- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Transfection of Babesia duncani: A Genetic Toolbox of This Pathogen to Advance Babesia Biology
Published: Vol 14, Iss 12, Jun 20, 2024 DOI: 10.21769/BioProtoc.5016 Views: 2065
Reviewed by: Alba BlesaRITU SOMAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2022

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Human babesiosis is a tick-borne disease caused by Babesia pathogens. The disease, which presents with malaria-like symptoms, can be life-threatening, especially in individuals with weakened immune systems and the elderly. The worldwide prevalence of human babesiosis has been gradually rising, prompting alarm among public health experts. In other pathogens, genetic techniques have proven to be valuable tools for conducting functional studies to understand the importance of specific genes in development and pathogenesis as well as to validate novel cellular targets for drug discovery. Genetic manipulation methods have been established for several non-human Babesia and Theileria species and, more recently, have begun to be developed for human Babesia parasites. We have previously reported the development of a method for genetic manipulation of the human pathogen Babesia duncani. This method is based on positive selection using the hDHFR gene as a selectable marker, whose expression is regulated by the ef-1aB promoter, along with homology regions that facilitate integration into the gene of interest through homologous recombination. Herein, we provide a detailed description of the steps needed to implement this strategy in B. duncani to study gene function. It is anticipated that the implementation of this method will significantly improve our understanding of babesiosis and facilitate the development of novel and more effective therapeutic strategies for the treatment of human babesiosis.
Keywords: BabesiosisGraphical overview
Background
Babesia are parasitic protozoa of the Apicomplexa phylum, which encompasses other important human pathogens such as the agents of malaria and toxoplasmosis. Babesia parasites are transmitted primarily by ticks and can infect various mammalian species [2]. In humans, babesiosis is caused by several Babesia species including B. microti, B. duncani, B. divergens, and B. venatorum [3]. Clinical manifestations of the disease include fever, hemolytic anemia, and in severe cases, complications such as cardiac failure, respiratory distress, and pulmonary issues, which can ultimately lead to death. Patients with underlying immunological disorders, undergoing immunosuppressive treatment, or having undergone splenectomy are particularly susceptible to experiencing severe symptoms and increased mortality if infected by Babesia [3–5]. B. duncani is found primarily in western United States and exhibits higher virulence in animal models compared to B. microti, leading to acute mortality in mice and in hamsters [6–9]. Morphologically, there are no significant distinguishing features between B. duncani and B. microti [10].
Gene editing has become an invaluable tool for the investigation of gene function and validation of potential drug targets [11]. Advancing our understanding of the fundamental biology of Babesia parasites requires the development of genetic manipulation techniques [12]. Previous studies have demonstrated successful genetic manipulation methods in various protozoan pathogens, including Babesia bovis, B. gibsoni, B. ovis, B. ovata, Theileria annulata, and Theileria parva [13–18]. Successful transfection of piroplasmids was achieved in B. bovis [1] using the elongation factor 1-aB (ef-1aB) promoter to drive expression of a reporter GFP-BSD consisting of a fusion between the green fluorescent protein (GFP) and the blasticidin S deaminase (BSD) [19], which was integrated into the ef-1αA region through homologous recombination. Subsequent studies employed a similar approach for stable transfection in B. gibsoni, B. ovata, and Babesia sp. Xinjiang [17,18,20]. While a genetic modification method has been reported for B. microti, it mainly relies on fluorescent tags due to the lack of effective in vivo drug screening markers, challenging the generation of gene-edited strains in B. microti [21]. Genetic manipulation of human Babesia parasites has also been facilitated by the availability of a continuous in vitro culture system in human red blood cells and the sequencing, assembly, and annotation of their genomes [5,9,22–24]. Unlike B. microti, for which only a short-term culture could be achieved, long-term and continuous in vitro culture of B. duncani has been established using hamster and human erythrocytes [24–27]. Furthermore, in B. duncani, an animal model of lethal infection has been optimized, thus paving the path for evaluating the importance of specific genes in vivo [8,27]. The first genetic modification of B. duncani [28] was achieved using a transient transfection technique to express the mCherry reporter in the parasite under the regulatory control of the ef-1αB promoter. Subsequently, a stable expression of eGFP was achieved in B. duncani using the human dihydrofolate reductase (hDHFR) gene selectable marker, which confers resistance to the antifolate WR99210. The development of genetic tools for the manipulation of B. duncani provides a unique opportunity to study gene function in B. duncani and to gain further insights into its biology and pathogenesis.
Development of the protocol
The electroporation method has been widely utilized for various apicomplexan parasites, including Toxoplasma gondii (the causative agent of toxoplasmosis) and Plasmodium spp. (the agents of human malaria). Electroporation is an effective means of introducing foreign DNA into these parasites for genetic manipulation and functional studies. Electroporation methods are indeed categorized into two main types based on the electrical parameters. The first is low voltage–long pulse, which employs a lower electrical voltage for a relatively longer pulse duration. This approach is typically considered gentler and is often used when cell viability is a primary concern. The high voltage–short pulse method involves using a high electrical voltage for a very short pulse duration. It is often referred to as the burst or high-field electroporation. This approach is more efficient in terms of introducing foreign material into cells but may have a greater impact on cell viability. The choice between these methods depends on the specific requirements of the experiment, such as the type of cells or organisms being electroporated and the desired level of transfection efficiency vs. cell survival. In B. duncani, as in previously reported cases in B. bovis [1], the high voltage–short pulse electroporation method results in higher transfection efficiency. To enhance transfection efficiency in B. duncani, our optimization efforts focused on changing the parameters of electroporation, including voltage, volume, and frequency, with the aim of achieving higher levels of transfection efficacy.
When comparing different electroporation instruments, we usually find variations in their effectiveness for transfection. We found that a higher transfection efficiency could be achieved using the Gemini SC Electroporation System from BTX, whereas transfection using the Bio-Rad Gene Pulser Xcell was not successful.
Materials and reagents
Biological materials
Chemically competent E. coli DH5α [F− supE44 ΔlacU169 (φ80 lacZDM15) hsdR17 (rk−mk) recA1 endA1 thi1 gyrA relA] is used for routine cloning and plasmid maintenance. The competent cells are available via ThermoFisher (catalog number: 18265017)
Hamster RBCs are used for B. duncani culture. The hamster RBCs are obtained from Syrian golden hamsters [Crl: LVG (SYR)] and can be prepared following the protocol.
B. duncani WA1 strain (ATCC PRA-302™) is expanded through in vitro cultivation and the protocol.
MilliQ water
Bacto tryptone (Becton Dickinson, catalog number: 211705)
Bacto yeast extract (Becton Dickinson, catalog number: 212750)
Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S5886)
Bacto agar (Becton Dickinson, catalog number: 214010)
Ampicillin sodium salt (Sigma-Aldrich, catalog number: A9518)
Phanta Max Super-Fidelity DNA Polymerase (Vazyme, catalog number: P505)
2× Rapid Taq Master Mix (Vazyme, catalog number: P222)
ClonExpress MultiS One Step Cloning Kit (Vazyme, catalog number: C113)
100 bp DNA ladder (Vazyme, catalog number: MD104-01)
EasyPure® Quick Gel Extraction kit (Trans, catalog number: EG-101)
EasyPure® Plasmid MiniPrep kit (Trans, catalog number: EM-101)
EndoFree Maxi plasmid kit (TIANGEN, catalog number: DP117)
TIANamp Geneomin DNA kit (TIANGEN, catalog number: DP304)
VP-SFM ATGTM (Thermo Fisher Scientific, Gibco, catalog number: 12559019)
Ala-Gln (MACKLIN, catalog number: A800584)
AlbuMax II (Thermo Fisher Scientific, Gibco, catalog number: 11021037)
Antimycotic (antibiotic) (Thermo Fisher Scientific, Gibco, catalog number: 15240-062)
WR99210 (MCE, catalog number: HY-116387)
MBP146-78 (TargetMol, catalog number: T7321)
Giemsa’s stain (Sigma-Aldrich, catalog number: G4507)
Immersion oil (Sigma-Aldrich, catalog number: 1046990100)
Trisodium citrate (Sigma-Aldrich, catalog number: V900443)
Citric acid (Sigma-Aldrich, catalog number: C0759)
Glucose (Sigma-Aldrich, catalog number: G8270)
Sodium dihydrogen phosphate (Sigma-Aldrich, catalog number: 5438400100)
Adenine (Sigma-Aldrich, catalog number: A2786)
Mannitol (Sigma-Aldrich, catalog number: M4125)
Potassium chloride (KCl) (Sigma-Aldrich, catalog number: S5886)
Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: C5670)
Dipotassium hydrogen phosphate (K2HPO4) (Sigma-Aldrich, catalog number: P8281)
Potassium dihydrogen phosphate (KH2PO4) (Sigma-Aldrich, catalog number: P5655)
4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES) (Sigma-Aldrich, catalog number:H4034)
Ethylene glycol-bis(2-aminoethylether)-N, N, N', N'-tetraacetic acid (EGTA) (Sigma-Aldrich, catalog number: E3889)
Magnesium chloride hexahydrate (MgCl2·6H2O) (Sigma-Aldrich, catalog number: M2393)
Saponin (Sigma-Aldrich, catalog number: S7900)
Absolute ethanol (Sigma-Aldrich, catalog number: 1009831011)
Alsever’s solution (Sigma-Aldrich, catalog number: A3551)
DMSO (Sigma-Aldrich, catalog number: D8418)
Glycerol (Sigma-Aldrich, catalog number: G5516)
Tris (MACKLIN, catalog number: T819512)
Ethylenediaminetetraacetic acid (EDTA) (MACKLIN, catalog number: E809068)
Acetic acid (MACKLIN, catalog number: A801295)
Potassium hydroxide (KOH, MACKLIN, catalog number: P816399)
Agarose (MACKLIN, catalog number: A800342)
GelRed (MACKLIN, catalog number: G917739)
Ethidium bromide (EB, MACKLIN, catalog number: E808961)
Sodium acetate buffer, 3 M, pH 5.2 (MACKLIN, catalog number: S885174)
Hoechst (Beyotime, catalog number: CH1024)
Solutions
LB medium (see Recipes)
LB plates (see Recipes)
Ampicillin stock solutions (100 mg/mL) (see Recipes)
TAE buffer (see Recipes)
VP-SFM complete media (see Recipes)
Red blood cell preservation solution (see Recipes)
Cytomix buffer (see Recipes)
20% Saponin stock solution (see Recipes)
5 mM WR99210 storage solution (see Recipes)
Freezing solution (see Recipes)
Recipes
LB medium
Prepare LB medium by dissolving the following ingredients (with the help of a magnetic stirrer) in MilliQ water: 10 g/L tryptone, 5 g/L yeast extract, and 5 g/L NaCl.
Aliquot the broth in clean bottles, autoclave at 121 °C for 15 min, and let it cool down before use.
LB medium can be stored at room temperature for up to six months.
LB plates
Prepare the LB medium as described above, aliquot in clean bottles, and add 15 g/L Bacto agar. Autoclave at 121 °C for 15 min and let cool down to ~50 °C at room temperature before the addition of antibiotics or other supplements (when needed). Mix well and pour ~20 mL into Petri dishes. Plates can be stored at 4 °C for up to one month.
Ampicillin stock solutions (100 mg/mL)
Weigh 1,000 mg of ampicillin sodium salt using an electronic scale and dissolve in 10 mL of MilliQ water. Filter the solution through a 0.2 μm filter in the laminar flow hood and prepare 1 mL aliquots in sterilized 1.5 mL microcentrifuge tubes. Antibiotic stock solutions can be stored at -20 °C for up to one year.
TAE buffer
The 50× TAE stock solution contains 2 mol/L Tris (242 g/L), 0.1 mol/L EDTA disodium salt (37.2 g/L), and 57.1 mL/L acetic acid. Add double-distilled water (ddH2O) to a final volume of 1 L, place the solution on the magnetic stirrer, and stir until it is completely dissolved. Once dissolved, adjust the pH to 8.0. The 50× TAE stock solution can be stored at room temperature for several years; dilute it with ddH2O to 1× TAE buffer before use.
VP-SFM complete media
Prepare VP-SFM complete medium by dissolving the following ingredients (with the help of a magnetic stirrer) in MilliQ water: 17.6 g/L VP-SFM ATGTM, 0.86 g/L Ala-Gln, 2 g/L AlbuMax II, and 10 mL/L 100× antibiotic/antimycotic. Filter the solution through a 0.2 μm filter in the laminar flow hood and prepare 50 mL aliquots in a sterilized centrifuge tube. Medium can be stored at 4 for up to six months.
Red blood cell preservation solution
Prepare red blood cell preservation solution by dissolving the following ingredients (with the help of a magnetic stirrer) in MilliQ water: 1.5 g/L trisodium citrate, 0.2 g/L citric acid, 7.93 g/L glucose, 0.94 g/L sodium dihydrogen phosphate, 0.14 g/L adenine, 4.97 g/L sodium chloride, and 14.57 g/L mannitol. Aliquot the solution in clean bottles, autoclave at 121 °C for 15 min, and let it cool down before use. Red blood cell preservation solution can be stored at 4 for up to six months.
Cytomix buffer
In a clean and sterile container or bottle, add approximately 800 mL of MilliQ water. Carefully add the dry ingredients in the following order while gently stirring to ensure proper dissolution: 6.7 g KCl, 0.012 g CaCl2, 1.30 g K2HPO4, 1.02 g KH2PO4, 4.46 g HEPES, 0.76 g EGTA, and 0.762 g MgCl2·6H2O; adjust the pH 7.5 with potassium hydroxide (KOH). After all the components are dissolved, carefully adjust the volume to 1 L using MilliQ water and mix thoroughly to ensure the solution is homogenous. Filter the solution through a 0.2 μm filter in the laminar flow hood and prepare 50 mL aliquots in a sterilized centrifuge tube. Medium can be stored at 4 for up to six months.
20% Saponin stock solution
Weigh 20 g of saponin using an electronic scale and dissolve in 100 mL of 1× PBS (8.0 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, 0.24 g/L KH2PO4, PH 7.4). Filter the solution through a 0.2 μm filter in the laminar flow hood and prepare 1 mL aliquots in sterilized 1.5 mL microcentrifuge tubes. Saponin stock solutions can be stored at −20 °C for up to one year. Dilute the saponin solution to 0.1% with PBS when lysing red blood cells.
5 mM WR99210 storage solution
Add 0.5067 mL of DMSO to 1 mg of WR99210 powder. Gently swirl to dissolve. Prepare small 10 μL aliquots and store at -20 °C for up to 12 months. Prepare a 5 μM working concentration by diluting it in DMSO at a 1:1,000 ratio before use.
Freezing solution
In a 50 mL centrifuge tube, add 15 mL of glycerol, followed by 35 mL of Alsever's solution. Mix the solution by vortex thoroughly and then filter it using a 0.2 μm filter. Freezing solution can be stored at 4 °C for up to six months.
Laboratory supplies
Micropipette refill tips (10, 200, and 1,000 µL)
1.5 mL Eppendorf tube (Pullen, catalog number: PL03001)
PCR tubes, 8 tubes per strip, 125 strips per unit (Crystalgen (GY), catalog number: L-2081)
PCR tubes, domed 8 caps per strip, 125 strips per unit (Crystalgen (GY), catalog number: L-2082)
24-well plates (Corning, catalog number: 3524)
96-well plates (Corning, catalog number: 3917)
15 mL centrifuge tube (PULLEN, catalog number: PL01005)
50 mL centrifuge tube (PULLEN, catalog number: PL01006)
1 mL syringe (Beyotime, catalog number: FS802)
Equipment
Micropipette set, Eppendorf Research plus (0.1–2.5, 2–20, 20–200, and 100–1,000 µL) (Eppendorf, catalog number: 3123000012, 3123000098, 3123000055, and 3123000063)
Laboratory glass bottles (250, 500, and 1,000 mL)
T100 thermal cycler (Bio-Rad, catalog number: 1861096)
Petri dishes (Ø × H: 92 × 16 mm, with ventilation cams)
Benchtop centrifuge (Eppendorf, catalog number: 5810R)
Electric thermostatic incubator (Shanghai Jinghong Instrument, catalog number: DNP-9272)
Floored large refrigerated shaking incubator (Shanghai Zhichu Instrument, catalog number: ZQLY-300S)
Medical icebox (Panasonic, catalog number: MPR-710)
Low temperature freezer (-20 °C) (Panasonic, catalog number: MDF-539)
Gemini SC electroporation system (BTX, catalog number: 45-2042)
pH meter (Sartorius, model: PB-11)
Gene Pulser/MicroPulser electroporation cuvettes, 0.2 cm gap (Bio-Rad, catalog number: 1652082)
Modular incubator chamber (Billups-Rothenberg, Inc, model: MIC-101)
Millipore 0.2 μM filters (Sigma-Aldrich, catalog number: SLGNDZ5)
Blue-light gel cutter (Sangon, model: G600312-000)
NanoDrop 2000 (Thermo Fisher, model: pedestal mode)
Water bath (Beyotime, model: E0530)
Gel imaging system (Bio-Rad, model: XXX)
OLYMPUS FRAME_BX63 scanning confocal microscope (OLYMPUS FRAME, model: BX63)
Cell counter (Watson, model: 177-112C)
Nalgene Mr. Frosty Freezing Container (Thermo Scientific, catalog number: 5100-0036)
Software
SnapGene (RRID: SCR_015052, https://www.snapgene.com, SnapGene 7.0.2)
Procedure
Plasmid design
Promoter region: For the promoter region, select the 250 base pairs located upstream of the transcription start site as shown in Figure 1A. Commonly used promoters for drug screening tags include ef-1aB promoter and enolase promoter.
Marker selection: Choose from commonly used fluorescent reporter genes such as eGFP, mCherry, or luciferase, as indicated in Figure 1A. For drug selection markers, commonly used options are human dihydrofolate-reductase (hDHFR) and puromycin-N-acetyltransferase (PAC). The addition of the drug WR99210 disables the Babesia DHFR enzyme and halts nucleic acid synthesis. By expressing hDHFR, which is resistant to the drug, the function of the babesia DHFR is replaced. PAC confers resistance to puromycin.
Terminators region: Downstream of the gene ORF (open reading frame), commonly used terminators include ef-1a, rap-1, and DHFR-TS (dihydrofolate reductase-thymidylate synthase). Most termination sites are between 200 and 600 bp in length.
Homologous arms: When performing gene insertion and replacement, it is essential to have a segment of homologous arms to target the gene of interest. Typically, the homologous arms consist of 750 bp located both upstream and downstream of the modification site, as illustrated in Figure 1B.
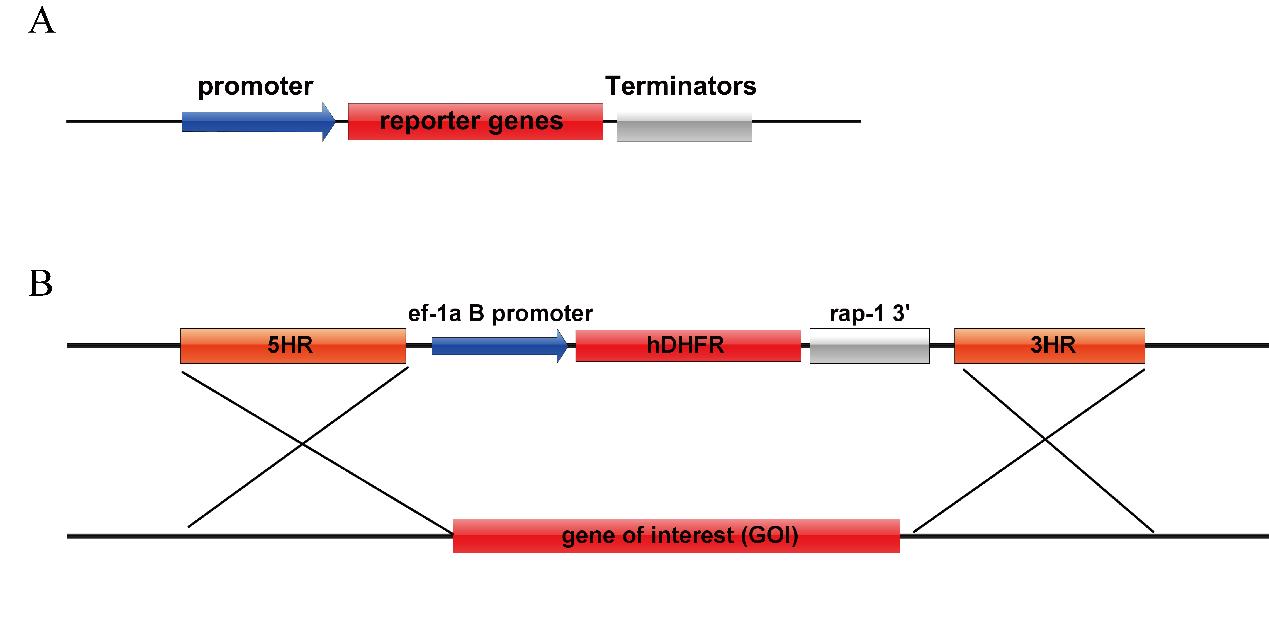
Figure 1. Vector maps common for B. duncani. A. Plasmid map for transient transfection. B. Plasmid for genetic modification.Design the oligonucleotides to construct the plasmid. Firstly, identify the gene of interest in the genome and extract 1,500 bp of flanking upstream and downstream of this gene. Select 750 bp of sequence upstream and downstream of the gene locus for the homologous arms. The genome of B. duncani has been uploaded to NCBI with the accession number GCA_028658345.1.
Design primers according to the ClonExpress MultiS One Step Cloning kit instructions for amplifying the homologous arms, drug selection marker, and linearized vector. Dilute the ordered primer (5 nmol) with nuclease-free H2O (500 μL) to a final concentration of 10 μM.
Amplify the homologous arms, drug selection marker, and linearized plasmid by PCR. The template for PCR of homologous arms is genomic DNA (20 ng) isolated using the TIANamp Geneomin DNA kit, while the templates for the drug selection marker and the linearized plasmid are plasmid DNA (1 ng) isolated with the EasyPure® Plasmid MiniPrep kit. Extract the DNA using the Tissue DNA kit following the manufacturer’s instructions. Prepare the PCR mixture according to Table 1.
Table 1. Phanta Max Super-Fidelity DNA Polymerase reaction system
Component Volume Final concentration Template 1 μL Primer-F 10 μM 1 μL 0.2 μM Primer-R 10 μM 1 μL 0.2 μM 2 × Phanta Max buffer 25 μL dNTP mix (10 mM each) 1 μL 0.2 mM Phanta Max Super-Fidelity DNA Polymerase 1 μL Nuclease-free H2O 20 μL Total 50 μL Perform the PCR reaction under the following cycling conditions (Table 2):
Table 2. Phanta Max Super-Fidelity DNA Polymerase reaction protocol settings
Cycle number Denature Anneal Extend 1 95 °C, 5 min 2–36 95 °C, 15 s 52 °C, 15 s 72 °C, 2 min 37 72 °C, 5 min PCR product purification: Purify the PCR products by using agarose gel electrophoresis [1.5% (wt/vol) in TAE buffer, supplemented with 1:10,000 (vol/vol) GelRed or ethidium bromide]. Add 6 μL of 10× DNA loading buffer to the 50 μL of PCR product and then load the mixture into a well of the agarose gel. Load 5 μL of 100 bp DNA ladder in a flanking well of the same gel. Run the gel in 1× TAE buffer at 120 V for 30 min. Excise the target DNA band with a blue-light gel cutter and extract the DNA fragment by using a EasyPure® Quick Gel Extraction kit, following the manufacturer’s instructions.
After extracting the DNA fragment, measure the concentration of extracted DNA fragment on a NanoDrop 2000.
Use the ClonExpress MultiS One Step Cloning kit to ligation the fragments obtained in step A5. According to the manufacturer's instructions, mix the DNA fragments. Prepare the ligation mixture according to the following table (Table 3):
Table 3. Ligation mixture system
Component Quantity Volume pBluescript fragment 60 ng Marker Selection fragment 72 ng 5HR fragment 30 ng 3HR fragment 30 ng 5× CE MultiS buffer 2 μL Exnase MultiS 1 μL ddH2O To 10 μL Total 10 μL Incubate the ligate mixture from step A7 at 37 °C for 45 min in a T100 thermal cycler.
Add the entire ligate mixture from Step A8 to 100 μL of DH5α chemically competent cells and incubate the Eppendorf tube containing the competent cells on ice for 30 min.
Heat-shock the competent cells in a 42 °C water bath for 90 s and then immediately keep them on ice for 1 min.
Add 500 μL of LB medium (without any antibiotics) into the tube, put the tube in a thermostatic shaker, and shake the tube at 580× g for 45 min at 37 °C.
Centrifuge the recovered bacteria at 1,500× g for 5 min at room temperature and then remove most of the supernatant by pipetting, leaving ~50 μL of LB medium to resuspend the bacteria. Evenly apply the bacterial solution to an LB agar plate with ampicillin (100 μg/mL) using a glass rod or beads, invert the plate, and incubate at 37 °C overnight.
After overnight incubation, single clones are visible on the LB agar plate. Pick up five individual clones and culture each in 1 mL of LB medium with ampicillin (100 μg/mL) individually. Shake them in a 37 °C shaker incubator at 580× g for 12–14 h.
Verifying spacer cloning by PCR (cPCR): To perform a cPCR in a 10 µL reaction volume, to check for transformants harboring the correctly cloned spacer inside the pBluescript, prepare the following reaction mix (Table 4):
Table 4. Rapid Taq Master Mix reaction system
Component Quantity (μL) Final concentration M13F 10 μM 0.25 0.25 μM M13R 10 μM 0.25 0.25 μM 2× Rapid Taq Master Mix 5 1× Bacterial from step A13 1 ddH2O 3.5 Total 10 Perform the PCR reaction under the following cycling conditions (Table 5):
Table 5. Rapid Taq Master Mix reaction protocol settings
Cycle number Denature Anneal Extend 1 95 °C, 5 min 2–36 95 °C, 15 s 52 °C, 15 s 72 °C, 2 min 37 72 °C, 5 min Pipette 5 μL of each PCR reaction along with the GeneRuler 100 bp DNA ladder on a 1.5% (wt/vol) agarose gel prepared in 1× TAE buffer and stained with ethidium bromide. Run the gel at 120 V for 30 min and visualize the bands using the Gel Jet Documentation System. A 3,500 nt band is expected.
Use EasyPure® Plasmid MiniPrep kit to extract the plasmids from the clones in Step A13, following the manufacturer’s instructions, and then send these plasmids for Sanger sequencing to confirm that the sequence of the constructed Pbs-TPX-1 KO is correct.
Use EndoFree Maxi Plasmid kit to extract the plasmids from the clones in Step A14, following the manufacturer’s instructions.
After extraction, perform ethanol precipitation to obtain a pellet of plasmid DNA. Ethanol precipitation is a DNA purification technique. Here are the specific steps:
Transfer the extracted DNA solution to a new centrifuge tube.
Add 0.1 volume of NaOAc buffer (3 M, pH 5.2) and 2.5 volumes of cold ethanol (either absolute ethanol or isopropanol). Mix the solution thoroughly.
Place the tube in a -20 freezer for 15 min or freeze it at -80 for at least 30 min to allow DNA precipitation.
Centrifuge the tube at 12,000–16,000× g for 10–15 min at 4 . This will cause the DNA to form a pellet at the bottom of the tube.
Carefully pour off the supernatant without disturbing the DNA pellet. Take care not to disrupt the pellet.
Wash the DNA pellet by adding 70% ethanol. Gently swirl the tube to ensure thorough washing.
Centrifuge again at 12,000–16,000× g for 5 min at 4 to remove the ethanol wash.
Carefully pour off the ethanol, ensuring that the DNA pellet is left to air-dry as much as possible.
Rinse the walls of the tube gently with wash solution to remove any residual ethanol.
Air-dry the DNA pellet at room temperature for 5–10 min or use a low-speed centrifuge to remove any remaining liquid.
Add an appropriate volume of solvent, such as Tris-EDTA buffer, to completely dissolve the DNA pellet.
Please note that during the ethanol precipitation method, it is important to use sterile reagents and maintain a sterile working environment to avoid DNA contamination. Additionally, handle the samples with care to prevent DNA damage or loss.
(Optional) Linearization: Plasmid linearization is achieved by using restriction enzymes to cleave the plasmid at specific recognition sites. Following linearization, the DNA fragments can be concentrated using the ethanol precipitation method.
Note: Linearizing the plasmid is an optional step; using circular plasmids can also yield the desired results. Linearized plasmids are more prone to recombine with the chromosome compared to non-linearized plasmids. Circular plasmids may exist in the parasite as episomal vector.
Syrian golden hamster erythrocyte collection
Anesthetize hamsters using a respiratory anesthesia device and administer isoflurane at a concentration of 2.5% (refer to Figure 2A).
Collect blood from the orbital vein of each hamster using a 1-mL syringe (refer to Figure 2B). Each hamster can provide 150 μL of blood with a 2-week interval between collections. After collecting the blood, immediately add it to an equal volume of red blood cell preservation solution and gently shake it until thoroughly mixed.
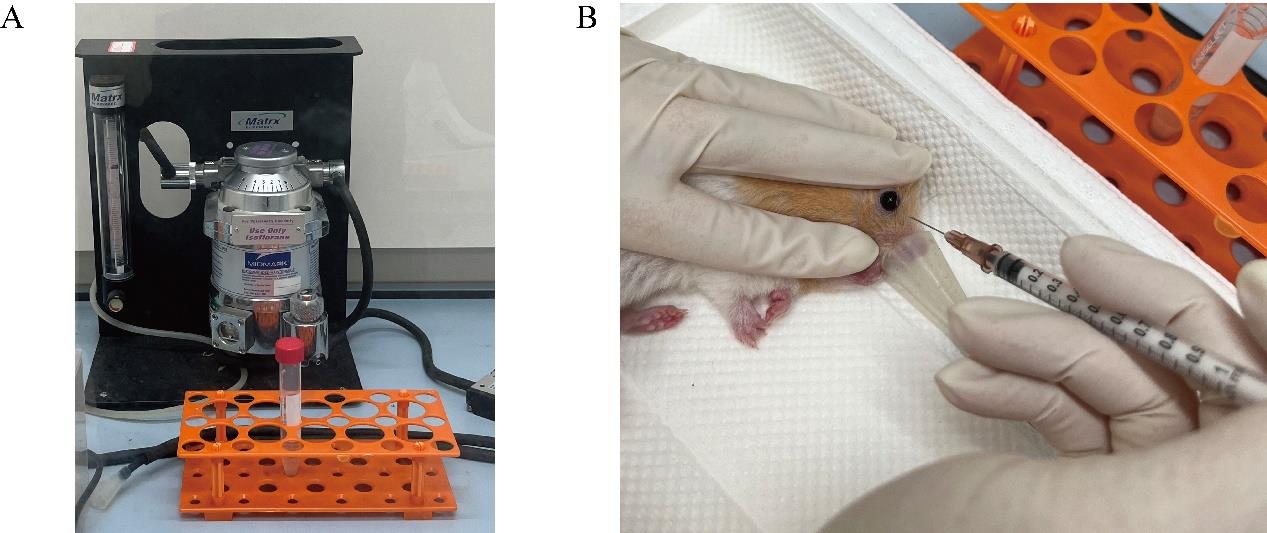
Figure 2. Representative image depicting Syrian golden hamster erythrocyte collection.To the collected blood, add twice the volume of red blood cell preservation solution. Place the mixture in a centrifuge at 500× g for 10 min at 4 . Carefully remove the upper layer of liquid after each centrifugation, repeating the process three times.
After completing the washing process, add an equal volume of red blood cell preservation solution to the washed red blood cells. Store the solution at 4 °C.
Note: The washed RBCs (50% hematocrit) can be immediately used for in vitro culture or stored at 4 °C for further use. The washed RBCs can be stored at 4 °C and used for up to two weeks in cell culture.
B. duncani culture
To perform a culture medium change for B. duncani, follow the steps outlined below:
Prewarm the culture medium: Before use, warm the culture medium to 37 . This ensures that the medium is at the optimal temperature for the growth of B. duncani.
Remove the culture supernatant: Carefully discard the culture supernatant from the culture vessel, ensuring not to disturb the settled red blood cells at the bottom.
Add fresh culture medium: Add an appropriate amount of prewarmed culture medium to the culture vessel to achieve the desired total volume. The exact volume required will depend on your specific culture system’s requirements.
Mix the red blood cells: Gently agitate the culture vessel to mix the red blood cells and the new culture medium by aspirator, ensuring that they are evenly distributed. It is important to avoid vigorous agitation that may potentially damage the red blood cells.
Medium change frequency: The frequency of medium changes depends on the parasitemia level, which refers to the percentage of infected red blood cells. If the parasitemia is below 5%, you can change the medium every 48 h. However, if parasitemia exceeds 5%, more frequent medium changes are necessary, typically every 24 h.
Note:
Culture system for B. duncani: B. duncani parasites can be cultured in different sizes of culture dishes or cell culture bottles. For instance, in a 24-well plate, each well contains 950 μL of culture medium and 50 μL of red blood cells. Refer to Table 6 below for the recommended amounts of culture medium and red blood cells for different culture containers.
Table 6. Culture system for B. duncani
Culture vessel Culture medium volume Red blood cell volume Culture medium replacement volume 96-well plate 95 μL 5 μL 70 μL 48-well plate 475 μL 25 μL 400 μL 24-well plate 950 μL 50 μL 800 μL 12-well plate 1,900 μL 100 μL 1,600 μL 6-well plate 4.75 mL 250 μL 4 mL T25-cell culture bottle 4.5 mL 500 μL 4 mL T75-cell culture bottle 27 mL 3 mL 24 mL Culturing environment: To achieve optimal growth, B. duncani requires 37 °C, saturated humidity, and a specific gas composition in the culturing environment. The recommended composition is 2% O2, 5% CO2, and 93% N2. There are two common methods to establish this environment:
Gas-infused culture flask: A culture flask designed for gas infusion can be utilized. The pre-mixed gas with the desired composition of 2% O2, 5% CO2, and 93% N2 is introduced into the flask, creating the appropriate culturing environment. The flask is typically equipped with a gas exchange system to regulate and maintain the desired gas levels.
Tri-gas incubator: Alternatively, a tri-gas incubator can be employed. This incubation system offers the capability to adjust gas proportions. It is equipped with controls to regulate the flow of gases and maintain the desired composition of 2% O2, 5% CO2, and 93% N2 within the enclosed chamber where the culture vessels are placed. The incubator provides a stable and controlled environment for the optimal growth of B. duncani.
Both methods ensure the availability of the required gas composition for the culturing process, enabling the optimal growth of B. duncani.
Continuous cell culture
Prewarm the medium at 37 °C.
Utilize a 24-well plate and maintain 1 mL cultures. Monitor the parasitemia, aiming to keep it between 4% and 8% (though it can reach as high as 30%).
Adjust the culture parasitemia to 1% and maintain the hematocrit at 5% by splitting the culture. For example, if the parasitemia is at 1 mL culture at 5% hematocrit, mix culture well and transfer 250 μL into a new well, diluting it down to 1%.
Add 750 μL of VP-SFM and add 37.5 μL of RBC to the new well.
To prepare blood smears from cultured B. duncani samples, please follow the steps below:
During medium change, tilt the culture dish and carefully remove the red blood cells from the bottom using a pipette.
Place two glass slides side by side, slightly overlapping each other.
Using a pipette, transfer a small drop of the collected red blood cells onto the center of the slide.
Quickly and smoothly spread the drop of blood across the slides using the edge of another slide at a 45-degree angle.
Allow the smears to air dry completely.
Once dried, fix the smears by immersing them in methanol for approximately 1–2 min.
Prepare the Giemsa stain according to the instructions provided by the manufacturer.
Submerge the dried smears in the Giemsa stain for the recommended staining duration, typically around 10–20 min.
Gently rinse the smears with distilled water to remove excess stain.
Allow the smears to air dry completely before examining them under a microscope.
By following these steps, you can create blood smears from the cultured B. duncani samples. These smears can then be further examined and analyzed using a microscope for diagnostic or research purposes.
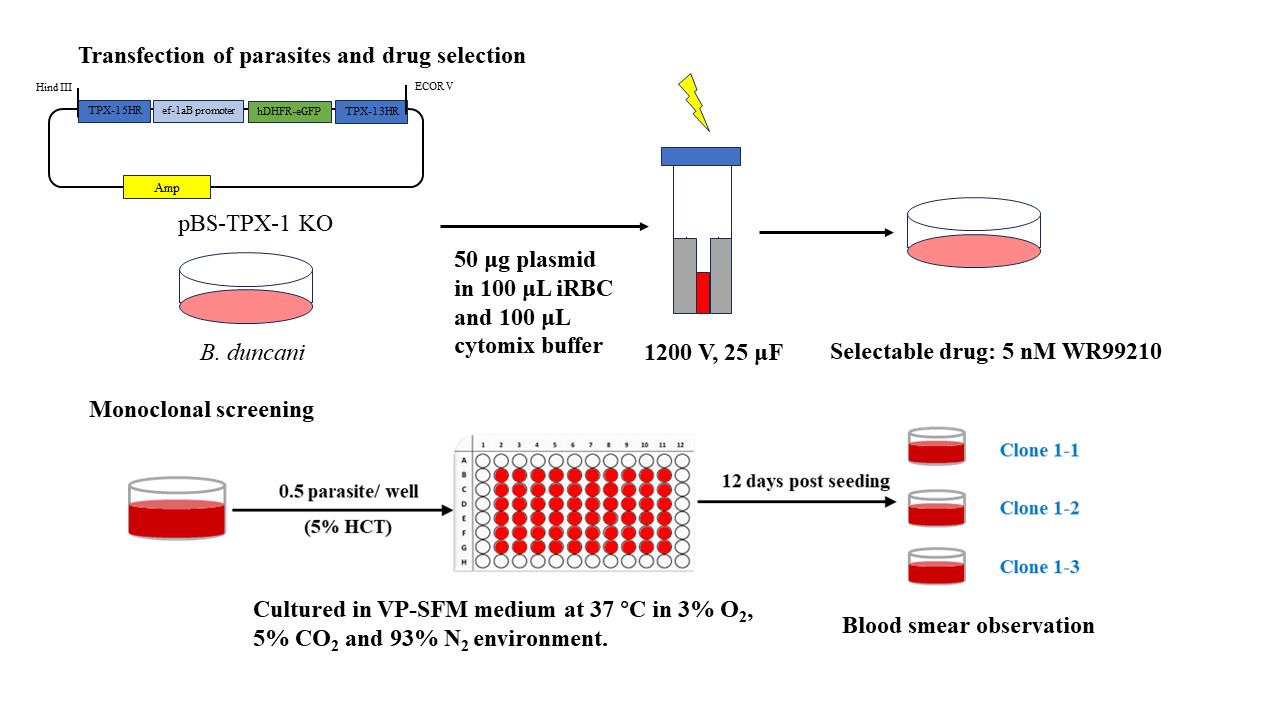
Transfection of B. duncani
Obtaining infected red blood cells: Three days prior to transfection, prepare 100 μL of B. duncani with an initial parasitemia of 1%. It is advisable to change the culture medium daily. On the day of transfection, the parasitemia should be between 15% and 20%.
(Optional) On the third day of culture, when the parasitemia is at 15%–20%, change the medium to one containing 1 μM of MBP146–78 and continue culturing at 37 for 12–24 h. This step is intended to synchronize the B. duncani parasites to the tetrad stage, which increases their survival rate after electroporation.
(CRITICAL STEP) Carefully remove the supernatant medium and resuspend 100 μL of infected red blood cells in 1 mL of cytomix buffer. Centrifuge the mixture at 1,500× g for 2 min at 4 and discard the supernatant buffer. Repeat this washing step twice with cytomix buffer, discarding the supernatant after each wash.
Prepare a 24-well plate and add 50 μL of red blood cells and 950 μL of culture medium. Place the 24-well plate in a 37 °C incubator and keep it aside.
Dissolve the DNA precipitate obtained in step A18 with 200 μL of cytomix buffer and take up 10 μL of the DNA mixture. Measure the DNA concentration by NanoDrop2000.
Combine the harvested parasite-infected red blood cells, DNA from step D5, and cytomix buffer in a sterile microcentrifuge tube. Prepare the electroporation mixture according to Table 7.
Table 7. Electroporation transfection for B. duncani
Component Amount (μL) Final concentration Parasite-infected red blood cells 100 DNA 50 μg 0.25 μg/μL Cytomix buffer Add to 200 total 200 Gently mix the contents and transfer the mixture to a 0.2 cm electroporation cuvette, ensuring there are no air bubbles. Keep the cuvette on ice.
Set the BTX electroporator to the following settings: 1,200 V, 25 μF, 200 Ω, and prepare for electroporation.
Place the electroporation cuvette in the electroporation apparatus. After clicking on Ω, the screen will display the set transfection conditions (refer to Figure 3A). Then, click on GO to initiate the electroporation. The screen will show the actual electroporation conditions during the process (refer to Figure 3B). The resulting time constant should range between 0.4 and 0.7 ms.
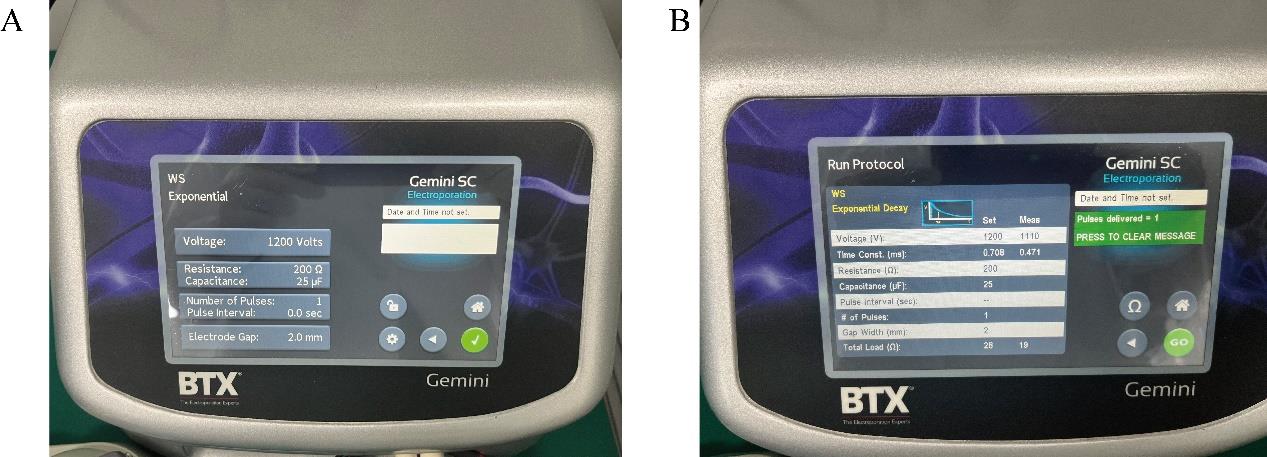
Figure 3. Representative image for electroporation. (A) Electroporation protocol setup. (B) Electroporation results.Repeat the operation described in step D9 for a second round of electroporation.
Note: Before each pulse, gently tap the electroporation cuvette to remove any generated air bubbles. During the second electroporation, splashing of the mixture might occur.
After completing the electroporation, transfer the whole mixture to the wells prepared in step D4 by transfer pipette (10–200 μL), gently mix, and place it in a cell culture incubator.
After 3 h following transfection, replace the prewarmed medium.
Genetic modification strain screening
Twenty-four hours after transfection, replace the culture medium with a medium containing 5 nM WR99210. Prepare blood smears as described in step C3 of B. duncani culture to calculate the transfection efficiency.
Subsequently, change the medium containing 5 nM WR99210 daily and add 25 μL of fresh red blood cells on the seventh day post-transfection.
After 7–12 days of drug selection, resuspend the culture and transfer 100 μL to a new well. Then, add 45 μL of uninfected red blood cells and 855 μL of medium.
(Optional) Withdraw 2 μL of the parasitized red blood cells for live cell fluorescence microscopy to confirm the expression of the fluorescent protein that co-expresses with the drug selection marker. For live-cell imaging, the parasite-infected blood was first washed twice with PBS using a centrifuge set at 1500× g for 2 min each time. The cells were then stained with 1 μg/mL Hoechst 33342 in PBS for a duration of 5 min. All images were captured and processed using identical settings in the OLYMPUS FRAME_BX63 scanning confocal microscope with a 100× numerical-aperture (NA) oil objective.
Note: Live cell fluorescence is optional. The expression of the fluorescent protein does not necessarily confirm the successful construction of the genetically modified parasite strain. Further confirmation through PCR is required.
Extract the remaining DNA from the parasitized red blood cells using the TIANamp Genomic DNA kit following the manufacturer's instructions.
Design primers that anneal to the flanking regions of interest. Confirm the presence of the desired genetic modification using PCR identification.
Note: To prevent false positives caused by plasmid DNA, set the forward primer approximately 200 bp upstream of 5H and the reverse primer approximately 200 bp downstream of 3H.
Analyze 5–10 μL of each PCR reaction along with an appropriate ladder (GeneRuler 100 bp DNA ladder) on a 1%–2.5% (wt/vol) agarose gel prepared in 1× TAE buffer and stained with ethidium bromide. Run the gel at 120 V for at least 30 min and visualize the size of the PCR product using the Gel Jet Documentation System.
Note: PCR1 is used to confirm the correct recombination of the 5’ end of the insert fragment with the genome, PCR2 is used to confirm the correct insertion of the 3’ end of the insert fragment, and PCR3 is used to confirm the deletion of the target gene. Refer to Figure 5B to see the presence of bands of varying sizes in the modified parasite strains, including those of the same size as the WT.
Monoclonal screening
Due to the variability in drug selection efficiency, achieving stable genetically modified parasite strains solely through drug selection is challenging. Therefore, clonal selection is necessary to obtain stable genetically modified parasite strains.
Cultivate the strains with the correct insertion (as confirmed by PCR in steps E5–E7 above) until the parasitemia reaches 5%.
Centrifuge the culture at 1,500× g for 5 min.
Take 1 μL of the red blood cell pellet (approximately 1 × 107 cells) and resuspend it in 1 mL of PBS. Mix well and use a cell counter to determine the cell count.
Dilute the parasitized red blood cells with PBS to achieve a concentration of 100 red blood cells per microliter (with approximately five parasites per microliter).
Add 6 mL of culture medium without WR99210 and 300 μL of fresh uninfected red blood cells to a 15 mL centrifuge tube and mix well.
Add 6 μL of the diluted solution (containing the parasite-infected red blood cells) to the mixture of culture medium and red blood cells, to achieve a total of 30 parasites in the mixture.
After mixing the solution, add 100 μL of the mixture to each well of a 96-well cell culture plate to achieve a concentration of 0.5 parasites per well.
Change the medium every three days, replacing 70 μL of the medium.
On the second medium change, use a culture medium containing 5% red blood cells.
On the fourth medium change, perform blood smears to observe the Babesia parasites.
CRITICAL STEP: Typically, around 20 wells out of the 60 should contain parasites. If more than 30 wells yield parasites, it is likely that the obtained parasites are not monoclonal.
Upon observing that the monoclonal parasite strains have grown to a parasitemia of 5%, mix the culture well and then transfer 10 μL to a new 96-well plate. Add 90 µL of culture medium containing 5% red blood cells to each well.
Place the clonal parasite strains in a cell culture incubator for further cultivation.
Transfer the remaining 90 μL of monoclonal culture medium to a clean PCR tube. Centrifuge for 2 min using a microcentrifuge.
Discard the supernatant and retain the red blood cells.
Add 100 μL of PBS containing 0.1% saponin to lyse the red blood cells for 5 min.
Centrifuge the Babesia parasites using a microcentrifuge for 5 min to pellet them at the bottom of the tube.
Discard the supernatant and add 20 μL of nuclease-free water. Mix well, then boil the mixture in a water bath for 10 min.
Centrifuge the boiled sample in a microcentrifuge for 5 min. The resulting supernatant contains the DNA of the clonal parasite strain.
To confirm that the clonal parasite strain has the correct insertion, perform three PCR reactions. Use the setup and thermal cycler program for the expected size of the mutant or the WT PCR fragment. Include a colony of the WT strain as a nonedited control.
Analyze 5–10 μL of each PCR reaction along with an appropriate ladder (GeneRuler 100 bp DNA ladder) on a 1%–2.5% (wt/vol) agarose gel prepared in 1× TAE buffer and stained with EB. Run the gel at 120 V for at least 30 min and visualize the size of the PCR product using the Gel Jet Documentation System.
Sequencing of PCR products of the expected size is conducted to confirm the correct insertion.
Transfer the parasite strain with the desired genetic modification from the 96-well plate to a new 24-well cell culture plate for amplification.
Freeze single clone strains when the parasitemia is at 20%. The method for freezing B. duncani is as follows:
Take the mixture of culture medium and red blood cells and transfer it to a new 1.5ml Eppendorf tube.
Use a centrifuge at 1,500× g for 2 min at 4 to separate the supernatant.
Carefully remove the supernatant to avoid losing the cell pellet. Add 100 µL of freezing solution to the pellet and mix to ensure an even distribution.
Transfer the mixture to a new cryogenic storage tube.
Place the storage tube in a Mr. Frosty™ Freezing container set at -80 °C to gradually decrease the temperature over two days. This helps prevent cell damage.
After completion of the freezing process, immediately transfer the storage tube to a liquid nitrogen container for long-term preservation of B. duncani parasites.
This method can be used to freeze B. duncani parasites for future experiments. In liquid nitrogen, the parasites can be stored for an extended period. However, it is essential to follow the appropriate biosafety and laboratory protocols to ensure the safe storage and handling of the samples.
General notes and troubleshooting
Expanding B. duncani for transfection
Because the transfection method for B. duncani results in a high mortality rate, increasing the parasite's infection rate can enhance the number of parasites post-transfection. By using VP-SFM medium and hamster red blood cells, the parasitemia of B. duncani can be raised to over 30%. Increasing the initial parasitemia to 2%–4% during passages ensures that sufficient parasitemia can be obtained for transfection by the third day.
Improve the post-electroporation survival rate of Babesia parasites
Electric transfection of B. duncani employs high voltage and low pulse times, which results in a substantial loss of parasites after electroporation. The reasons for parasite death include both the stimulus from high voltage and, significantly, the rupture of red blood cells caused by electroporation. As a result, only fully developed tetrad-stage parasites can continue to grow. Thus, increasing the proportion of tetrad-stage Babesia parasites is essential to enhance their survival rate following electroporation. Given that Babesia replication is asynchronous, obtaining synchronized tetrad-stage parasites through regular culturing is challenging. While increasing parasitemia does raise the proportion of tetrads to some extent, the impact on post-electroporation survival is not significant. Babesia parasites egress via the cGMP pathway, and the use of a cGMP-dependent protein kinase (PKG) inhibitor can block egress, allowing for the synchronization of Babesia parasites to the tetrad stage. Compound C1 (MBP146-78) is a known PKG inhibitor, and its effect is reversible; removing C1 permits the normal growth of the parasites. The use of compound C1 to treat B. duncani synchronizes them to the tetrad stage, increasing post-electroporation survival rates from 2.5%–5% to 10%–15%, significantly improving subsequent screening efficiency.
Data analysis
Live-cell imaging for B. duncani
Fusing the drug selection marker with GFP allows for the observation of drug selection marker expression in B. duncani parasites using fluorescence microscopy (refer to Figure 4). Through fluorescence microscopy, the expression of the green fluorescent protein in the cytoplasm of B. duncani parasites can be detected. It is important to note that observing the expression of the fluorescent protein does not guarantee that the drug selection marker has recombined correctly as expected; there may still be cases of single-side homologous recombination.
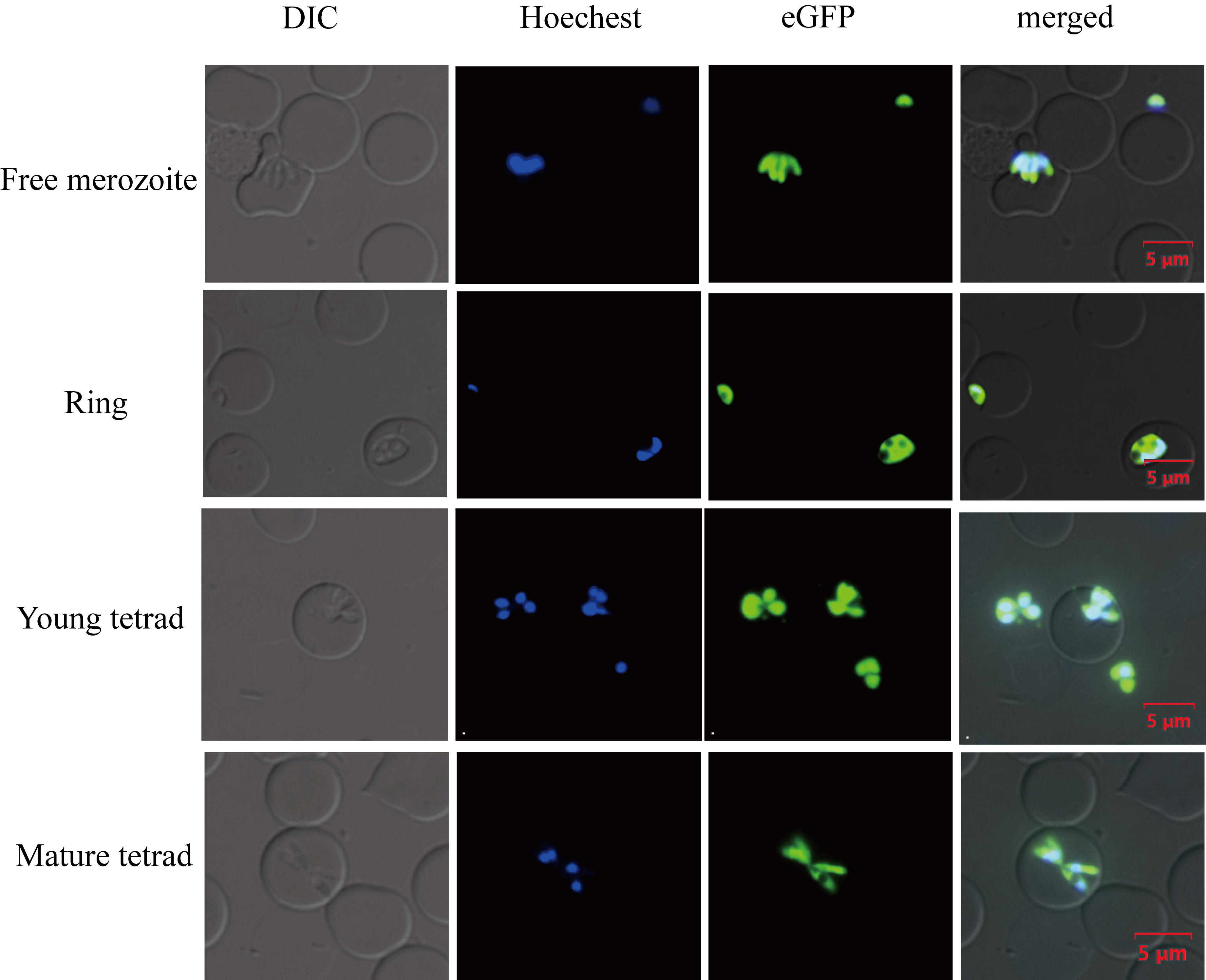
Figure 4. Live-cell imaging for B. duncani. Green fluorescence corresponds to the transfected parasite expressing eGFP, Hoechst staining represents the nucleus of the parasite, and DIC (differential interference contrast microscope) image shows a parasitized red blood cell (RBC). Merged image represents the overlap of all images. Scale bar = 5 μm.Confirming gene modifications through PCR
PCR is particularly useful for detecting the presence of homologous integration events using a combination of a plasmid-specific oligonucleotide (not specific to the gene-targeting sequence) and one directed to the genomic sequence located immediately outside of the gene-targeting fragment found in the plasmid. To confirm the correct insertion of the drug selection marker into the target gene locus, amplify the upstream and downstream sequences connected to the drug selection marker. The presence of such a product (which should be sequenced for confirmation) demonstrates that homologous integration has indeed occurred.
Screen the selected monoclonal parasite strains through PCR1 and PCR2 to confirm the precise insertion of the drug selection marker into the TPX-1 gene locus in B. duncani. Additionally, PCR3 is used to verify the deletion of the TPX-1 gene (refer to Figure 5).
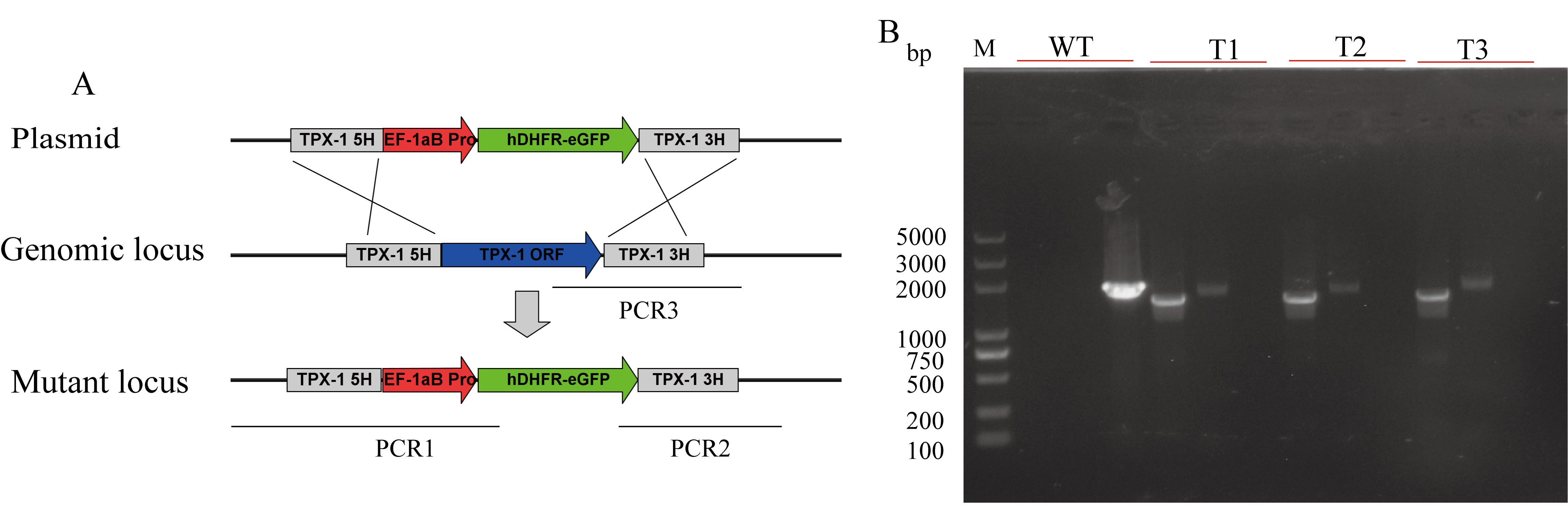
Figure 5. Targeted disruption of the B. duncani TPX-1 gene. A. Plasmid construct for disruption of the B. duncani TPX-1 gene. B. PCR confirmation of the disruption of the B. duncani TPX-1 gene. Monoclonal strains T1, T2, and T3 were identified by PCR1, PCR2, and PCR3, with WT strain used as the control.
Limitations
Optimizing the transfection efficiency in intracellular parasites like B. duncani can be challenging due to the need for external DNA to traverse both the red blood cell and the parasite's cell membrane. Achieving a transfection efficiency of 0.5%–1% through optimization is a notable accomplishment. However, it is important to consider that the use of higher voltage can reduce the post-transfection survival rate of the parasites, typically to approximately 2.5%–5%. This lower survival rate can prolong the time needed to select edited parasite strains, often requiring approximately one month. These challenges highlight the need for continued refinement and optimization of transfection methods for this parasite.
In addition, this protocol specifically outlines a method for gene editing through homologous recombination. To further advance research on genes that are crucial for the growth and development of B. duncani, it would be beneficial to develop conditional knockout systems such as Di-Cre. These conditional knockout systems can allow for the precise control and study of genes that are vital to the life cycle of the parasite.
Validation of protocol
This protocol or parts of it has been used and validated in the following research article(s):
Wang, S. et al. (2022). Establishment of a Transient and Stable Transfection System for Babesia duncani Using a Homologous Recombination Strategy. Front Cell Infect Microbiol.
Acknowledgments
Funding was provided by grant 2022YFD1801700 from the National Key Research and Development Program of China, grants 32172879 and 31930108 from the National Natural Science Foundation of China, funds from the Top-notch Young Talent Supporting Program (L.H.), and grant 2262022DKYJ001 from the Fundamental Research Funds for the Central Universities. We also sincerely appreciate Choukri Ben Mamoun from Department of Infectious Diseases, School of Medicine, Yale University, for his help in editing the manuscript.
Ethical considerations
This study was approved by the Scientific Ethic Committee of Huazhong Agricultural University (permit number: HZAUMO-2017-040). All mice were handled in accordance with the Animal Ethics Procedures and Guidelines of the People’s Republic of China.
References
- Suarez, C. E. and McElwain, T. F. (2010). Transfection systems for Babesia bovis: a review of methods for the transient and stable expression of exogenous genes. Vet. Parasitol. 167(2–4): 205–215. https://doi.org/10.1016/j.vetpar.2009.09.022.
- Yabsley, M. J. and Shock, B. C. (2013). Natural history of Zoonotic Babesia: Role of wildlife reservoirs. Int. J. Parasitol. Parasites Wildl 2: 18–31. https://doi.org/10.1016/j.ijppaw.2012.11.003.
- Renard, I. and Ben Mamoun, C. (2021). Treatment of Human Babesiosis: Then and Now. Pathogens 10(9). https://doi.org/10.3390/pathogens10091120.
- Vannier, E. G., Diuk-Wasser, M. A., Ben Mamoun, C. and Krause, P. J. (2015). Babesiosis. Infect. Dis. Clin. North. Am. 29(2): 357–370. https://doi.org/10.1016/j.idc.2015.02.008.
- Virji, A. Z., Thekkiniath, J., Ma, W., Lawres, L., Knight, J., Swei, A., Roch, K. L. and Mamoun, C. B. (2019). Insights into the evolution and drug susceptibility of Babesia duncani from the sequence of its mitochondrial and apicoplast genomes. Int. J. Parasitol. 49(2): 105–113. https://doi.org/10.1016/j.ijpara.2018.05.008.
- Wozniak, E. J., Lowenstine, L. J., Hemmer, R., Robinson, T. and Conrad, P. A. (1996). Comparative pathogenesis of human WA1 and Babesia microti isolates in a Syrian hamster model. Lab. Anim. Sci. 46(5): 507–515.
- Chiu, J. E., Renard, I., Pal, A. C., Singh, P., Vydyam, P., Thekkiniath, J., Kumar, M., Gihaz, S., Pou, S., Winter, R. W., et al. (2021). Effective Therapy Targeting Cytochrome bc(1) Prevents Babesia Erythrocytic Development and Protects from Lethal Infection. Antimicrob. Agents Chemother. 65(9): e0066221. https://doi.org/10.1128/AAC.00662-21.
- Pal, A. C., Renard, I., Singh, P., Vydyam, P., Chiu, J. E., Pou, S., Winter, R. W., Dodean, R., Frueh, L., Nilsen, A. C., et al. (2022). Babesia duncani as a Model Organism to Study the Development, Virulence, and Drug Susceptibility of Intraerythrocytic Parasites In Vitro and In Vivo. J. Infect. Dis. 226(7): 1267–1275. https://doi.org/10.1093/infdis/jiac181.
- Singh, P., Lonardi, S., Liang, Q., Vydyam, P., Khabirova, E., Fang, T., Gihaz, S., Thekkiniath, J., Munshi, M., Abel, S., et al. (2023). Babesia duncani multi-omics identifies virulence factors and drug targets. Nat. Microbiol. 8(5): 845–859. https://doi.org/10.1038/s41564-023-01360-8.
- Villatoro, T. and Karp, J. K. (2019). Transfusion-Transmitted Babesiosis. Arch. Path. Lab. 143(1): 130–134. https://doi.org/10.5858/arpa.2017-0250-RS.
- Suarez, C. E., Bishop, R. P., Alzan, H. F., Poole, W. A. and Cooke, B. M. (2017). Advances in the application of genetic manipulation methods to apicomplexan parasites. Int. J. Parasitol. Parasites Wildl. 47(12): 701–710. https://doi.org/10.1016/j.ijpara.2017.08.002.
- Suarez, C. E. and Noh, S. (2011). Emerging perspectives in the research of bovine babesiosis and anaplasmosis. Vet. Parasitol. 180(1–2): 109–125. https://doi.org/10.1016/j.vetpar.2011.05.032.
- Adamson, R., Lyons, K., Sharrard, M., Kinnaird, J., Swan, D., Graham, S., Shiels, B. and Hall, R. (2001). Transient transfection of Theileria annulata. Mol. Biochem. Parasitol. 114(1): 53–61. https://doi.org/10.1016/s0166-6851(01)00238-9.
- Swensen, J. S., Xiao, Y., Ferguson, B. S., Lubin, A. A., Lai, R. Y., Heeger, A. J., Plaxco, K. W. and Soh, H. T. (2009). Continuous, real-time monitoring of cocaine in undiluted blood serum via a microfluidic, electrochemical aptamer-based sensor. J. Am. Chem. Soc. 131(12): 4262–4266. https://doi.org/10.1021/ja806531z.
- Asada, M., Tanaka, M., Goto, Y., Yokoyama, N., Inoue, N. and Kawazu, S. (2012). Stable expression of green fluorescent protein and targeted disruption of thioredoxin peroxidase-1 gene in Babesia bovis with the WR99210/dhfr selection system. Mol. Biochem. Parasitol. 181(2): 162–170. https://doi.org/10.1016/j.molbiopara.2011.11.001.
- De Goeyse, I., Jansen, F., Madder, M., Hayashida, K., Berkvens, D., Dobbelaere, D. and Geysen, D. (2015). Transfection of live, tick derived sporozoites of the protozoan Apicomplexan parasite Theileria parva. Veterinary parasitology 208(3–4): 238–241. https://doi.org/10.1016/j.vetpar.2015.01.013.
- Hakimi, H., Yamagishi, J., Kegawa, Y., Kaneko, O., Kawazu, S. and Asada, M. (2016). Establishment of transient and stable transfection systems for Babesia ovata. Parasit Vectors 9: 171. https://doi.org/10.1186/s13071-016-1439-z.
- Liu, M., Adjou Moumouni, P. F., Asada, M., Hakimi, H., Masatani, T., Vudriko, P., Lee, S. H., Kawazu, S. I., Yamagishi, J. and Xuan, X. (2018). Establishment of a stable transfection system for genetic manipulation of Babesia gibsoni. Parasit Vectors 11(1): 260. https://doi.org/10.1186/s13071-018-2853-1.
- Mamoun, C. B., Gluzman, I. Y., Goyard, S., Beverley, S. M. and Goldberg, D. E. (1999). A set of independent selectable markers for transfection of the human malaria parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. U S A 96(15): 8716–8720. https://doi.org/10.1073/pnas.96.15.8716.
- Wang, J., Wang, X., Guan, G., Yang, J., Liu, J., Liu, A., Li, Y., Luo, J. and Yin, H. (2021). Stable transfection system for Babesia sp. Xinjiang. Parasit. Vectors 14(1): 463. https://doi.org/10.1186/s13071-021-04940-x.
- Jaijyan, D. K., Govindasamy, K., Singh, J., Bhattacharya, S. and Singh, A. P. (2020). Establishment of a stable transfection method in Babesia microti and identification of a novel bidirectional promoter of Babesia microti. Sci. Rep. 10(1): 15614. https://doi.org/10.1038/s41598-020-72489-3.
- Cornillot, E., Hadj-Kaddour, K., Dassouli, A., Noel, B., Ranwez, V., Vacherie, B., Augagneur, Y., Bres, V., Duclos, A., Randazzo, S., et al. (2012). Sequencing of the smallest Apicomplexan genome from the human pathogen Babesia microti. Nucleic. Acids Res. 40(18): 9102–9114. https://doi.org/10.1093/nar/gks700.
- Garg, A., Stein, A., Zhao, W., Dwivedi, A., Frutos, R., Cornillot, E. and Ben Mamoun, C. (2014). Sequence and annotation of the apicoplast genome of the human pathogen Babesia microti. PLoS One 9(10): e107939. https://doi.org/10.1371/journal.pone.0107939.
- Abraham, A., Brasov, I., Thekkiniath, J., Kilian, N., Lawres, L., Gao, R., DeBus, K., He, L., Yu, X., Zhu, G., et al. (2018). Establishment of a continuous in vitro culture of Babesia duncani in human erythrocytes reveals unusually high tolerance to recommended therapies. J. Biol. Chem. 293(52): 19974–19981. https://doi.org/10.1074/jbc.AC118.005771.
- McCormack, K. A., Alhaboubi, A., Pollard, D. A., Fuller, L. and Holman, P. J. (2019). In vitro cultivation of Babesia duncani (Apicomplexa: Babesiidae), a zoonotic hemoprotozoan, using infected blood from Syrian hamsters (Mesocricetus auratus). Parasitol. Res. 118(8): 2409–2417. https://doi.org/10.1007/s00436-019-06372-0.
- Jiang, W., Wang, S., Li, D., Zhang, Y., Luo, W., Zhao, J. and He, L. (2023). Continuous In Vitro Culture of Babesia duncani in a Serum-Free Medium. Cells 12(3). https://doi.org/10.3390/cells12030482.
- Kumari, V., Pal, A. C., Singh, P. and Mamoun, C. B. (2022). Babesia duncani in Culture and in Mouse (ICIM) Model for the Advancement of Babesia Biology, Pathogenesis, and Therapy. Bio Protoc 12(22). https://doi.org/10.21769/BioProtoc.4549.
- Wang, S., Li, D., Chen, F., Jiang, W., Luo, W., Zhu, G., Zhao, J. and He, L. (2022). Establishment of a Transient and Stable Transfection System for Babesia duncani Using a Homologous Recombination Strategy. Front Cell Infect. Microbiol. 12: 844498. https://doi.org/10.3389/fcimb.2022.844498.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Wang, S., Wang, J., Li, D., Chen, F., Luo, W., Zhao, J. and He, L. (2024). Transfection of Babesia duncani: A Genetic Toolbox of This Pathogen to Advance Babesia Biology. Bio-protocol 14(12): e5016. DOI: 10.21769/BioProtoc.5016.
Category
Microbiology > Microbial genetics > Genome editing
Molecular Biology > DNA > Transfection
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.