- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CRISPR-Cas9 Protocol for Efficient Gene Knockout and Transgene-free Plant Generation

Published: Vol 14, Iss 11, Jun 5, 2024 DOI: 10.21769/BioProtoc.5012 Views: 3446
Reviewed by: Noelia ForesiPooja VermaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

May 2023

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
Gene editing technologies have revolutionized plant molecular biology, providing powerful tools for precise gene manipulation for understanding function and enhancing or modifying agronomically relevant traits. Among these technologies, the CRISPR-Cas9 system has emerged as a versatile and widely accepted strategy for targeted gene manipulation. This protocol provides detailed, step-by-step instructions for implementing CRISPR-Cas9 genome editing in tomato plants, with a specific focus in generating knockout lines for a target gene. For that, the guide RNA should preferentially be designed within the first exon downstream and closer to the start codon. The edited plants obtained are free of transgene cassette for expression of the CRISPR-Cas9 machinery.
Key features
• Two sgRNAs employed.
• Takes 6–12 months to have an edited transgene-free plant.
• Setup in tomato.
Keywords: CRISPR-Cas9Graphical overview
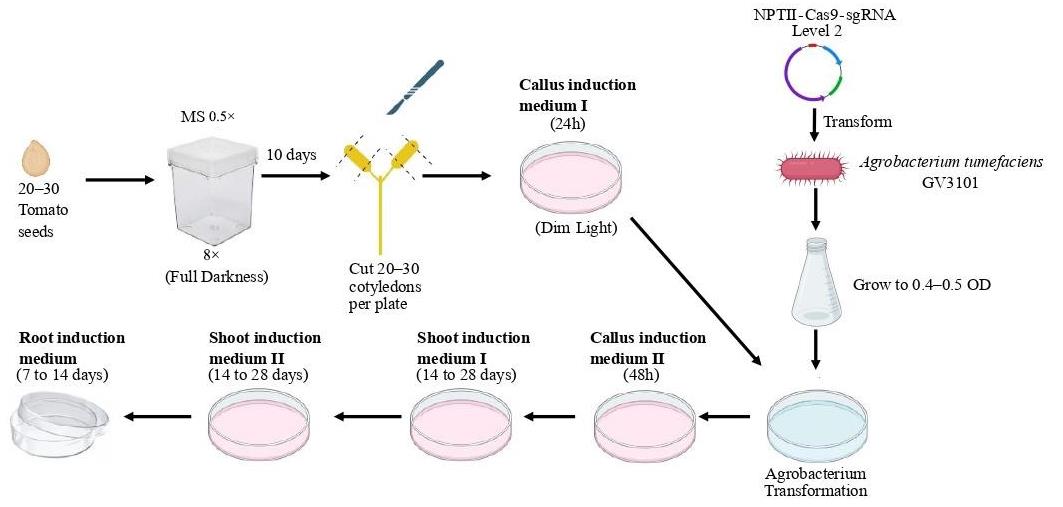
Agrobacterium-mediated transformation and plant regeneration in Solanum lycopersicum for gene editing by CRISPR-Cas9 method.
Background
Tomato (Solanum lycopersicum) is an important model organism for crop improvement studies. CRISPR-Cas9 provides an effective tool for uncovering the function complexity of tomato genes. The main objective of this protocol is to establish a robust strategy for the production of knockout lines in tomato plants. Knockouts involve targeted disruption of a specific gene, resulting in loss of function. This specific variation allows researchers to examine the role of individual genes in a variety of biological processes, from development to environmental responses. It could also be used for plant genetic improvement. To achieve disruption of target genes, the CRISPR-Cas9 system utilizes small guide RNAs (sgRNAs) that direct the Cas9 endonuclease to specific genomic regions. In this system, sgRNAs have been carefully designed to target a region preferably within the first exon downstream of the ATG codon of the gene of interest. To improve efficiency, two sgRNAs are used in this protocol. The selection of sgRNAs is based on their specificity, avoiding off-targets, and ensuring efficiency. The protocol outlines a detailed workflow, including cloning of sgRNAs, assembly of expression cassettes in a vector, and Escherichia coli and Agrobacterium transformation. Then, we provide a detailed protocol for tomato transformation and screening of edited plants in order to generate homozygous edited plants free of transgene. We rely on works previously published by Nekrasov et al. [1] and Van Eck et al. [2], along with our own personal experience [3], to create a comprehensive and effective protocol for generating knock-out plants in tomato in 6–12 months. The protocol was used to demonstrate that phospholipase C2 knock-out tomato plants are more resistant to Botrytis cinerea than wild-type plants, with less ROS, an increase in jasmonic acid, and a reduction in salicylic acid–response marker genes [3].
Materials and reagents
Biological materials
Escherichia coli DH5α (Thermo Fisher, catalog number: EC0112)
Tomato (Solanum lycopersicum cv. MoneyMaker Cf-0) (in-house propagated)
Agrobacterium tumefaciens, strain: GV3101 (GoldBio, catalog number: CC-207-5x50)
pZG23C04 commercial vector (ZGene Biotech Inc, Taibei, Taiwan)
pICSL01009::AtU6p was a gift from Sophien Kamoun (Addgene plasmid #46968; http://n2t.net/addgene:46968; RRID: Addgene_46968)
pICH47751 was a gift from Sylvestre Marillonnet (Addgene plasmid #48002; http://n2t.net/addgene:48002; RRID: Addgene_48002)
pICH47761 was a gift from Sylvestre Marillonnet (Addgene plasmid #48003; http://n2t.net/addgene:48003; RRID: Addgene_48003)
pICSL11024 (pICH47732::NOSp-NPTII-OCST) was a gift from Jonathan D Jones (Addgene plasmid #51144; http://n2t.net/addgene:51144; RRID: Addgene_51144)
pICH47742::2x35S-5'UTR-hCas9(STOP)-NOST was a gift from Sophien Kamoun (Addgene plasmid #49771; http://n2t.net/addgene:49771; RRID: Addgene_49771)
pICH41780 was a gift from Sylvestre Marillonnet (Addgene plasmid #48019; http://n2t.net/addgene:48019; RRID: Addgene_48019)
pAGM4723 was a gift from Sylvestre Marillonnet (Addgene plasmid #48015; http://n2t.net/addgene:48015; RRID: Addgene_48015)
Reagents
Agar (Britania, catalog number: B010406)
Agarose (GenBiotech, catalog number: RU1010)
Ampicillin (Amp) (GenBiotech, catalog number: A-0104-5)
Acetosyringone (Sigma-Aldrich, catalog number: D134406)
Bleach
BpiI (BbsI) (Thermo Scientific, catalog number: ER1011)
Buffer G (Thermo Scientific, catalog number: ER1011)
BsaI HF (New England BioLabs, catalog number: R3733S)
DNA Marker 100 bp (Inbio Highway, catalog number: K0177)
DNA Marker 100 bp (PBL, catalog number: MA0201)
DNA Marker Lambda/HindIII (Inbio Highway, catalog number: K0180)
Ethanol 96%
Plastic centrifuge tube, 15 and 50 mL (Henso Medical)
Gentamicin (GenBiotech, catalog number: G400-50)
HindIII (Promega, catalog number: R6041)
Buffer E (Promega, catalog number: R6041)
IPTG (GenBiotech, catalog number: I2481C5)
IAA (Merck, catalog number: 353)
Kanamycin (GenBiotech, catalog number: K0126-5)
Kinetin (Sigma, catalog number: K0753)
Kit dNTPS (Inbio Highway, catalog number: K1404)
Magenta boxes, W × L × H: 77 mm × 77 mm × 97 mm (V8505 MagentaTM vessel)
MS + Gamborg B5 vitamins (Duchefa-Biochemi, catalog number: P1278801)
NaCl (J.T. Baker, catalog number: 3624-i9)
PCR purification kit (Inbio Highway, catalog number: K1206)
Phytoagar (Sigma, catalog number: P8169)
Purification plasmid DNA kit (Qiagen, catalog number: 12123)
Rifampicin (GenBiotech, catalog number: R-120-1)
Sterile water
Sucrose (Cicarelli, catalog number: 841214)
Taq DNA polymerase (INBIO HIGHWAY, catalog number: K1007)
Taq DNA Buffer (INBIO HIGHWAY, catalog number: K1007)
MgCl2 (INBIO HIGHWAY, catalog number: K1007)
T4 ligase (Thermo Fisher, catalog number: EL0011)
T4 ligase buffer (Thermo Fisher, catalog number: EL0011)
Thiamine HCl (Sigma, catalog number: T-3902)
Timentin (GoloBio, catalog number: T-104-25)
Pipette tips (Henso Medical)
Tryptone (OXOID, catalog number: LP0042B)
X-Gal (GenBiotech, catalog number: I4281C5)
Yeast extract (OXOID, catalog number: LP0021)
2,4-D (Sigma, catalog number: D7299)
L trans-Zeatin (Golobio, catalog number: Z-105-50)
Eppendorf (Henso Medical)
Primers
Cas9_6F: 5' ACTAGCCTTGTGGCCCTACC 3'
Cas9_6R: 5' TCGATCTAGTAACATAGATGACACC 3'
RB_F1: 5' GGATAAACCTTTTCACGCCC 3'
Solutions
½ MS (see Recipes)
CIM I (see Recipes)
CIM II (see Recipes)
SIM I (see Recipes)
SIM II (see Recipes)
RIM (see Recipes)
LB (see Recipes)
Recipes
½ MS
2.15 g/L MS + Gamborg B5 vitamins
10 g/L sucrose
8 g/L agar
pH = 5.8
CIM I
4.3 g/L MS + Gamborg B5 vitamins
30 g/L sucrose
5.2 g/L Phytoagar
Sterilize and add 1 mg/L thiamine HCl, 1 mg/L 2,4-D, and 0.2 mg/L kinetin.
CIM II
4.3 g/L MS + Gamborg B5 vitamins
30 g/L sucrose
5.2 g/L Phytoagar
Sterilize and add 1 mg/L thiamine HCl, 1 mg/L 2,4-D, 0.2 mg/L Kinetin, and 200 μM acetosyringone
SIM I
4.3 g/L MS + Gamborg B5 vitamins
30 g/L sucrose
5.2 g/L Phytoagar
Sterilize and add 1 mg/L thiamine HCl, 2 mg/L trans-Zeatin, 100 mg/L kanamycin, and 250 mg/L timentin.
SIM II
4.3 g/L MS + Gamborg B5 vitamins
30 g/L sucrose
5.2 g/L Phytoagar
Sterilize and add 1 mg/L thiamine HCl, 1 mg/L trans-Zeatin, 100 mg/L kanamycin, 250 mg/L timentin, and 0.1 mg/L IAA.
RIM
4.3 g/L MS + Gamborg B5 vitamins
30 g/L sucrose
5.2 g/L Phytoagar
Sterilize and add 50 mg/L kanamycin, 250 mg/L timentin, and 1 mg/L IAA.
LB
10 g/L tryptone
5g/L NaCl
5g/L yeast extract
Laboratory supplies
Petri dishes (9 cm × 1.5 cm) (Biopetri)
Petri dishes (9 cm × 2.5 cm) (Biopetri)
Scalpel
Equipment
37 °C oven (SAN JOR, model: SE60A)
E. coli pulser transformation apparatus (Bio-Rad, model: 155103)
Heat dry bath (Benchmark, model: BSH1001-E)
Veriti 96-well thermal cycler (Applied Biosystems, model: 9902)
DNA electrophoresis apparatus (Bio-Rad, model: 55656)
Microcentrifuge Sorvall Legend Micro 17R (Thermo Fisher Scientific, model: 75002440)
30 °C oven (SAN JOR, model: SL300)
Orbital Shaker Thermo Forma (Thermo Fisher Scientific, model: 320REL)
PIPETMAN L Starter Kit, 4 Pipette Kit, P2L, P20L, P200L, P1000L (Gilson, model: F167370)
Cultivation room at 25 °C, photoperiod 16:8 h light/dark
Software and datasets
CRISPR2 (http://crispr.hzau.edu.cn/CRISPR2)
Cas-OFFinder (http://www.rgenome.net/cas-offinder/)
Solgenomics (https://solgenomics.net/)
Bioedit sequence alignment editor
Procedure
Design small guide RNAs (sgRNAs) for the target gene
Search for the DNA sequence of interest in the Sol Genomics database.
Design two sgRNAs in the first exon using the web page CRISPR-P 2.0 and Cas-OFFinder for plants (http://crispr.hzau.edu.cn/CRISPR2; http://www.rgenome.net/cas-offinder/). Choose sgRNAs based on specificity (without off-targets for chains of 20 and 12 nt), with 40%–50% GC content, without poly(T) repeats, and with an activity score greater than 0.5. The chosen sgRNA must be a good choice for both online design tools.
Design primers to amplify a region of approximately 600–700 bp where sgRNAs are included (see General note 1).
Check if the selected region shows no allelic variations in the sgRNAs regions. To determine allelic variants, extract DNA from 10 tomato plants, amplify the target region, and sequence it. It is normal for different cultivars to have nucleotide differences from reference genomes.
Level 1 vector assembly (based on Nekrasov et al. [1])
Design primers. To the guide sequence (20 bp in underlined capital letters chosen in section A), add flanking sequences as follows. BsaI site is indicated in italic letters, the transcribed sequence is represented in capital letters.
Forward sgRNA primer:
tgtggtctcaATTNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAG
And a reverse sgRNA primer:
gtcggtctcaAGCGCTCAAGAGGATAAAACCTCAC
Construct plasmid level 1 using Golden Gate cloning.
Perform sgRNAs assembly by using the primers and amplifying with the commercial CRISPR/Cas9 vector pZG23C04 as a template (Tables 1 and 2). The resulting PCR product (210 bp, Figure 1) will have the following sequence (primer sequences in bold, BsaI cutting site in italic letters, the 20 bp guide sequence in underlined capital letters, and transcribed sequence in capital letters).
tgtggtctcaATTNNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTTACTAGTTTTGATCTTGAAAGATCTTTTATCTTTAGAGTTAAGAACTCTTTCGTATTTTGGTGAGGTTTTATCCTCTTGAGCGCTtgagaccgac
Table 1. PCR conditions
Reagent Volume (µL) Template DNA (CRISPR/Cas9 vector pZG23C04, 100 ng/µL) 1 MgCl2(25 mM) 4.2 10× BufferTaq 5 10 mM dNTPs 1.6 Forward primer (10 mM) 2.5 Reverse primer (10 mM) 2.5 Taq DNA Polymerase (5,000 U/mL) 0.5 H2O 32.5 Table 2. PCR program
Step Time, temperature Cycles Initial denaturation 30 s, 98 °C 1 Denaturation 10 s, 98 °C 35 Annealing 30 s, 60 °C Extension 30 s, 72 °C Final extension 3 min, 72 °C 1 Final hold ∞ 4 °C 1 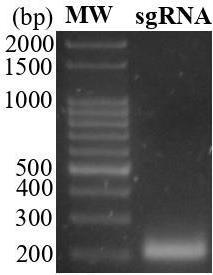
Figure 1. sgRNA verification. Five microliters of the PCR product was loaded onto a 2% agarose gel. MW: 100 bp marker.Run 5 µL of PCR products on a 2% agarose gel.
Desalt 45 µL of PCR products using a PCR purification kit.
Assemble the level-1 sgRNAs expression cassette using Golden Gate Assembly. Cut-ligate the PCR products (sgRNA1 and sgRNA2) into different level-1 vectors, one into pICH47751 (CarbR) and the other into pICH47761 (CarbR) (Tables 3 and 4, Figure 2A). In the same cut-ligation reaction, the sgRNAs are placed under the control of the U6 promoter into a level-1 vector.
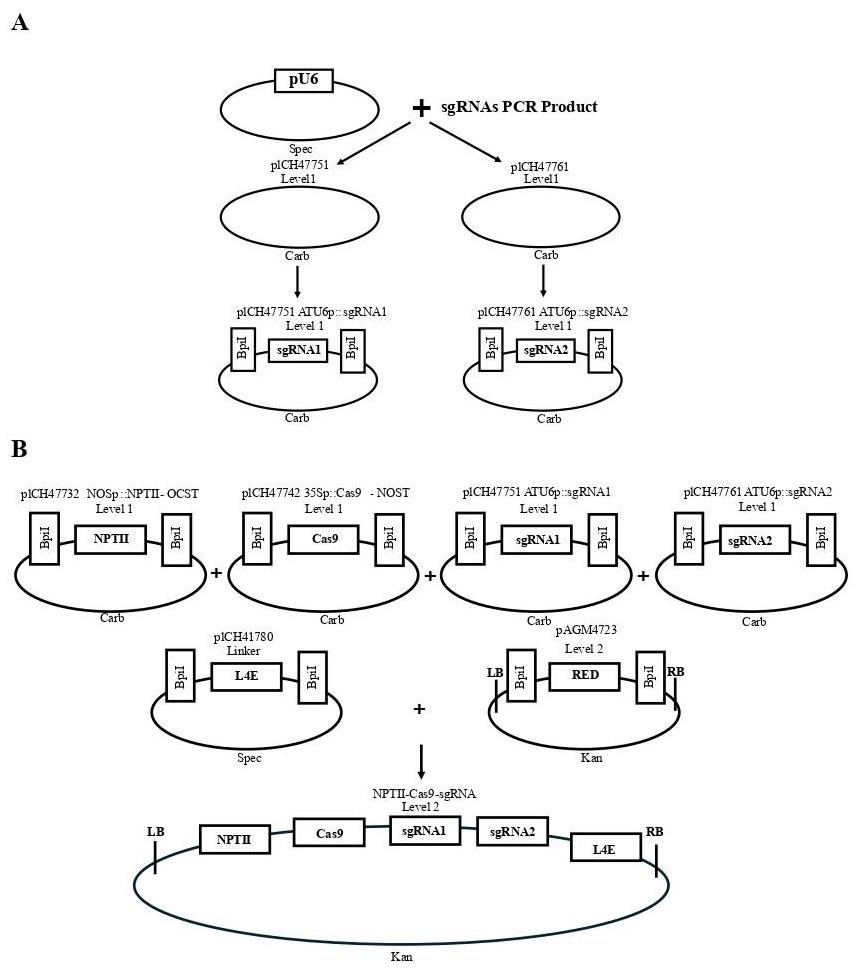
Figure 2. Cloning strategies. A. Insertion of two sgRNAs under the control of the U6 promoter into a level-1 vector. B. Insertion of the U6-sgRNAs, a selectable marker (NPTII), and a Cas9 cassette into a level 2 expression vector.Table 3. Cut-ligation conditions
Reagent Volume 5× T4 ligase buffer 4 µL BsaI-HF (10,000 U/mL) 1 µL pICH47751 or pICH47761 300 ng pICSL01009::AtU6p 100 ng sgRNA PCR desalted (100 µg/µL) 5 µL T4 ligase (5 U/μL) 3 µL H2O to 20 µL Table 4. Assembly reaction
Time, temperature Cycles 5 min, 37 °C 50 5 min, 20 °C 10 min, 50 °C 1 10 min, 80 °C 1 Transform 50 µL of chemically competent E. coli cells with the product from the cut-ligation reactions.
i. Place -80 °C competent E. coli cells on ice for 30 min.
ii. Incubate 50 µL of competent E. coli cells with 10 µL of each cut-ligation product on ice for 20 min.
iii. Heat shock the cells at 42 °C for 45 s.
iv. Add 700 µL of LB.
v. Incubate for 7 min on ice and then 1h at 37 °C.
vi. Plate transformation onto LB plates with 100 µg/mL Amp, 20 mg/mL X-Gal, and 100 µL of 0.1 M IPTG. Incubate at 37 °C overnight.
Pick the white colonies from each cloning reaction and plate them onto the master plate LB-Amp (100 µg/mL). Conduct a colony PCR to check for the presence of sgRNAs. Use the same primer design as in stepB1 and the PCR conditions and program mentioned in step B2 (Tables 2 and 3). Run the PCR products on a 2% agarose gel.
Select a positive colony from each cloning reaction and use it to inoculate 5 mL of LB-Amp. Incubate overnight at 37 °C on a shaker.
Purify each level-1 construct using a Qiagen plasmid DNA purification kit following manufacturer’s protocol.
Check level-1 construction by BpiI digestion at 37 °C for 1 h (Table 5). Run the reaction on a 2% agarose gel; expect to observe a 240 bp band released from the plasmid (Figure 3).
Table 5. Digest conditions
Reagent Volume (µL) H2O 12 10× Buffer G 2 Level 1 construct 5 BpiI (10 U/ µL) 1 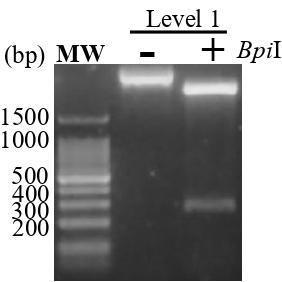
Figure 3. Level-1 construct. The purified level-1 construct was digested or not with BpiI at 37 °C for 1 h and the product was run on 2% agarose gel. -: without BpiI; +: with BpiI; MW: marker 100 bp.Verify the level-1 constructs through sequencing using the RB_F1 primers.
Level 2 assembly
Combine the assembled sgRNAs with a selectable marker (NPTII) and a Cas9 cassette into a level 2 Acceptor (Tables 6 and 7, Figure 2B).
Table 6. Cut-ligation conditions
Reagent Volume 10× T4 ligase buffer 2 µL BpiI (10 U/μL) 1.5 µL plCH47751::U6p::sgRNA1 300 ng plCH47761::U6p::sgRNA2 300 ng pICSL11024 (pICH47732::NOSp-NPTII-OCST) 300 ng pICH47742::2x35S-5’UTR-hCas9(STOP)-NOST 300 ng plCH41780 linker 100 ng pAGM4723 300 ng T4 ligase (5 U/μL) 2 µL H2O to 20 µL Table 7. Assembly reaction
Time, temperature Cycles 5 min, 37 °C 50 5 min, 20 °C 10 min, 50 °C 1 10 min, 80 °C 1 Transform 50 µL of chemically competent E. coli cells with 10 µL of the cut-ligation product as previously described. Select on LB-Kanamycin (Kan) agar plates. Pick five white colonies and inoculate them in 5 mL of LB-Kan overnight culture at 37 °C on a shaker (see General note 2).
Purify the level-2 construct using a Qiagen plasmid DNA purification kit following the manufacturer’s protocol.
Check level-2 construction by HindIII digestion at 37 °C for 1 h (Table 8). Run the reaction on a 2% agarose gel; expect to observe bands around 4 kb, 1.8 kb, 1.4 kb, 500 bp, and 400 bp released from the plasmid (Figure 4). Additionally, verify by sequencing using the RB_F1 primer.
Table 8. Digest conditions
Reagent Volume (µL) H2O 12 10× Buffer E 2 Level 2 construct 5 HindIII (10 U/µL) 1 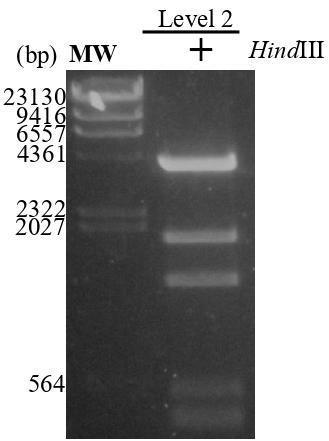
Figure 4. Level-2 construct. The purified level-2 plasmid was digested with HindIII at 37 °C for 2 h and the product was run on 2% agarose gel (expected bands: around 4 kb, 1.8 kb, 1.4 kb, 500 bp, and 400 bp). MW: marker; Lambda/HindIII.
Agrobacterium transformation
Thaw the electrocompetent Agrobacterium on ice.
Incubate 2 μL of the level-2 construct with Agrobacterium for 30 min on ice.
Transfer to the electroporation cuvette (see General note 3).
Electroporate at 2.5 kV until the apparatus beeps.
Add 1 mL of LB into the cuvette, resuspend, and transfer back to an Eppendorf tube.
Incubate for 1 h at 30 °C.
Plate on LB agar plates with kanamycin (50 μg/mL), gentamicin (25 μg/mL), and rifampicin (100 μg/mL).
Grow for 48 h at 30 °C.
Plant transformation (adapted from Van Eck et al. [2])
Sterilize seeds:
Immerse 1 g of seeds in 25 mL of 70% ethanol for 2 min.
Wash the seeds with sterile water.
Add 25 mL of 20% bleach for 20 min.
Rinse the seeds three times with sterile water.
Sow the seeds in Magenta boxes with ½ MS (20–30 seeds/box). Use eight Magenta boxes per transformation.
Leave the seeds at 4 °C for two days (from Friday, day 1, to Monday).
Put the boxes at 25 °C in darkness for eight days (from Monday to Thursday, day 11).
On the morning of day 10 (Monday), release a liquid culture Agrobacterium by picking a single colony from a fresh plate (see General note 4) into 4 mL of LB with the appropriate antibiotics. Simultaneously, release a liquid culture with only LB (control) and LB + antibiotics. Put the liquid culture of Agrobacterium on a shaker at 28 °C at 12 rcf.
On Day 11, transfer the germinated plants to autoclaved glass plates containing autoclaved filter paper moistened with water in a laminar flow hood.
Cut each cotyledon at both ends with a scalpel (see General note 5). The explant is usually generated around each wound, facilitating the identification of independent transformation events in the same cotyledon.
Transfer the cotyledons to a plate with CIM I (callus induction medium I) to induce callus formation. Place them with the abaxial side facing the culture medium. Transfer the cotyledons to eight plates (30–40 cotyledons per plate).
Place the plates in dim light at 25 °C for 24 h (see General note 6).
Prepare four Erlenmeyer flasks, each with 250 mL of LB plus the appropriate antibiotics. Inoculate them with 1, 2, 5, and 10 µL of the liquid culture of Agrobacterium released previously in stepE5 (do not discard the controls). Incubate the flasks at 28 °C in a shaker at 12 rcf for 14–16 h.
On day 12, measure the OD600 of Agrobacterium cultures. The ideal OD is between 0.5 and 0.6, but any value within the range of 0.4–0.9 can be used.
Divide the Erlenmeyer flask that best fits the reference OD into two 15 mL Falcon tubes and centrifuge at 1600 rcf for 10 min.
Discard the supernatant and resuspend the pellet in a sterile solution of 10 mL of 10mM MgSO4.
Pour the Agrobacterium suspension into a sterile Petri dish (15 mL/dish).
Immerse the cotyledons in this suspension for approximately 10 min, stirring gently by hand.
Extract the cotyledons from the suspension and place them shortly on sterile filter paper (avoid drying out the explants). Immediately, transfer them to plates previously prepared with CIM II (callus induction medium II).
Leave 20 cotyledons uninfected to use as controls.
Place the plates at 25 °C with dim light for 48 h.
On day 14, transfer the explants to plates prepared with SIM I (shoot induction medium I) for the induction of shoot formation. Place 15–20 explants per plate. Transfer 10 of the cotyledons uninfected with Agrobacterium to medium without antibiotics and the other 10 to medium with antibiotics. Place the plates in a plant room at 25 °C with a light/dark cycle of 16:8 h for 14–18 days.
On day 28–32, transfer the explants to fresh agar every 15 days. When green tissue appears on the explant (Figure 5A), use a scalpel to cut and separate the remains of cotyledons and untransformed material (brown or yellow explants) and transfer them to plates prepared with SIM II (shoot induction medium II). Let the remaining explants continue in SIM I until they also generate green tissue.
Note: At this stage, individual explants cannot be identified.

Figure 5. Plant regeneration. Plant regeneration from transformed explant in different stages. A. SIM I. B. SIM II. C. RIM. Scale bar: 0.1 cm.On day 45–60, it is expected the emergence of sprouts (Figure 5B), indicative of independent events. Cut the sprouts when they reach a sufficient height to be detached from the base without carrying remnants of explants or cutting the apex. Transfer the cut sprouts to a tall Petri dish filled with RIM (root induction medium). Simultaneously, transfer 10 positive controls and 10 negative controls. Obtain the controls from plates without incubation with Agrobacterium, without kanamycin. Transfer them to RIM without (negative) or with kanamycin (positive). Positive controls (without kanamycin) should regenerate, and negative controls should not.
Around day 55–70 (10–12 days in RIM), check for root development in the sprouts (Figure 5C). Sprouts with successful transformation should have roots growing within the medium (see General note 7). In non-transformed cases, the root either does not grow during this period or grows over the medium (escaping the medium). Identify the sprouts with successful transformation and transplant them into the ground.
Once the generated plants have developed their first pair of true leaves (approximately 20 days), conduct total gDNA extraction from leaves by CTAB method according to Capron et al. [4].
Utilize this gDNA as a template for PCR to assess the presence of the plasmid, utilizing specific primers for the vector (e.g., CAS9). In positive cases, perform another PCR with primers designed in step A3. Run 5 μL of the PCR product on a 2% agarose gel, where a double band may be observed if a large deletion occurs. Use the remaining portion for Sanger sequencing, where frameshift (as explained later) can be observed in case of editing.
Examine the plant sequences to identify those displaying a frameshift in the reading frame within the sgRNA area (indicative of heterozygotes). In Figure 6, observe how the sequencing follows a normal pattern and suddenly a frameshift occurs between base 420 and 430, indicated by an arrow.
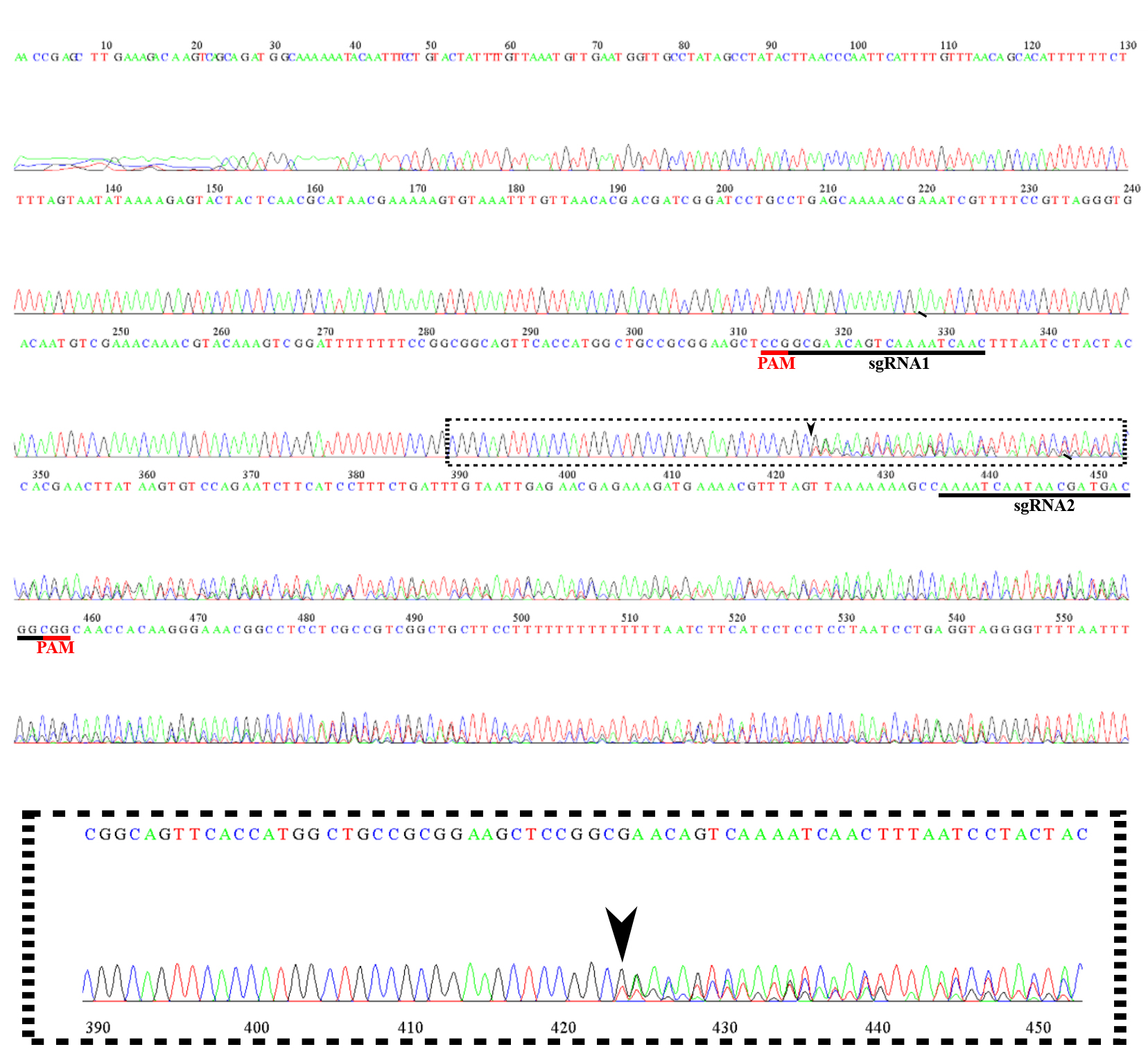
Figure 6. Chromatogram of edited plant DNA as an example. Extraction of T0 plant gDNA, followed by PCR amplification. The resulting PCR products were sequenced, revealing a frameshift (indicated with an arrow) in the reading frame in the sgRNA2 region (note in the zoom-in that the chromatogram gets at least two peaks of different bases at the same position). Annotation includes the positions of sgRNA1 and sgRNA2, as well as the corresponding protospacer adjacent motifs (PAMs).If a frameshift is not found, perform a sequence alignment to confirm no editing or that editing occurred on both DNA strands, although this is a rare occurrence.
Proceed with the plants exhibiting these characteristics.
Search for homozygous edited lines.
Germinate 10 plants from T1 generation and extract gDNA as in step E23. Perform PCR with primers designed in step A3. Sequence PCR product.
Note: In this step, homozygous, heterozygous, and wt lines could be obtained for the deletion.
Perform a sequence alignment (ClustalW) where it is possible to observe the deletion in homozygous lines (Figure 7).

Figure 7. Sequence alignment. Sequence comparison of wt and KO 1 and 2 for PLC2 in T1 generation plants used in Perk et al. [3].Conduct a PCR analysis using specific primers for CAS9 (Cas9_F and Cas9_R). It is possible to observe homozygous lines without the transgene in the T1 generation (indicated by a negative PCR result for CAS9). If it is not possible to find a transgene-free line, continue with the next generation until it is found; it will lead to an edited and transgene-free plant.
Validation of protocol
This protocol was used to generate the KO lines employed in CRISPR/Cas9-mediated phospholipase C 2 knock-out tomato plants more resistant to Botrytis cinerea by Perk et al. [3].
General notes and troubleshooting
General notes
Primers must hybridize at least 100 bp from the sgRNAs, so Sanger sequencing would be optimal in sgRNAs regions.
Note that orange colonies carry the empty vector.
Electroporation cuvettes should be pre-chilled on ice.
The Agrobacterium plate should not be more than 1 week old.
The leaves should not be enlarged or opened.
Plastic white bags can be used to dim the light.
If roots have not appeared by day 12, discard the plants.
Acknowledgments
This work was supported by Bayer (Grants 4traits) and ANPCyT (PICT 2017 No0601).
Competing interests
The authors declare that they have no conflict of interest.
References
- Nekrasov, V., Wang, C., Win, J., Lanz, C., Weigel, D. and Kamoun, S. (2017). Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci Rep7(1): 482.
- Van Eck, J., Keen, P. and Tjahjadi, M. (2019). Agrobacterium tumefaciens-mediated transformation of tomato. In: Kumar, S., Barone, P. and Smith, M. (Eds.). Transgenic plants. Methods in Molecular Biology. vol 1864 (pp 225–234). Humana Press, New York.
- Perk, E. A., Arruebarrena Di Palma, A., Colman, S., Mariani, O., Cerrudo, I., D’Ambrosio, J. M., Robuschi, L., Pombo, M. A., Rosli, H. G., Villareal, F., et al. (2023). CRISPR/Cas9-mediated phospholipase C 2 knock-out tomato plants are more resistant to Botrytis cinerea. Planta 257(6): 117.
- Capron, A., Gourgues, M., Neiva, L. S., Faure, J. E., Berger, F., Pagnussat, G., Krishnan, A., Alvarez-Mejia, C., Vielle-Calzada, J. P., Lee, Y. R., et al. (2008). Maternal Control of Male-Gamete Delivery in Arabidopsis Involves a Putative GPI-Anchored Protein Encoded by the LORELEI Gene. Plant Cell. 20(11): 3038–3049.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Perk, E. A., Laxalt, A. M. and Cerrudo, I. (2024). CRISPR-Cas9 Protocol for Efficient Gene Knockout and Transgene-free Plant Generation. Bio-protocol 14(11): e5012. DOI: 10.21769/BioProtoc.5012.
Category
Plant Science > Plant transformation
Plant Science > Plant molecular biology > DNA > DNA recombination
Biological Sciences > Biological techniques > CRISPR/Cas9
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.