- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Linearly Amplified Single-Stranded RNA-Derived Transcriptome Sequencing (LAST-seq)
Published: Vol 14, Iss 11, Jun 5, 2024 DOI: 10.21769/BioProtoc.4998 Views: 2157
Reviewed by: Alka MehraClara Morral Martinez
Original research article
The authors used this protocol in:

Aug 2023
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Single-cell RNA sequencing (scRNA-seq) stands as a cutting-edge technology widely used in biological and biomedical research. Existing scRNA-seq methods rely on reverse transcription (RT) and second-strand synthesis (SSS) to convert RNA to cDNA before amplification. However, these methods often suffer from limited RT/SSS efficiency, which compromises the sensitivity of RNA detection. Here, we develop a new method, linearly amplified single-stranded RNA-derived transcriptome sequencing (LAST-seq), which directly amplifies the original single-stranded RNA without prior RT and SSS and offers high-sensitivity RNA detection and a low level of technical noise in single-cell transcriptome analysis. LAST-seq has been applied to quantify transcriptional bursting kinetics in human cells, advancing our understanding of chromatin organization’s role in regulating gene expression.
Key features
• An RNase H/DNA polymerase-based strategy to attach the T7 promoter to single-stranded RNA.
• T7 promoter mediated IVT on single stranded RNA template at single cell level.
Keywords: In vitro transcriptionGraphical overview
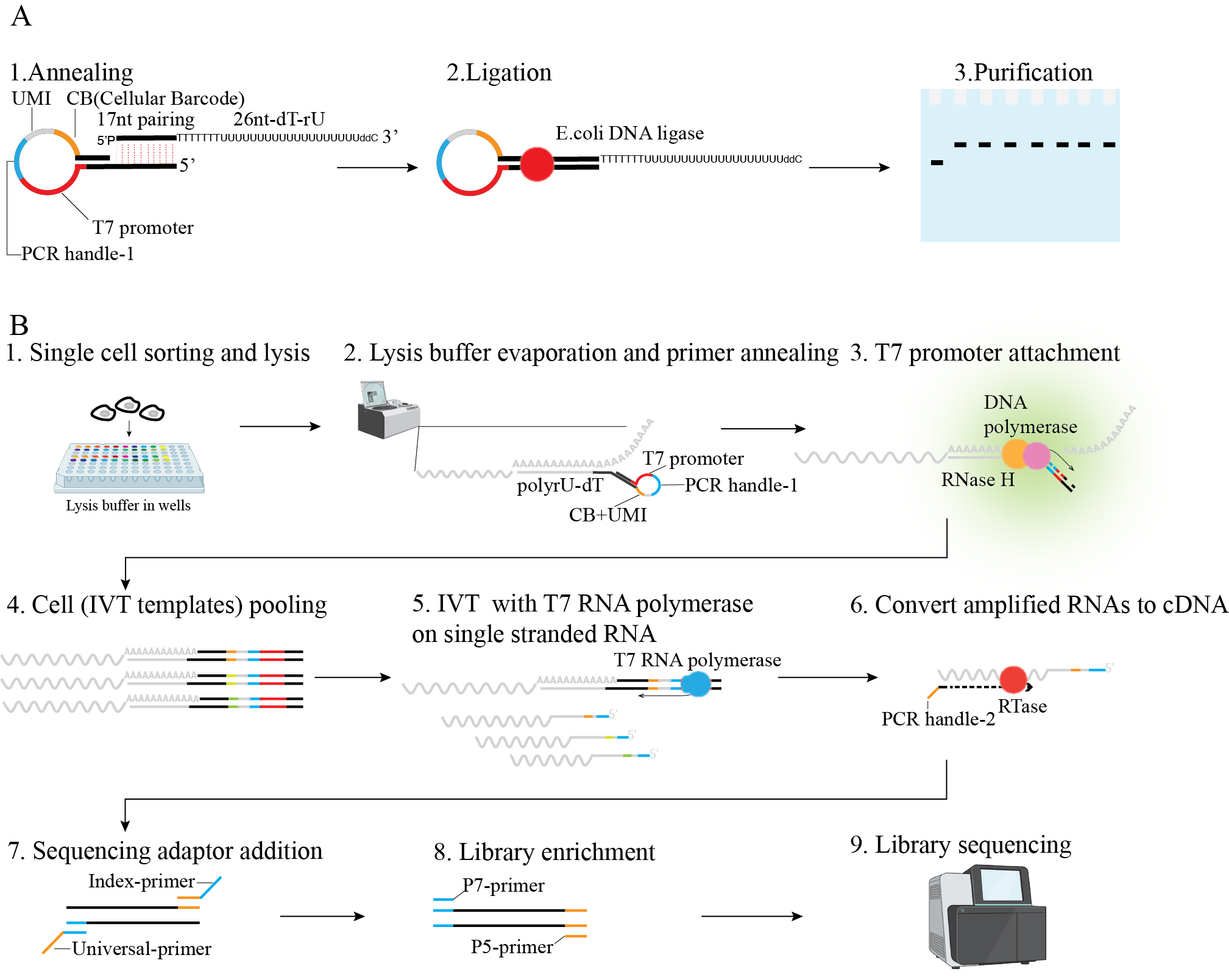
Figure 1. Scheme of linearly amplified single-stranded RNA-derived transcriptome sequencing (LAST-seq). A. LAST primer design and preparation. B. LAST-seq workflow.
Background
Single-cell transcriptome analyses have been fueled by the advancement in scRNA-seq methods. Recent technical progress has focused on enhancing digital counting through unique molecular identifiers (UMIs) [1–3], increasing cellular throughput while reducing the cost [4–10], optimizing individual protocol steps [1,2,11,12], and miniaturization [11,13,14].
Despite these improvements, the fundamental chemistry involving reverse transcription (RT) and second-strand synthesis (SSS) before single-stranded RNA amplification remains unchanged. While RT relies on reverse transcriptase, current scRNA-seq methods utilize various SSS strategies with limited efficiency, including terminal transferase [15,16] or template switching [1,5,6,9,11,12,17,18], to create cDNA priming sites for subsequent PCR, conversion from RNA/cDNA hybrid to double-stranded DNA by RNase H and DNA Pol for in vitro transcription [8,14,19], random annealing to the single-stranded cDNA for extension [2], and direct Tn5 tagmentation of the RNA/cDNA hybrid molecules [20]. As a result, the inherent limitations of RT/SSS efficiency in existing scRNA-seq methods compromise the single-molecule capture efficiency of the original RNA molecules in single cells, leading to reduced measurement accuracy and increased technical noise.
To address this challenge, we developed a novel scRNA-seq method, linearly amplified single-stranded RNA-derived transcriptome sequencing (LAST-seq). Unlike previous methods reliant on inefficient RT/SSS before RNA amplification, LAST-seq directly amplifies original ssRNA molecules in single cells in a linear fashion without prior RT/SSS. This approach achieves a high single-molecule capture efficiency and reduced technical noise compared with existing scRNA-seq methods [21]. Using LAST-seq, we characterized gene expression noise and transcriptional bursting kinetics and investigated the role of chromatin organization in regulating gene expression [21].
Materials and reagents
Reagents
E. coli DNA ligase (New England BioLabs, catalog number: M0205S)
10× TBE (Tris/boric acid/EDTA) buffer (Bio-Rad, catalog number: 1610770)
TEMED (Bio-Rad, catalog number: 1610800)
Ammonium persulfate (APS) (Bio-Rad, catalog number: 1610700)
40% Acrylamide/Bis Solution, 29:1 (Bio-Rad, catalog number: 1610146)
Urea (Bio-Rad, catalog number: 1610731)
Sodium acetate (NaOAc) (3 M) pH 5.5 (Thermo Fisher Scientific, catalog number: AM9740)
NovexTM TBE-urea sample buffer (Thermo Fisher Scientific, catalog number: LC6876)
Formamide (MilliporeSigma, catalog number: 11814320001)
Tris (1 M), pH 8.0 (Thermo Fisher Scientific, catalog number: AM9855G)
GenEluteTM-LPA (MilliporeSigma, catalog number: 56575)
SYBRTM Gold nucleic acid gel stain (Thermo Fisher Scientific, catalog number: S11494)
Trypsin-EDTA (Thermo Fisher Scientific, catalog number: 25200056)
DMEM (Thermo Fisher Scientific, catalog number: 10564011)
Fetal bovine serum (MilliporeSigma, catalog number: F2442)
Phosphate-buffered saline (PBS), 1× without calcium and magnesium (Corning, catalog number: 21-040-CV)
TritonTM X-100 (MilliporeSigma, catalog number: T8787)
SUPERase•InTM RNase inhibitor (Thermo Scientific, catalog number: AM2694)
Deoxynucleotide (dNTP) solution mix (New England BioLabs, catalog number: N0447S)
RNase H (New England BioLabs, catalog number: M0297S)
Klenow fragment (3'→5' exo-) (New England BioLabs, catalog number: M0212L)
Ribonucleotide solution mix (New England BioLabs, catalog number: N0466S)
T7 RNA polymerase (New England BioLabs, catalog number: M0658S)
DL-dithiothreitol solution (DTT) (MilliporeSigma, catalog number: 646563)
MgCl2 (Thermo Fisher Scientific, catalog number: AM9530G)
RNA MagClean DX (Aline Bioscience, catalog number: C-1005-5/50)
Nuclease-free water (Thermo Fisher Scientific, catalog number: AM9937)
SuperScriptTM IV reverse transcriptase (Thermo Fisher Scientific, catalog number: 18090050)
Q5® high-fidelity 2× Master Mix (New England BioLabs, catalog number: M0492S)
PCRClean DX (Aline Bioscience, catalog number: C-1003-5)
QubitTM dsDNA HS Assay Kit (Thermo Fisher Scientific, catalog number: Q32854)
QubitTM assay tubes (Thermo Fisher Scientific, catalog number: Q32856)
Agilent High Sensitivity DNA kit (Agilent, catalog number: 5067-4626)
Oligos (Integrated DNA Technologies)
(Optional) High Output v2.5 reagent kit (Illumina, catalog number: 20024906)
Solutions
MIX solution for 10% TBE-Urea acrylamide gel (see Recipes)
Recipes
MIX solution for 10% TBE-Urea acrylamide gel
Components Volume 40% Acrylamide 29:1 125 mL Urea 210 g Formamide 100 mL 10× TBE 50 mL Nuclease-free water X Total 500 mL Note: The MIX can be stored at 4 °C for at least six months.
Laboratory supplies
Pipette tips (Neptune Scientific, model: S3)
AlumaSeal® CS films for cold storage (MilliporeSigma, catalog number: Z722634-100EA)
Hard-Shell® 96-well PCR plates (Bio-Rad, catalog number: HSP9601)
Axygen® 0.2 mL Maxymum Recovery® thin-wall PCR tubes (Corning, catalog number: PCR-02-L-C)
DNA LoBind® tubes (Eppendorf, catalog number: 022431021)
QubitTM assay tubes (Thermo Fisher Scientific, catalog number: Q32856)
(Optional) x-tracta gel extraction tool (MilliporeSigma, catalog number: Z722390-100EA)
Notes:
Pipette tips used in this protocol should be low-retention, RNase-free, and DNase-free, with an aerosol filter.
PCR tubes and plates used in this protocol should be low-retention, RNase-free, and DNase-free.
Equipment
GILSON® Pipetman (GILSON®, model: D10-D1000)
Freezer (VWR, model: VWR ULT Freezer 528 Eco Premium)
Countess 3 (Thermo Fisher Scientific, model: FL model)
Centrifuge 5418 R (Eppendorf, model: 5418 R)
PCR Cooler (Eppendorf, catalog number: 022510509)
VWR® Mini Centrifuge (VWR, catalog number: 76269-064)
Vortex-Genie® 2 mixer (Fisher Scientific, catalog number: SI-0236)
Vacufuge plus, Centrifuge Concentrator (Eppendorf, model: basic with A-2-VC rotor)
Eppendorf 5810R (Eppendorf, model: 5810R)
ThermoMixer C (Eppendorf, model: 5382)
Bio-Rad C1000 Touch Thermal Cycler (Bio-Rad, model: 1851148)
AirClean Systems 600 PCR workstation (AirClean Systems, model: 600)
Mini-PROTEAN® Tetra Cell (Bio-Rad, catalog number: 1658004)
Mini-PROTEAN® Tetra Cell Casting Module (Bio-Rad, catalog number: 1658021)
UViewTM Mini Transilluminator (Bio-Rad, catalog number: 1660531)
NanoDrop (Thermo Fisher Scientific, catalog number: ND-ONE-W)
PowerPacTM Basic Power Supply (Bio-Rad, catalog number: 1645050)
Qubit 4 Fluorometer (Thermo Fisher Scientific, catalog number: Q33238)
2100 Bioanalyzer Instrument (Agilent, model: G2939A)
High-performance computing cluster (NIH, Biowulf, model: NA)
BD FACSAria IIu (BD Bioscience, model: NA)
Magnetic rack (Diagenode, catalog number: B04000001)
Roto mini plus (Benchmark, catalog number: R2020)
Software and datasets
bcl2fastq, v2.20, free: https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.htmL
cutadapt, v 1.15, free: https://cutadapt.readthedocs.io/en/stable/
zUMIs, v.2.9.7, free: https://github.com/sdparekh/zUMIs
The datasets are available at https://github.com/lyuj2022/LAST-seq.
Procedure
LAST-seq primer preparation
Cast 10% TBE-Urea acrylamide gel.
Assemble the 1.5 mm gel-casting module according to the manufacturer’s manual.
Prepare the gel mixture in a 50 mL conical tube as below (Table 1).
Table 1. 10% TBE-Urea acrylamide gel solution
Components Volume MIX 10 mL 30% APS 33.2 µL TEMED 4 µL Slowly pour the gel solution into the gel-casting module filling all the space between the short plate and spacer plate and avoiding introducing bubbles. Then, insert a 10-well comb. Wait for 30 min for gel polymerization.
Note: The gel solution is prepared for casting one TBE-Urea acrylamide gel, with 10 wells for 10 LAST primers. Alternatively, one can use a commercially available 10% precast TBE-urea gel that fit the Mini-PROTEAN® Tetra Cell.
Assemble the mixture in a 96-well plate for each hairpin primer (BC1-BC80, see Table S1 for hairpin primers) as below (Table 2).
Table 2. Annealing mixture
Components Volume Working concentration 100 µM hairpin primer 25 µL 50 µM 10× E. coli DNA ligase reaction buffer 5 µL 1× Nuclease-free water 20 µL Seal the plate with AlumaSeal® CS film and place the plate in a thermocycler with a lid temperature set to 70 °C. Then, heat the plate to 65 °C for 5 min and slowly cool down to 25 °C (-0.5 °C/cycle).
Notes:
The hairpin primer contains a 5' end docking site (20 nt) for loop anchoring and dTrU tail annealing, a T7 promoter (20 nt) for IVT and partial loop anchoring, a PCR handle (21 nt of TruSeq Read2) to dock index primers, a UMI/barcode (8 nt/6 nt) to tag mRNA copies and individual cells, and a 3' end docking site (11 nt) for loop anchoring. After annealing, the primer will form a loop and leave a 5' end overhang (Figure 1A). The hairpin primer can be stored at -20 °C for at least one year.
A higher lid temperature than 75 °C will detach the AlumaSeal® CS films from the plate.
Assemble the ligating mixture in a new 96-well plate as below (Table 3), seal the plate, and incubate at 16 °C for 30 min.
Table 3. Ligating mixture
Components Volume Working concentration 50 µM hairpin primer 2 µL 5 µM 100 µM dTrU tail primer 1 µL 5 µM 10× E. coli DNA ligase reaction buffer 2 µL 1× E. coli DNA Ligase 1 µL 0.5 U/µL Nuclease-free water 14 µL Note: The dTrU primer contains a linker (17 nt) to join the hairpin primer, a poly-dT-rU region (7 nt–19 nt) to capture mRNA, and a 3' end blocker to prevent primer extension (Figure 1A).
Set up the electrophoresis apparatus and carefully remove the comb, avoiding damaging the wells. Pre-run the gel in 1× TBE buffer at 300 V for 10 min, then rinse wells with buffer using a P200 pipette.
Mix 20 µL of ligated mixture and 20 µL of 2× TBE-Urea sample buffer. Load 40 µL sample per well. Run the gel at 150 V for 45 min.
Purify the ligated LAST primer by ethanol precipitation.
Carefully open the glass chamber and take out the gel. Soak the gel in 20 mL of 1× TBE containing 1× SYBR Gold for 20 min.
Place the gel on the UV transilluminator. While the oligo is illuminated with UV, use a clean razor blade to cut the oligo band of interest (~130 nt in length) of each well, keeping the size of the gel slice as small as possible. Then, transfer the gel slice to 1.5 mL Eppendorf tubes.
Note: Alternatively, one can use x-tracta gel extraction tool to extract the band of interest.
Crush the gel slice with disposable tips, add 400 µL of 0.3 M sodium acetate per gel slice, and incubate the tubes on dry ice for 15 min.
Note: Incubating gel slices on dry ice for 15 min will freeze the buffer and gel, which will enlarge the meshes of the acrylamide gel, facilitating primer diffusion from the gel while the buffer and gel are unfrozen.
Rotate the tubes at 10× rpm at room temperature for 4 h.
Centrifuge the tube at 20,000× g for 15 min and transfer the supernatant to a 1.5 mL tube.
Add 2.5-fold volume of cold 100% ethanol and 3 µL of 5 µg/µL GenElute-PLA. Place tubes at -20 °C overnight.
Note: GenElute-PLA is short for Linear PolyAcrylamide, which is a neutral carrier for precipitating nucleic acids with ethanol.
Centrifuge the tube at 20,000× g for 30 min at 4 °C and discard the supernatant.
Wash the white pellet twice with 75% cold ethanol and centrifuge at 20,000× g for 10 min at 4 °C.
Dry the pellet in a clean PCR workstation for 20 min and resuspend the pellet (invisible) with 10 µL of TE.
Quantify the LAST primer using NanoDrop and adjust the concentration to 40 ng/µL (equal to ~1 µM).
Note: The UV absorbance at 260 nm is proportional to the nucleic acid concentration. The primer consists of 19 ribonucleotides and 111 deoxyribonucleotides, and thus we used 34 as the conversion factor for the LAST primer. The ultimate yield is approximately 0.6 µg.
Transfer primers to a 96-well plate, seal the plate with AlumaSeal® CS films, and store the plate at -80 °C for up to one year.
Preparation of lysis buffer
Add 2 µL of 1 µM LAST primers (BC1-BC80) into wells (each well for a unique LAST primer) of a new 96-well plate containing 48 µL of 10 mM Tris buffer and 0.5% RNase inhibitor, resulting in 40 nM LAST primer per well.
Assemble the lysis buffer in a new 96-well plate with the components as below (Table 4), resulting in 20 µL of lysis buffer per well. Then, use a P10 multi-pipette to aliquot 2 µL of lysis buffer per well to new 96-well plates. Seal plates with AlumaSeal® CS films and store at -80 °C.
Table 4. Lysis buffer
Components Volume Working concentration Nuclease-free water 10 µL 1% Triton X-100 2 µL 0.1 % 40 nM LAST primer 1 µL 2 nM/µL 2 U/µL RNase inhibitor 1 µL 0.1 U/µL 2 mM dNTP 5 µL 0.5 mM 600 pg RNA carrier 1 µL 30 pg/µL Notes:
Each 96-well plate contains 80 LAST primers in 80 wells and thus can barcode up to 80 single cells.
The working volume of lysis buffer per well is 2 µL. Here, we assembled 20 µL of lysis buffer per well (per LAST primer) and aliquoted them to multiple plates for future use.
Preparation of single-cell suspension
Remove the medium of HEK293T cells in a 10 cm Petri dish and quickly rinse cells twice with 5 mL of 37 °C prewarmed PBS.
Immediately add 1 mL of 0.25% Trypsin-EDTA to a 10 mm Petri dish, and then incubate in a 5% CO2 humidified incubator at 37 °C for 2 min.
Neutralize the Trypsin-EDTA by adding 2 mL of DMEM containing 10% fetal bovine serum and rinse the dish by pipetting to collect all cells. Transfer 3 mL of cell suspension to a 15 mL conical tube.
Spin down cells at 300× g for 3 min at 4 °C.
Aspirate the supernatant, resuspend cells with 1 mL of cold PBS, and place the tube on ice.
Count cells using an automated cell counter and adjust the concentration with cold PBS to 1 × 107 cells/mL.
Pass cells through a 40 µm cell strainer, keep cells on ice, and proceed with single-cell sorting.
Single-cell sorting
Take out 96-well plates containing lysis buffer, keep them sealed, and place them on ice until loading them into the sorter.
Use a small portion of your samples or flow cytometry compensation beads as a pilot sample to align the sorter to an empty 96-well plate sealed with film. Make sure, by visual inspection, that the droplets have been ejected in the center of each well.
Replace the plate and the pilot sample with an unsealed plate containing lysis buffer and your sample of interest, respectively.
Sort single cells into individual wells. Seal the plates with AlumaSeal® CS films and centrifuge them at 1,000× g for a few seconds at 4 °C.
Note: Store plates at -80 °C if you do not proceed immediately.
Lysis buffer evaporation
Place the 96-plate in the thermal cycler and set the lid to 70 °C. Heat the plate at 65 °C for 5 min and immediately put it on a PCR cooler for 2 min.
Peel off the film carefully and vacuum-centrifuge the plates at 1,400 rpm for 25 min at 30 °C in a PCR workstation. Prepare the tagging buffer per well as below (Table 5), while waiting for the evaporation, and keep the buffer on ice.
Table 5. Tagging buffer
Components Volume Working concentration Nuclease-free water 1.875 µL 10× NEB2 0.25 µL 1× 20 U/µL RNase inhibitor 0.125 µL 1 U/µL 0.5 U/µL RNase H 0.1 µL 0.02 U/µL 5 U/µL Klenow fragment (3'→5' exo-) 0.15 µL 0.3 U/µL
Notes:
Place the vacuum-centrifuge in a UV-treated PCR workstation to minimize the impact of environmental RNase contamination.
After evaporation, the lysis buffer is invisible. Make sure the lysis buffer is completely evaporated. The purpose of using vacuum-centrifugation is to increase the primer concentration by evaporating the lysis buffer, thereby achieving high annealing efficiency with a small quantity of LAST primer.
The tagging buffer recipe is for one well of a 96-well plate. When preparing the tagging mixture, scale up the volume according to the number of wells.
T7 promoter attachment and single-stranded RNA in vitro transcription (IVT)
Add 2.5 µL of tagging buffer to each well and seal the plate with film. Spin the plate for seconds and vortex the plate using a ThermoMixer at 1,000× rpm for 1 min.
Spin the plate for a few seconds and place it in a thermal cycler at 37 °C for 30 min. Meanwhile, prewarm RNA beads (RNA MagClean DX) and RNA beads buffer (spin down 550 µL of RNA beads to obtain 520 µL of buffer) to room temperature for at least 30 min. Prepare 2.5 µL of rinsing buffer per well as below (Table 6):
Table 6. Rinsing buffer
Components Volume Working concentration Nuclease-free water 2.125 µL 10× NEB2 0.25 µL 1× 20 U/µL RNase Inhibitor 0.125 µL 1 U/µL Notes:
In this process, RNase H nicks the RNA polyA tail at the DNA–RNA hybrid region formed by annealed polyA and poly-dT-rU. Subsequently, DNA polymerase (Klenow exo-) extends the remaining polyA end of this RNA using the LAST primer as a template, thereby attaching T7 promoter, UMI/barcode to each RNA molecule.
The attachment efficiency of T7 promoter is approximately 65%, which is measured by assembling the LAST-seq primer and FAM-labeled RNA template at the molar ratio of 1.5:1, followed by nicking and extension reaction. The original RNA template would be nicked and elongated to ~150 nt after T7 promoter attachment. The FAM intensity is linearly and positively correlated to the number of molecules and thus could be used to estimate the percentage of molecules with T7 promoter attached (Figure 2).
The purpose of rinsing buffer is to rinse wells, therefore minimizing the residual IVT template in wells.
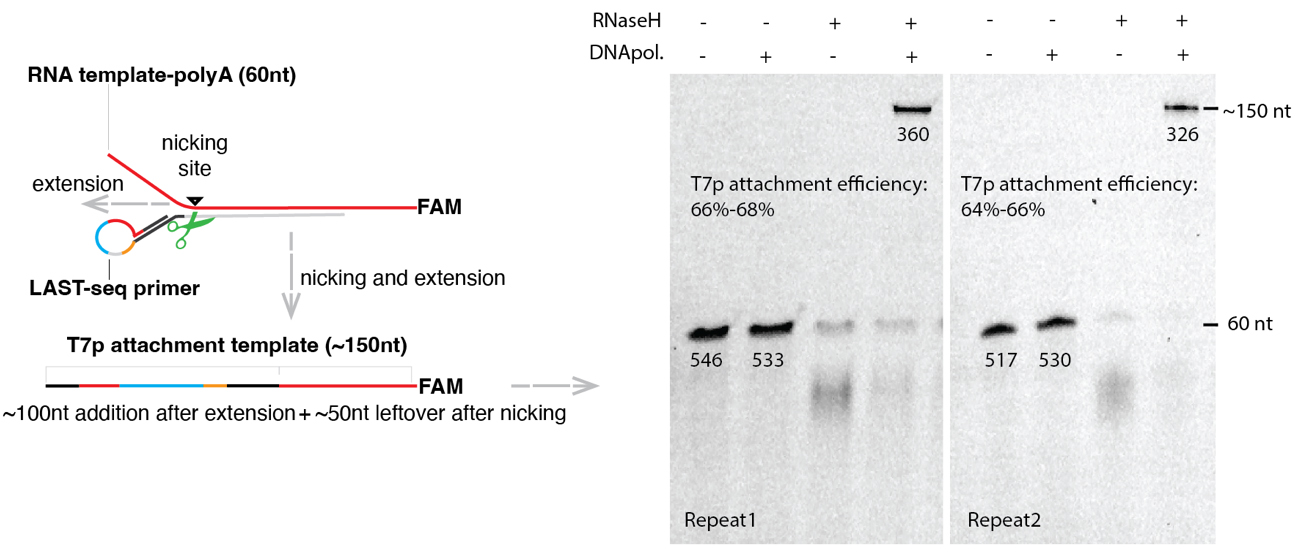
Figure 2. Strategy to measure the efficiency of T7 promotor attachment
After incubation, quickly spin the plate, place it on a PCR cooler, and carefully peel off the film.
Transfer the tagging buffer from each well to a 1.5 mL Eppendorf tube.
Add 2.5 µL of rinsing buffer to each well and quickly spin the plate. Transfer the rinsing buffer to the same 1.5 mL Eppendorf tube.
Add 200 µL of beads and 520 µL of beads buffer to the tube and mix well by pipetting; then, incubate at room temperature for 15 min. Meanwhile, thaw all reagents needed for IVT (see Reagents in Table 7).
Table 7. IVT buffer
Components Volume Working concentration Nuclease-free water 26 µL 10× RNA polymerase buffer 4 µL 1× 25 mM NTP 2 µL 1.25 mM 20 U/µL RNase inhibitor 2 µL 1 U/µL 100 mM DTT 2 µL 5 mM 100 mM MgCl2 2 µL 5 mM 50 U/µL T7 RNA polymerase 2 µL 2.5 U/µL After incubation, place the tube on the magnet for 2 min or until the solution clears. Then, discard the supernatant without disturbing beads.
Add 1 mL of freshly prepared 80% ethanol and incubate for 30 s, then discard the supernatant.
Repeat step F8.
Remove all residual liquid using a P10 pipette. Dry beads for up to 15 min until the pellet is not shiny. Meanwhile, assemble the IVT buffer as below and keep it on ice:
Resuspend beads with 40 µL of IVT buffer and transfer the mixture to a 200 µL PCR tube.
Incubate the tube in a thermal cycler at 37 °C for 14 h, then keep it at 4 °C.
Notes:
IVT efficiency of assembled single-stranded RNA template could reach a few 100-fold. Detailed information can be found in the original research article [21] (Supplementary Figure 2).
Before proceeding to step F13, prewarm RNA beads and beads buffer at room temperature for at least 30 min.
Place the tube on a magnetic stand for 2 min, then transfer the supernatant to a new PCR tube.
Add 32 µL of RNA beads and 40 µL of beads buffer, mix well, and incubate at room temperature for 15 min.
Place the tube on a magnetic stand for at least 5 min until the liquid clears. Then, discard the supernatant without disturbing beads.
Add 200 µL of freshly prepared 80% ethanol and incubate for 30 s, then discard the supernatant.
Repeat step F16.
Dry the beads for 3–5 min until the pellet is not shiny.
Resuspend beads with 7 μL of nuclease-free water. Pipette the entire volume up and down 10 times to mix thoroughly. Incubate at room temperature for 2 min.
Place the tube on a magnetic stand for 2 min. Then, transfer 6 µL of supernatant to a new PCR tube and keep it on ice.
Library preparation and sequencing
Reverse transcription
Mix 6 µL of RNA elution above with 1 µL of annealing buffer as below (Table 8). Incubate the tube in a thermal cycler at 70 °C for 2 min, then transfer it to the PCR cooler immediately.
Table 8. Annealing buffer
Components Volume Working concentration 25 mM random primer 0.5 µL 1.25 µM 10 mM dNTP 0.5 µL 0.5 mM Note: Random primer consists of a PCR handle (21 nt of TruSeq Read1) and a random hexamer.
Add 3 µL of RT buffer as below (Table 9) to 7 µL of the mixture above, mix well, and spin down.
Table 9. RT buffer
Components Volume Working concentration 5× SSIV Buffer 2 µL 1× 20 U/µL RNase inhibitor 0.25 µL 0.5 U/µL 0.4 M DTT 0.25 µL 10 mM 200 U/µL SuperScriptTM IV reverse transcriptase 0.5 µL 10 U/µL Incubate the tube at a thermal cycler and run the program as below.
Program: 23 °C for 10 min; 50 °C for 15 min; 80 °C for 10 min; hold at 4 °C
PCR for adaptor addition
Add the 10 µL of reaction product above to the PCR mixture as below (Table 10), mix well, and run the PCR program below. While waiting for the PCR, prewarm the PCRClean beads at room temperature for 30 min.
Table 10. PCR mixture
Components Volume Working concentration Q5 high-fidelity 2× Master Mix 50 µL 1× 10 µM universal primer 5 µL 0.5 µM 10 µM index primer 5 µL 0.5 µM Nuclease-free water 30 µL Program:
Step 1: 98 °C for 30 s;
Step 2 for 8 cycles: 98 °C for 10 s; 69 °C for 15 s; 72 °C for 20 s;
Step 3: 72 °C for 2 min;
Hold at 4 °C.
Note: Eighty barcoded LAST primers can only barcode 80 cells in a 96-well plate. In cases where users want to profile more than 80 cells, they can assign different index primers to cDNAs from different 96-well plates, thereby further scaling up the cellular throughput. As a result, for less than 80 cells, users can use the cellular barcode of LAST to distinguish cells; for more than 80 cells, users can distinguish each cell through the combination of barcodes from LAST primers and index primers. See Table S1 for index primers.
Add 90 µL of PCRClean beads to the PCR product, mix well, and incubate at room temperature for 5 min.
Place the tube on the magnetic stand for 5 min. Discard the supernatant without disturbing the beads.
Add 200 µL of freshly prepared 80% ethanol, incubate beads for 30 s, and then discard the supernatant.
Repeat step G2d.
Spin briefly and place the tube on the magnetic stand. Remove any remaining ethanol using a P10 pipette.
Resuspend beads with 51.5 µL of nuclease-free water and incubate for 2 min.
Place the tube on a magnetic stand for 2 min. Then, transfer 50 µL of supernatant to a new PCR tube.
Add 45 µL of beads to the PCR product, mix well, and incubate at room temperature for 5 min.
Repeat steps G2c–f.
Resuspend beads with 31.5 µL of nuclease-free water and incubate for 2 min.
Place the tube on a magnetic stand for 2 min. Then, transfer 30 µL of supernatant to a new PCR tube.
Add 27 µL of beads to the PCR product, mix well, and incubate at room temperature for 5 min.
Repeat steps G2c–f.
Resuspend beads with 11.5 µL of nuclease-free water and incubate at room temperature for 2 min.
Place the tube on a magnetic stand for 2 min. Then, transfer 10 µL of supernatant to a new PCR tube.
PCR for library enrichment.
Add the 10 µL product above to the PCR mixture as below (Table 11), mix well, and run the PCR program below.
Table 11. PCR mixture
Components Volume Working concentration Q5 High-Fidelity 2× Master Mix 12.5 µL 1× 10 µM P5 primer 1.25 µL 0.5 µM 10 µM P7 primer 1.25 µL 0.5 µM Program:
Step 1: 98 °C for 30 s;
Step 2 for 8 cycles: 98 °C for 10 s; 65 °C for 15 s; 72 °C for 20 s;
Step 3: 72 °C for 2 min;
Hold at 4 °C.
Notes:
P5 and P7 primers are adaptors for Illumina sequencing. They were used to amplify and enrich our final library. Their sequences are provided in Table S1.
PCR cycles could vary when using different cell lines or pooling various numbers of cells. We suggest using 10–12 cycles of PCR for 80 cells.
Add 90 µL of beads to the PCR product, mix well, and incubate at room temperature for 5 min.
Place the tube on the magnetic stand for 5 min. Discard the supernatant without disturbing the beads.
Add 200 µL of freshly prepared 80% ethanol and incubate beads for 30 s, then discard the supernatant.
Repeat step G3d.
Spin briefly and place the tube on the magnetic stand. Remove any remaining ethanol and dry beads for 3 min or until the pellet is not shiny.
Resuspend beads with 11.5 µL of nuclease-free water and incubate for 2 min.
Place the tube on a magnetic stand for 2 min. Then, transfer 10 µL of supernatant to a new PCR tube.
Quantify the library using the QubitTM dsDNA HS Assay Kit.
Note: The library concentration is approximately 1 ng/µL for eight cycles of PCR, which is sufficient for sequencing on NEXTseq550.
Check library distribution using 2100 Bioanalyzer Instrument (Figure 3) and sequence library using a High Output v2.5 reagent kit (Illumina) on NEXTseq550, with Read1 sequenced for 50 cycles and Read2 sequenced for 14 cycles.
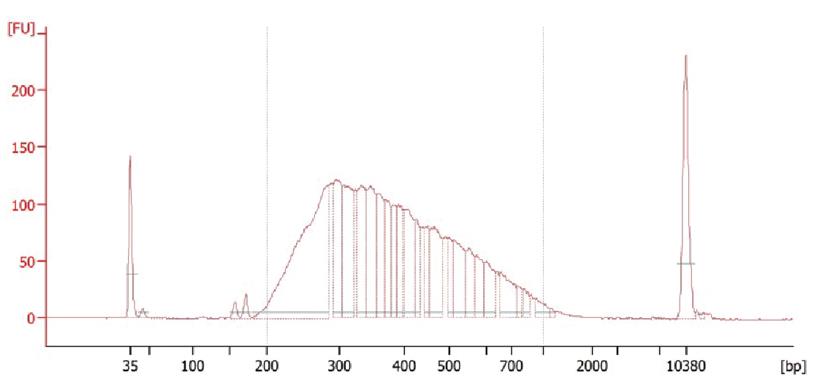
Figure 3. Distribution of linearly amplified single-stranded RNA-derived transcriptome sequencing (LAST-seq) library. The bioanalyzer data showing a library distribution for the transcriptome of 10 pooling HEK293T cells.Notes:
To check library distribution, load 1 µL (~1.0 ng/µL) library to the high-sensitivity DNA chip and run the bioanalyzer according to its manual.
Read1 contains RNA information. Read2 contains the UMI (8 nt) and barcode (6 nt) information.
Data analysis
Convert BCL files to fastq files using bcl2fastq v2.20.
Trim reads to remove bases of low quality at 3' end and discard reads with a length of Read1 and Read2 less than 22 nt and 14 nt, respectively, using cutadapt v 1.15 with parameters ‘--nextseq-trim=20 -m 22:14’.
Use zUMIs v.2.9.7 [22] for reads QC, read mapping, downsampling, and UMI counting. The resultant dataset is an expression matrix containing reads and UMI counts for genes (in rows) in each cell (in columns).
Check batch effect using principal component analysis or correct batch effect using sva R package (optional).
The related scripts (Rawdataprocess01 and 02) are available at https://github.com/lyuj2022/LAST-seq and are implemented in a Linux system.
Note: After reads trimming, more than 95% of Read1 and Read2 are in length of 50 nt and 14 nt, respectively. LAST-seq requires paired-end sequencing to obtain both RNA sequence (Read1) and UMI/barcode sequence (Read2).
Validation of protocol
This protocol was validated in:
Lyu, J., Chen, C. LAST-seq: single-cell RNA sequencing by direct amplification of single-stranded RNA without prior reverse transcription and second-strand synthesis. Genome Biol 24, 184 (2023) [21].
General notes and troubleshooting
Troubleshooting
| Problems | Solutions |
|---|---|
| Low yield of library | Perform additional rounds of PCR on your library with P5/P7 primers. |
| Small peaks dominate the library distribution | Make sure the LAST primer concentration is approximately 40 ng/µL before dilution. A higher concentration of LAST primer could lead to a large portion of smaller peak in the library. |
Acknowledgments
We thank Ferenc Livak, Subhadra Banerjee, and Caiyi Li from the CCR Flow Cytometry Core for single-cell sorting, and other members of the Chen lab for discussions. The HEK293T cell line is a gift from Dr. Peter D. Aplan. This study used the Biowulf Linux cluster at the National Institutes of Health. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute (1ZIABC011884).
Competing interests
The authors declare no competing interests.
References
- Hagemann-Jensen, M., Ziegenhain, C., Chen, P., Ramsköld, D., Hendriks, G. J., Larsson, A. J. M., Faridani, O. R. and Sandberg, R. (2020). Single-cell RNA counting at allele and isoform resolution using Smart-seq3. Nat Biotechnol. 38(6): 708–714.
- Hashimshony, T., Senderovich, N., Avital, G., Klochendler, A., de Leeuw, Y., Anavy, L., Gennert, D., Li, S., Livak, K. J., Rozenblatt-Rosen, O., et al. (2016). CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17: 77.
- Islam, S., Zeisel, A., Joost, S., La Manno, G., Zajac, P., Kasper, M., Lönnerberg, P. and Linnarsson, S. (2014). Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 11(2): 163–166.
- Cao, J., Packer, J. S., Ramani, V., Cusanovich, D. A., Huynh, C., Daza, R., Qiu, X., Lee, C., Furlan, S. N., Steemers, F. J., et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science. 357(6352): 661–667.
- Gierahn, T. M., Wadsworth, M. H., Hughes, T. K., Bryson, B. D., Butler, A., Satija, R., Fortune, S., Love, J. C. and Shalek, A. K. (2017). Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods. 14(4): 395–398.
- Han, X., Wang, R., Zhou, Y., Fei, L., Sun, H., Lai, S., Saadatpour, A., Zhou, Z., Chen, H., Ye, F., et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell. 172(5): 1091–1107 e1017.
- Jaitin, D. A., Kenigsberg, E., Keren-Shaul, H., Elefant, N., Paul, F., Zaretsky, I., Mildner, A., Cohen, N., Jung, S., Tanay, A., et al. (2014). Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science. 343(6172): 776–779.
- Klein, A. M., Mazutis, L., Akartuna, I., Tallapragada, N., Veres, A., Li, V., Peshkin, L., Weitz, D. A. and Kirschner, M. W. (2015). Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell. 161(5): 1187–1201.
- Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R., Kamitaki, N., Martersteck, E. M., et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 161(5): 1202–1214.
- Rosenberg, A. B., Roco, C. M., Muscat, R. A., Kuchina, A., Sample, P., Yao, Z., Graybuck, L. T., Peeler, D. J., Mukherjee, S., Chen, W., et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 360(6385): 176–182.
- Hahaut, V., Pavlinic, D., Carbone, W., Schuierer, S., Balmer, P., Quinodoz, M., Renner, M., Roma, G., Cowan, C. S., Picelli, S., et al. (2022). Fast and highly sensitive full-length single-cell RNA sequencing using FLASH-seq. Nat Biotechnol. 40(10): 1447–1451.
- Picelli, S., Björklund, Ã. K., Faridani, O. R., Sagasser, S., Winberg, G. and Sandberg, R. (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods. 10(11): 1096–1098.
- Hagemann-Jensen, M., Ziegenhain, C. and Sandberg, R. (2022). Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat Biotechnol. 40(10): 1452–1457.
- Salmen, F., De Jonghe, J., Kaminski, T. S., Alemany, A., Parada, G. E., Verity-Legg, J., Yanagida, A., Kohler, T. N., Battich, N., van den Brekel, F., et al. (2022). High-throughput total RNA sequencing in single cells using VASA-seq. Nat Biotechnol. 40(12): 1780–1793.
- Sheng, K., Cao, W., Niu, Y., Deng, Q. and Zong, C. (2017). Effective detection of variation in single-cell transcriptomes using MATQ-seq. Nat Methods. 14(3): 267–270.
- Tang, F., Barbacioru, C., Wang, Y., Nordman, E., Lee, C., Xu, N., Wang, X., Bodeau, J., Tuch, B. B., Siddiqui, A., et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 6(5): 377–382.
- Islam, S., Kjällquist, U., Moliner, A., Zajac, P., Fan, J. B., Lönnerberg, P. and Linnarsson, S. (2011). Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 21(7): 1160–1167.
- Ramsköld, D., Luo, S., Wang, Y. C., Li, R., Deng, Q., Faridani, O. R., Daniels, G. A., Khrebtukova, I., Loring, J. F., Laurent, L. C., et al. (2012). Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 30(8): 777–782.
- Hashimshony, T., Wagner, F., Sher, N. and Yanai, I. (2012). CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2(3): 666–673.
- Di, L., Fu, Y., Sun, Y., Li, J., Liu, L., Yao, J., Wang, G., Wu, Y., Lao, K., Lee, R. W., et al. (2020). RNA sequencing by direct tagmentation of RNA/DNA hybrids. Proc Natl Acad Sci USA. 117(6): 2886–2893.
- Lyu, J. and Chen, C. (2023). LAST-seq: single-cell RNA sequencing by direct amplification of single-stranded RNA without prior reverse transcription and second-strand synthesis. Genome Biol. 24(1): 184.
- Parekh, S., Ziegenhain, C., Vieth, B., Enard, W. and Hellmann, I. (2018). zUMIs - A fast and flexible pipeline to process RNA sequencing data with UMIs. GigaScience. 7(6): giy059.
Supplementary information
The following supporting information can be downloaded here:
- Table S1.
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Lyu, J. and Chen, C. (2024). Linearly Amplified Single-Stranded RNA-Derived Transcriptome Sequencing (LAST-seq). Bio-protocol 14(11): e4998. DOI: 10.21769/BioProtoc.4998.
Category
Molecular Biology > RNA > RNA sequencing
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.