.
- Díez, P., González-González, M., Lourido, L., Dégano, R., Ibarrola, N., Casado-Vela, J., LaBaer, J. and Fuentes, M. (2015). NAPPA as a Real New Method for Protein Microarray Generation. Microarrays 4(2): 214–227.
- Ramachandran, N., Hainsworth, E., Bhullar, B., Eisenstein, S., Rosen, B., Lau, A. Y., Walter, J. C. and LaBaer, J. (2004). Self-Assembling Protein Microarrays. Science. 305(5680): 86–90.
- Ramachandran, N., Raphael, J. V., Hainsworth, E., Demirkan, G., Fuentes, M. G., Rolfs, A., Hu, Y. and LaBaer, J. (2008). Next-generation high-density self-assembling functional protein arrays. Nat Methods. 5(6): 535–538.
- Yazaki, J., Galli, M., Kim, A. Y., Nito, K., Aleman, F., Chang, K. N., Carvunis, A. R., Quan, R., Nguyen, H., Song, L., et al. (2016). Mapping transcription factor interactome networks using HaloTag protein arrays. Proc Natl Acad Sci USA. 113(29): E4238–E4247.
- Manzano-Román, R. and Fuentes, M. (2019). A decade of Nucleic Acid Programmable Protein Arrays (NAPPA) availability: News, actors, progress, prospects and access. J Proteomics. 198: 27–35.
- Bhattarai, M., Wang, Q., Chen, H. and Faik, A. (2024). New Insights on Beta-Glycan Synthases Using in Vitro Gt-Array (I-GT-ray) Platform. Available at SSRN: https://ssrn.com/abstract=4753263 or http://dx.doi.org/10.2139/ssrn.4753263
- Bhattarai, M., Wang, Q., Javaid, T., Venkataraghavan, A., Al Hassan, M. T., O’Neill, M., Tan, L., Chen, H. and Faik, A. (2024). Streamlining assays of glycosyltransferases activity using in vitro GT-array (i-GT-ray) platform: Application to family GT37 fucosyltransferases. J Biol Chem. 300(3): 105734.
- Javaid, T., Bhattarai, M., Venkataraghavan, A., Held, M. and Faik, A. (2024). Specific protein interactions between rice members of the GT43 and GT47 families form various central cores of putative xylan synthase complexes. Plant J. 118(3): 856–878.
- Sandhu, A. P. S., Randhawa, G. S. and Dhugga, K. S. (2009). Plant Cell Wall Matrix Polysaccharide Biosynthesis. Mol Plant. 2(5): 840–850.
- Amos, R. A. and Mohnen, D. (2019). Critical Review of Plant Cell Wall Matrix Polysaccharide Glycosyltransferase Activities Verified by Heterologous Protein Expression. Front Plant Sci. 10: e00915.
- Ishikawa, M., Kuroyama, H., Takeuchi, Y. and Tsumuraya, Y. (2000). Characterization of pectin methyltransferase from soybean hypocotyls. Planta. 210(5): 782–791.
- Faik, A., Price, N. J., Raikhel, N. V. and Keegstra, K. (2002). An Arabidopsis gene encoding an α-xylosyltransferase involved in xyloglucan biosynthesis. Proc Natl Acad Sci USA. 99(11): 7797–7802.
- Mortimer, J. C., Miles, G. P., Brown, D. M., Zhang, Z., Segura, M. P., Weimar, T., Yu, X., Seffen, K. A., Stephens, E., Turner, S. R., et al. (2010). Absence of branches from xylan in Arabidopsis gux mutants reveals potential for simplification of lignocellulosic biomass. Proc Natl Acad Sci USA. 107(40): 17409–17414.
- Rennie, E. A., Hansen, S. F., Baidoo, E. E., Hadi, M. Z., Keasling, J. D. and Scheller, H. V. (2012). Three Members of the Arabidopsis Glycosyltransferase Family 8 Are Xylan Glucuronosyltransferases. Plant Physiol. 159(4): 1408–1417.
- Ericsson, U. B., Hallberg, B. M., DeTitta, G. T., Dekker, N. and Nordlund, P. (2006). Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 357(2): 289–298.
- Denisov, I. G. and Sligar, S. G. (2017). Nanodiscs in Membrane Biochemistry and Biophysics. Chem Rev. 117(6): 4669–4713.
- Denisov, I. G., Grinkova, Y. V., Lazarides, A. A. and Sligar, S. G. (2004). Directed Self-Assembly of Monodisperse Phospholipid Bilayer Nanodiscs with Controlled Size. J Am Chem Soc. 126(11): 3477–3487.
- Omadjela, O., Narahari, A., Strumillo, J., Mélida, H., Mazur, O., Bulone, V. and Zimmer, J. (2013). BcsA and BcsB form the catalytically active core of bacterial cellulose synthase sufficient for in vitro cellulose synthesis. Proc Natl Acad Sci USA. 110(44): 17856–17861.
- Goverde, C. A., Pacesa, M., Goldbach, N., Dornfeld, L. J., Balbi, P. E. M., Georgeon, S., Rosset, S., Kapoor, S., Choudhury, J., Dauparas, J., et al. (2024). Computational design of soluble and functional membrane protein analogues. Nature. 631(8020): 449–458.
- Goverde, C. A., Wolf, B., Khakzad, H., Rosset, S. and Correia, B. E. (2023). De novo protein design by inversion of the AlphaFold structure prediction network. Protein Sci. 32(6): e4653.
- Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., Wicky, B. I. M., Courbet, A., de Haas, R. J., Bethel, N., et al. (2022). Robust deep learning–based protein sequence design using ProteinMPNN. Science. 378(6615): 49–56.
Article Information
Publication history
Received: May 4, 2024
Accepted: Aug 1, 2024
Available online: Aug 16, 2024
Published: Sep 20, 2024
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bhattarai, M., Javaid, T., Venkataraghavan, A. and Faik, A. (2024). In Vitro GT-array (i-GT-ray), a Platform for Screening of Glycosyltransferase Activities and Protein–Protein Interactions. Bio-protocol 14(18): e5066. DOI: 10.21769/BioProtoc.5066.
- Bhattarai, M., Wang, Q., Javaid, T., Venkataraghavan, A., Al Hassan, M. T., O’Neill, M., Tan, L., Chen, H. and Faik, A. (2024). Streamlining assays of glycosyltransferases activity using in vitro GT-array (i-GT-ray) platform: Application to family GT37 fucosyltransferases. J Biol Chem. 300(3): 105734.
Category
Biochemistry > Protein
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.