- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Efficient Genetic Transformation and Suicide Plasmid-mediated Genome Editing System for Non-model Microorganism Erwinia persicina
(§ Technical contact) Published: Vol 14, Iss 6, Mar 20, 2024 DOI: 10.21769/BioProtoc.4956 Views: 2481
Reviewed by: Samik BhattacharyaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2023
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
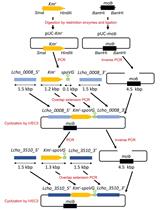
Abstract
Erwinia persicina is a gram-negative bacterium that causes diseases in plants. Recently, E. persicina BST187 was shown to exhibit broad-spectrum antibacterial activity due to its inhibitory effects on bacterial acetyl-CoA carboxylase, demonstrating promising potential as a biological control agent. However, the lack of suitable genetic manipulation techniques limits its exploitation and industrial application. Here, we developed an efficient transformation system for E. persicina. Using pET28a as the starting vector, the expression cassette of the red fluorescent protein–encoding gene with the strong promoter J23119 was constructed and transformed into BST187 competent cells to verify the overexpression system. Moreover, suicide plasmid–mediated genome editing systems was developed, and lacZ was knocked out of BST187 genome by parental conjugation transfer using the recombinant suicide vector pKNOCK-sacB-km-lacZ. Therefore, both the transformation and suicide plasmid–mediated genome editing system will greatly facilitate genetic manipulations in E. persicina and promote its development and application.
Key features
• Our studies establish a genetic manipulation system for Erwinia persicina, providing a versatile tool for studying the gene function of non-model microorganisms.
• Requires approximately 6–10 days to complete modification of a chromosome locus.
Graphical overview
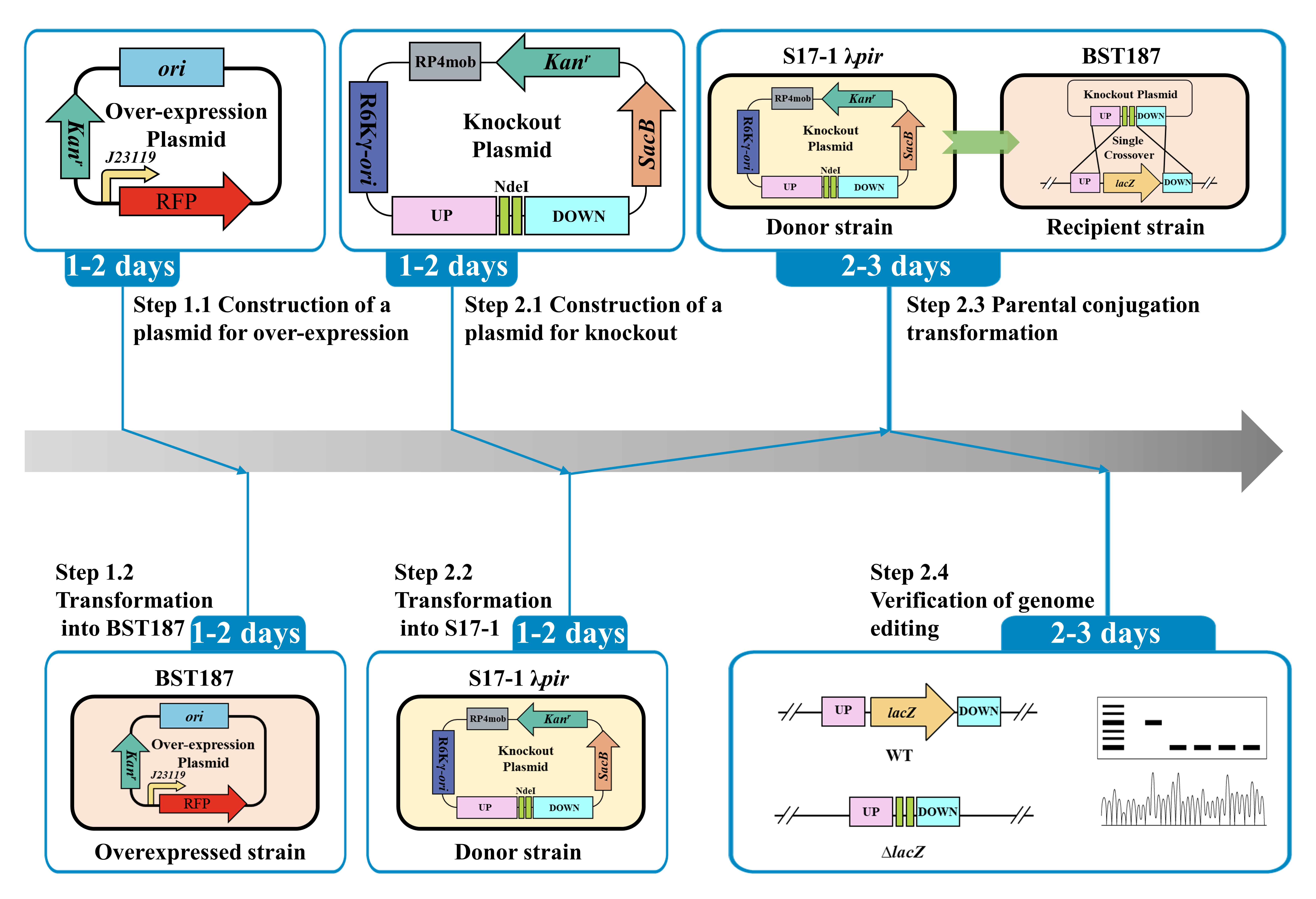
Background
Erwinia persicina belongs to the phylum Proteobacteria, class Gammaproteobacteria, order Enterobacteriales, family Enterobacteriaceae, and genus Erwinia [1]. E. persicina has been identified as a soft rot pathogen that causes pink-pigmented soft rot on plant hosts including garlic, onions, barley, Cucurbita pepo, celery, and alfalfa [2–8]. However, E. persicina has exhibited great potential as a biological control agent to inhibit Salmonella enterica contamination in the sprout food industry [9], Fusarium head blight (FHB) severity in wheat [10], and a variety of soil-borne pathogens (Alternaria solani, Sclerotinia sclerotiorum, Rhizoctonia solani, and Fusarium proliferatum) in potato dextrose agar plates [11]. Recently, E. persicina BST187 was isolated from the rhizosphere of tomato and exhibited broad-spectrum antibacterial activity due to its inhibitory effects on bacterial acetyl-CoA carboxylase [12].
The construction and application of chassis modification of E. persicina has not yet been studied, except by Zhao et al. [13]. Efficient genetic transformation and genome editing systems will be beneficial for exploring the pathogenic mechanisms of E. persicina to provide a fundamental basis for control strategies. Moreover, tools developed for the genetic manipulation of E. persicina will provide excellent cell factories for diverse industrial applications such as andrimids.
In this study, the details of the BST187 chassis modification system are described. We initially identify the design flaws with the pET series of expression plasmids used for BST187, as it lacks T7 RNA polymerase-encoding genes and T7 promoter cannot be used for the overexpression. Then, we demonstrate that the constitutive promoter J23119 could be used for the overexpression of the red fluorescent protein (RFP)-encoding gene. Finally, the whole preparation method and transformation parameters of E. persicina competent cells are determined. Moreover, the knockout and integration system of target genes into the genome of E. persicina strains are conducted by double-crossover homologous recombination using the suicide vector. A schematic diagram of suicide plasmid–mediated genome editing is shown in Figure 1.
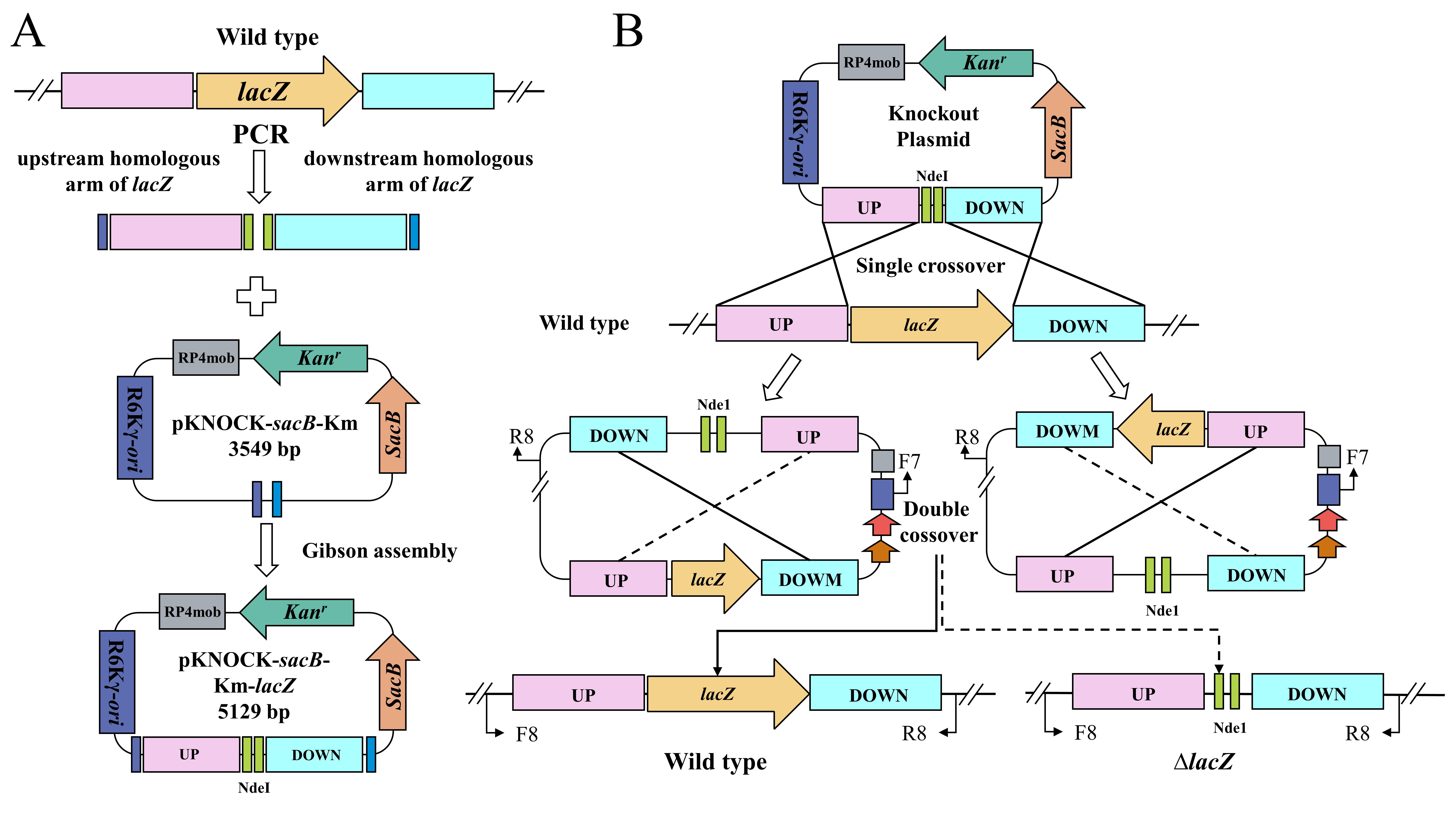
Figure 1. Schematic diagram of suicide plasmid–mediated genome editing. A. Construction of lacZ knockout plasmid. B. Parental conjugation transformation and recombination exchanges.
Materials and reagents
Biological materials
Erwinia persicina BST187 [13]
pGD plasmid [13]
pET28a-Cas9 plasmid [13]
Escherichia coli DH5α chemically competent cells (Biomed, catalog number: BC102-02)
pKNOCK-sacB-km [13]
Escherichia coli DH5α λpir chemically competent cells (ZOMANBIO, catalog number: ZK227)
Escherichia coli S17-1 λpir chemically competent cells (ANGYUBIO, catalog number: G6055-10)
Reagents
KOD OneTM PCR Master Mix (TOYOBO, catalog number: KMM-201)
Agarose (GenStar, catalog number: VA10252)
Nucleic acid dye (Biosharp, catalog number: BS354B)
StarMarker D5000 (GenStar, catalog number: M030-10)
StarMarker D8000 (GenStar, catalog number: M031-05)
New Gel Mini Purification kit (ZOMANBIO, catalog number: ZPN202-3)
EZ-HiFi Seamless Cloning kit (GenStar, catalog number: T196-20)
Super Taq DNA Polymerase (GenStar, catalog number: A012-100)
Fast Plasmid Miniprep kit (ZOMANBIO, catalog number: ZPK101-3)
Chemicals
Tryptone (OXOID, catalog number: LP0042B)
Yeast extract powder (OXOID, catalog number: LP0021B)
Agar powder (Solarbio, catalog number: A8190)
Kanamycin sulfate (Solarbio, catalog number: BS-K8020-5)
Ampicillin (Solarbio, catalog number: BS-A8180-5)
Sucrose (Sigma-Aldrich, catalog number: V900116)
NaCl (SCR, catalog number: 10019318)
KCl (SCR, catalog number: 10016318)
Na2HPO4·12H2O (Macklin, catalog number: S818120-500 g)
KH2PO4 (Macklin, catalog number: P815662-500 g)
Tris-base (Solarbio, catalog number: SJ025)
Glacial acetic acid (Macklin, catalog number: A801295-500 mL)
EDTA (Aladdin, catalog number: E116428-500 g)
CaCl2 (Macklin, catalog number: C804986-500 g)
Glycerol (DAMAO, catalog number: 2294)
Solutions
Kanamycin (50 mg/mL) (see Recipes)
Ampicillin (100 mg/mL) (see Recipes)
50% glycerol (see Recipes)
0.1 M CaCl2 (see Recipes)
0.85% NaCl (see Recipes)
50× TAE electrophoresis buffer (see Recipes)
LB medium (see Recipes)
10× Phosphate-buffered saline (PBS) (see Recipes)
Recipes
Kanamycin (50 mg/mL)
Dissolve 500 mg of kanamycin sulfate in 10 mL of ddH2O. Filter sterilize using a 0.22 µm syringe filter and store at -20 °C before use.
Ampicillin (100 mg/mL)
Dissolve 1,000 mg of ampicillin in 10 mL of ddH2O. Filter sterilize using a 0.22 µm syringe filter and store at -20 °C before use.
50% glycerol
Make the 50% glycerol solution by diluting 100% glycerol in ddH2O and autoclave.
0.1 M CaCl2
Dissolve 1.47 g of CaCl2·2H2O in ddH2O to a final volume of 100 mL and autoclave the solution.
0.85% NaCl
Dissolve 0.85 g of NaCl in ddH2O to a final volume of 100 mL and autoclave the solution.
50× TAE electrophoresis buffer (1,000 mL)
Note: Use 1 M HCl or 1 M NaOH to adjust the pH to 8.5 and dilute to 1× with ddH2O before use.
Reagent Final concentration Quantity Tris-base 2 M 242 g Glacial acetic acid 1 M 57.1 mL EDTA 100 mM 37.2 g H2O n/a To 1,000 mL LB medium (1,000 mL)
Add 1 mL of kanamycin (50 mg/mL) or ampicillin (100 mg/mL) into 1 L of LB broth or agar after autoclaving when the media has cooled to 50–55 °C.
Note: Sucrose solution should be autoclaved at 115 °C for 30 min.
Reagent Final concentration Quantity Tryptone 1% 10 g Yeast extract powder 0.5% 5 g NaCl 1% 10 g Agar powder 1.5% 15 g (agar plate) Sucrose 15% 150 g (sucrose plate) H2O n/a To 1,000 mL 10× Phosphate-buffered saline (PBS) (1,000 mL)
Use 1 M HCl or 1 M NaOH to adjust the pH to ~6.8. The pH of solution should be adjusted to 7.4 when diluted to 1× PBS.
Dilute 10× PBS to 1× PBS and then sterilize the solution by autoclaving for 15 min at 121 °C. Store it at 4 . Dissolve 1 mL of 1× PBS in 99 mL of ddH2O (0.01 M) and sterilize by autoclaving for 15 min at 121 °C before use.
Reagent Final concentration Quantity Na2HPO4·12H2O 100 mM 35.814 g KH2PO4 18 mM 2.4496 g NaCl 1.37 M 80.0669 g KCl 27 mM 2.0129 g H2O n/a To 1,000 mL
Laboratory supplies
Petri plates, 90 mm × 15 mm (Biosharp, catalog number: BS-90-D)
1.5 mL Eppendorf tubes (Biosharp, catalog number: BS-15-M)
0.2 mL PCR tubes (LABSELECT, catalog number: PST-0208-FT-C)
Pipette tips (Biosharp, catalog numbers: BS-10-T, BS-200-T, BS-1000-T)
Cell spreader (Biologix, catalog number: 65-1001)
Parafilm (Parafilm, catalog number: PM996)
12 mL Polypropylene test tube (Biosharp, catalog number: BS-PPT-12-S)
10 mL syringes (MOTUO, catalog number: MT-ZSQ-04)
Syringe filter (0.22 µm) (Merck Millipore, catalog number: C134954)
Sterile 50 mL conical tubes (LABSELECT, catalog numbers: CT-002-50A)
96-well cell culture plate (Corning, catalog numbers: CLS351172)
Blue cap reagent bottle (SHUNIU, catalog numbers: 250 mL, 500 mL)
Equipment
Pipettes (Eppendorf, model: 3121000023, 3121000090, 3120000267)
Ultrapure Water Systems (ZHIANG, model: Best-D)
PCR instruments (BIO-GENER, model: GET3XG)
Microwave oven (Midea, model: 2105210600)
DNA electrophoresis apparatus (Tanon, model: EPS-300)
Gel image system (Tanon, model: Tanon 1600R)
UV glue cutting instrument (Miulab, model: DUT-48)
Electrothermal constant temperature (MIULAB, model: DTC-100)
Water bath pot (Shanghai Yiheng, model: DK-8D)
Liquid nitrogen container (DONGYA, model: YZD-50)
Clean bench (AIRTECH, model: SW-CJ-1FD)
Ice machine (XUEKE, model: IMS-70)
Constant temperature oscillation incubator (Shanghai Zhichu, model: ZQZY-78AES)
Vortex (DLAB, model: VJ218AG0003297)
4 °C refrigerator (Zhongke Duling, model: MPC-5V656)
-20 °C ultra-low temperature freezer (Zhongke Duling, model: MDF-25V278W)
-80 °C ultra-low temperature freezer (Haier, model: DW86L626)
High-pressure sterilization pot (BioNation, model: CT-90A)
Centrifuge 5424R (Eppendorf, model: CENTRIFUGE 5424R)
High-speed freezing centrifuge (Eppendorf, model: CENTRIFUGE 5810R)
Electronic balance (RADWAG, model: WTC-2000)
Ultra-micro ultravioletvisible spectrophotometer (BIO-DL, model: Micro drop 5803032204)
Baking oven (CIMO, model: DHG-9143BS-II)
Oven incubator (CIMO, model: SPX-250BSH-II)
Microplate reader (Molecular Devices, model: SMP7)
pH meter (Sartorius, model: PB-10)
Software and datasets
SnapGene® (SnapGene, https://www.snapgene.com/)
Tanon 1600 Gel Image System (Tanon)
SoftMax Pro 7 (Molecular Devices, http://www.moleculardevices.com)
Procedure
Genetic transformation
Construct red fluorescent protein (RFP) overexpression cassette
Use Gibson assembly (a method for seamlessly joining multiple DNA fragments with overlapping ends, enabling the construction of larger and more complex DNA molecules [14]) to construct the pET28a-PJ23119::rfp plasmid, which contains an RFP reporter gene under the control of the constitutive promoter J23119. The vector map was generated by SnapGene, and the detailed procedure is as follows:
Amplify the target fragments by polymerase chain reaction (PCR) using KOD. Amplify the RFP gene (765 bp) from pGD plasmid with the primers F1/R1. Amplify the plasmid backbone (3,651 bp) from pET28a plasmid with the primers F2/R2. The components and procedures of PCR are described as follows (Table 1 and Table 2).
Note: Colony PCR.
Table 1. Components for PCR reaction of KOD
Components Volume Final concentration KOD OneTM PCR Master Mix 10 µL 1× Forward primer (10 µM) 1 µL 0.5 µM Forward primer (10 µM) 1 µL 0.5 µM Template 1 µL - ddH2O to 20 µL - Table 2. Thermocycling conditions for PCR reaction of KOD
Ta: The annealing temperature (Ta) chosen for PCR depends directly on the length and composition of the primers. The details of the primers are listed in Table S1.Step Temperature (°C) Duration No. of cycles Initial denaturation 98 3 min 1 Denaturation 98 10 s 35 Annealing Ta 5 s Extension 68 10 s per kb Final extension 68 2 min 1 Hold 4 ∞ - Purify PCR products by 1% agarose gel electrophoresis in 1× TAE electrophoresis buffer followed by gel extraction using a gel purification kit.
Measure the absorbance at 260 nm and estimate DNA concentration.
Use EZ-HiFi Seamless Cloning kit to construct the plasmid. The recombination reaction mixture (10 µL) contains 5 µL of 2× HiFi Seamless cloning mix and 5 µL of target fragment mix (vector: insert molar ratio of 1:3; the details of the calculations are listed in Table S2). Incubate the combined reaction mixture at 50 °C for 30 min and immediately place on ice or hold at -20 °C until use.
Transform 10 µL of the ligation reaction into 100 µL of E. coli DH5α competent cells by the heat-shock method following the manufacturer’s instructions. This heat treatment (usually approximately 42 °C) increases the permeability of the bacterial cell membrane, allowing the foreign DNA to enter the cells. Plate the transformation mix on LB agar medium (K+) and incubate the plate at 37 °C overnight.
Pick colonies with a 2.5 µL pipette tip and transfer the picked colony into 500 µL of liquid LB (K+). Incubate the 1.5 mL tubes on a rotary shaker (200 rpm) at 37 °C for 12 h.
Confirm the transformants through PCR using Super Taq DNA Polymerase with primers F3/R3 and capture the gel image with Tanon 1600 Gel Image System (Tanon). The expected fragment size generated by PCR is 1,131 bp (Figure 2). The components and procedures of the PCR are described as follows (Table 3 and Table 4).
Note: A volume of 2–5 µL of PCR product should be loaded on the gel.
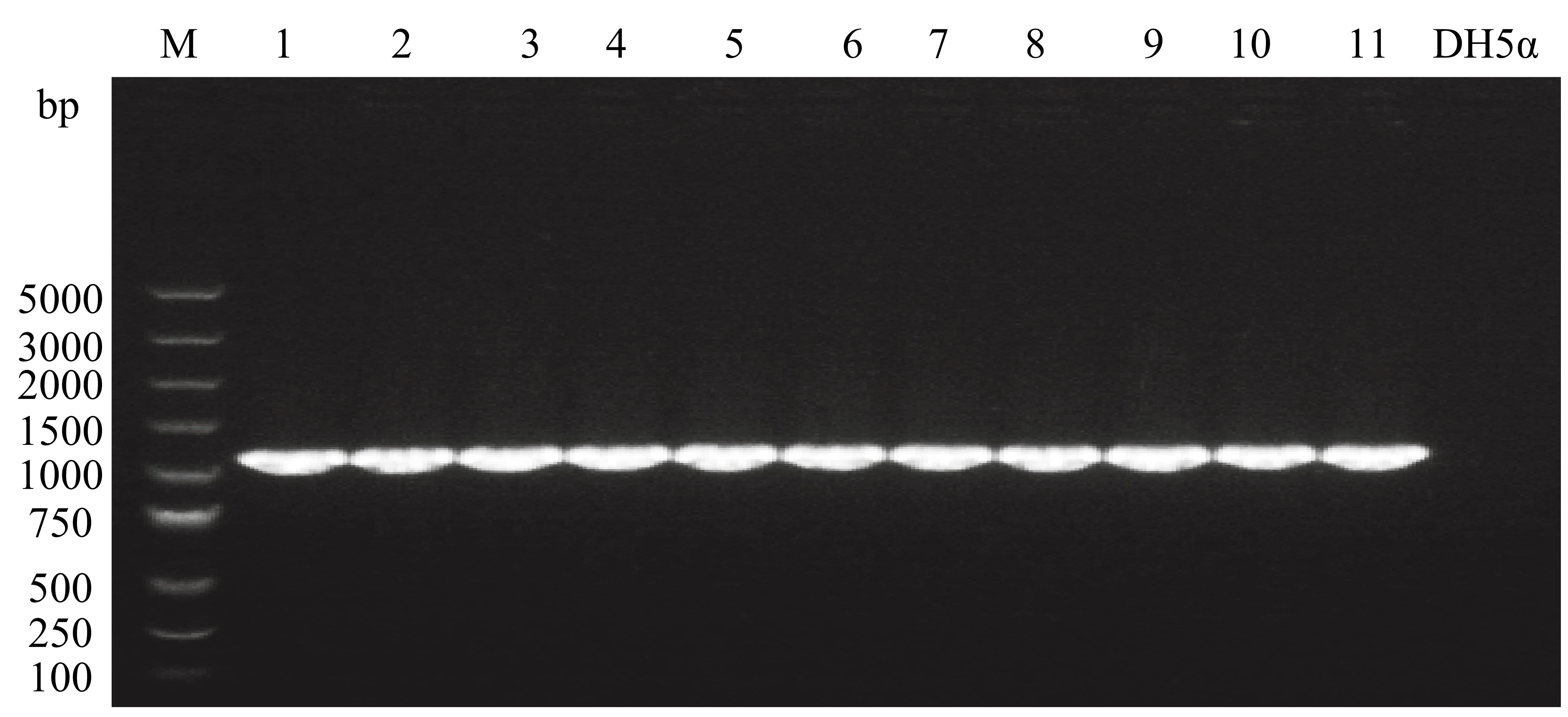
Figure 2. PCR amplification from the E. coli DH5α transformant carrying pET28a-PJ23119::rfpTable 3. Components for PCR reaction of Super Taq DNA Polymerase
Components Volume Final concentration 2× SuperTaq PCR StarMix 5 µL 1× Forward primer (10 µM) 0.5 µL 0.5 µM Forward primer (10 µM) 0.5 µL 0.5 µM Template 1 µL - ddH2O to 10 µL - Table 4. Thermocycling conditions for PCR reaction of Super Taq DNA Polymerase
Step Temperature (°C) Duration No. of cycles Initial denaturation 95℃ 2 min 1 Denaturation 95℃ 15 s 35 Annealing 58℃ 15 s Extension 72℃ 30 s per kb Final extension 72℃ 5 min 1 Hold 4 ∞ - Use sanger sequencing to evaluate the accuracy of the assembly (GENEWIZ Company). The sequencing data can be analyzed by SnapGene.
Cultivate the correct colonies and extract plasmids using the Fast Plasmid Miniprep kit. Store the pET28a-PJ23119::rfp plasmid at -20 °C until use.
E. persicina competent cells preparation
E. persicina BST187 was initially grown in 100 mL of LB broth and incubated at 28 °C for 12 h under shaking at 200 rpm to an optical density at 600 nm (OD600) of 0.4–0.8 (use SoftMax Pro 7).
Transfer the cultures into two precooled 50 mL conical sterile polypropylene centrifuge tubes and place them on ice for 20 min.
Centrifuge the cultures at 1,503× g for 5 min at 4 °C.
Resuspend the cell pellets in 5 mL of precooled 0.1 M CaCl2 solution and incubate on ice for 20 min.
Centrifuge the cells at 1,503× g for 5 min at 4 °C.
Resuspend the cell pellets in 2 mL of precooled 0.1 M CaCl2 and 1 mL of precooled 50% glycerol solution. Both components were added together in a single step.
Mix gently by pipetting up and down or flicking the tube 4–5 times. Store 100 µL of the yielding competent cells in 1.5 mL centrifuge tubes at -80 °C for a further transformation step.
Cell transformation
Take competent cells from -80 °C and thaw on ice (approximately 3–5 min). Add 10 µL of overexpression plasmids (approximately 50 ng/µL) into 100 µL of competent cells suspension and mix by tapping the tube gently.
Place competent cells on ice for 30 min.
Heat the mixture at 42 °C for 90 s; then, immediately transfer it to ice and incubate for 2 min.
Add 500 µL of LB broth into the mixture and incubate in a shaker at 200 rpm for 60 min at 28 °C.
Centrifuge the mixture at 1,878× g for 1 min.
Discard 500 µL of supernatant and spread 100 µL of the mixture on LB agar medium (K+). Incubate the plates for more than 24 h at 28 °C to obtain transformants.
Pick colonies into 1 mL of LB broth (A++K+) and incubate on a rotary shaker (200 rpm) at 28 °C for 12–24 h. Centrifuge the cells at 4,509× g for 5 min and examine them under a UV glue-cutting instrument for easy visibility with the naked eye (Figure 3).
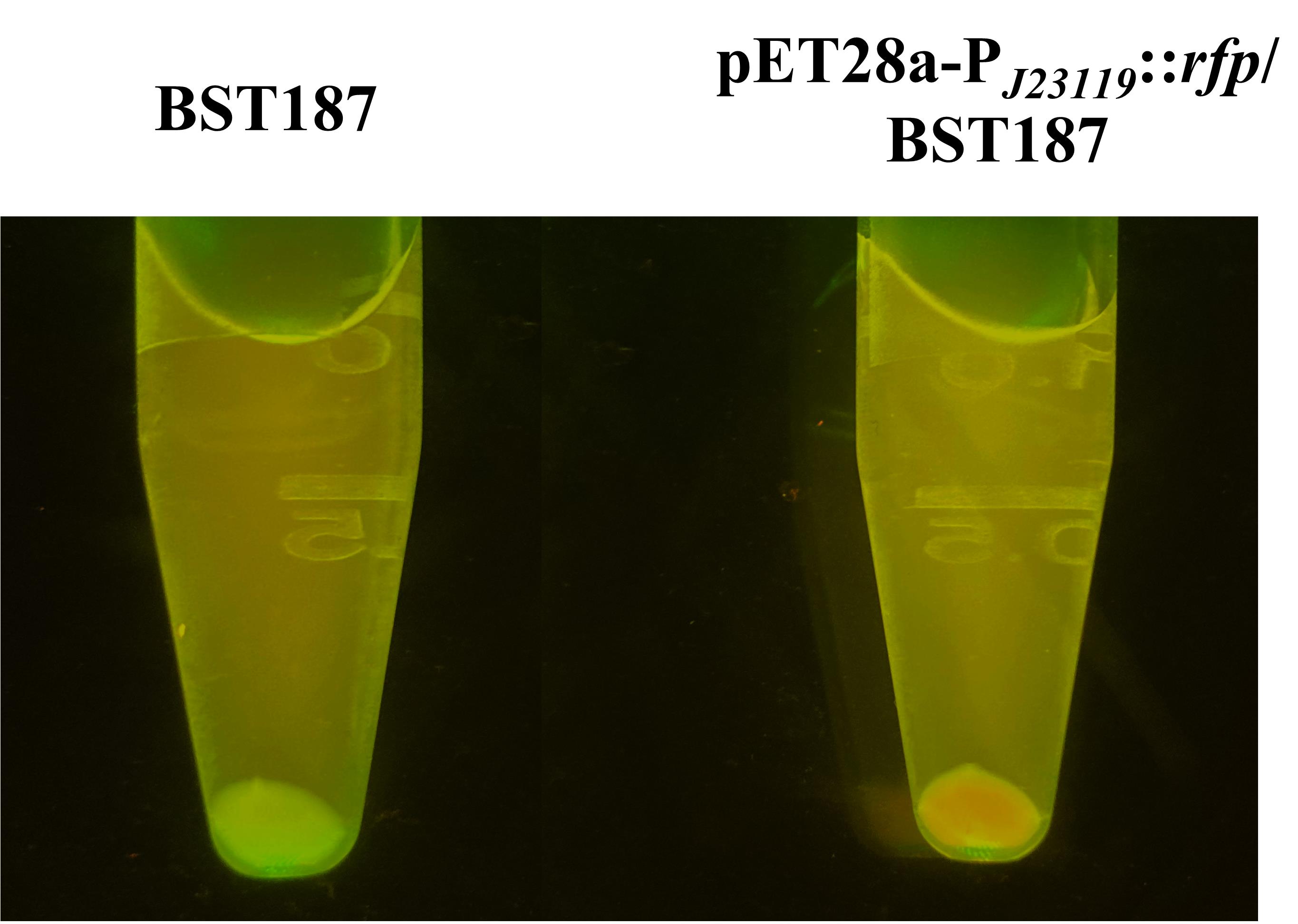
Figure 3. BST187 transformant carrying pET28a-PJ23119::rfp
Suicide plasmid–mediated genome editing
Construction of the lacZ knockout plasmid
Gene knockout was conducted by replacing the target genes in the chromosome with NdeI digestion site fragment (CATATG CATATG). A schematic diagram of vector construction for gene knockout and chromosomal integration is shown in Figure 1. The vector map was generated by SnapGene and detailed procedure is as follows:
Amplify the three fragments individually (upstream of lacZ, downstream of lacZ, and pKNOCK-sacB-km) by PCR using KOD. The specific primer pairs used to amplify each of the fragments are listed in Table S1, and PCR is described above.
Gibson assembly method is described above; transform 10 µL of the ligation reaction into 100 µL of E. coli DH5α λpir competent cells by the heat-shock method. See General note 3.
Verify the transformants via PCR with primers F7/R7 (use Tanon 1600 Gel Image System). The expected fragment size generated by PCR is 2,081 bp (Figure 4). The components and procedures of PCR are described above.
Cultivate the correct colonies and extract plasmids using the Fast Plasmid Miniprep kit. Store the pKNOCK-sacB-km-lacZ plasmid at -20 °C until use.
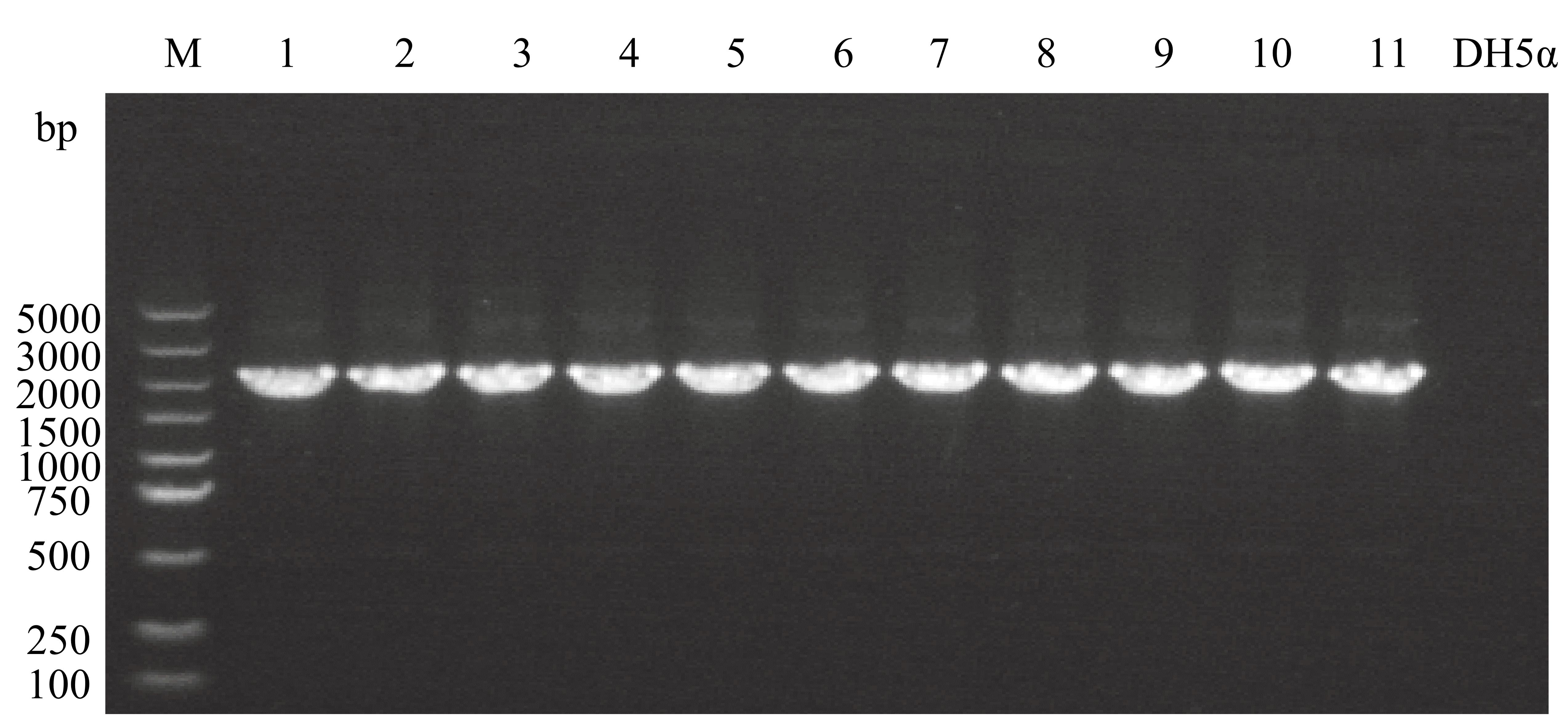
Figure 4. PCR amplification from E. coli DH5α λpir transformants carrying pKNOCK-sacB-km-lacZConstruction of the donor strains
Transform 10 µL of the lacZ knockout plasmid into 100 µL of E. coli donor strain S17-1 λpir competent cells by the heat-shock method following the manufacturer’s instructions. See General note 3.
Parental conjugation transformation
Preparation of donor strain: Inoculate E. coli S17-1 λpir in 5 mL of LB broth (K+) overnight at 37 °C with shaking at 200 rpm.
Preparation of recipient: Inoculate E. persicina BST187 in 5 mL of LB broth (A+) overnight at 28 °C with shaking at 200 rpm.
Centrifuge both cultures at 2,254× g for 5 min and remove the supernatant.
Resuspend the cell pellets with 3 mL of 0.01 M PBS buffer and centrifuge again to remove the supernatant. Repeat twice the steps of PBS resuspension and centrifugation to remove the supernatant.
Resuspend the cell pellets in 0.01 M PBS buffer solution to adjust the OD600 to 0.6–0.8 (use SoftMax Pro 7).
Combine donor strain S17-1 and recipient strain BST187 at a 1:1 volume ratio and incubate the mixture at 28 °C for approximately 10–12 h at 200 rpm.
Centrifuge 1 mL of conjugation suspension at 1,878× g for 2 min and reserve 100 µL of the supernatant to resuspend the pellets.
Plate the 100 µL resuspend pellets on LB agar medium (A++K+) and incubate the plate at 28 °C for more than 24 h.
Recombination exchanges
First recombination exchange: Pick single colonies (Figure 5A) inoculated in 500 µL of LB broth (A++K+) with shaking at 200 rpm for 10–12 h at 28 °C. See General note 4.
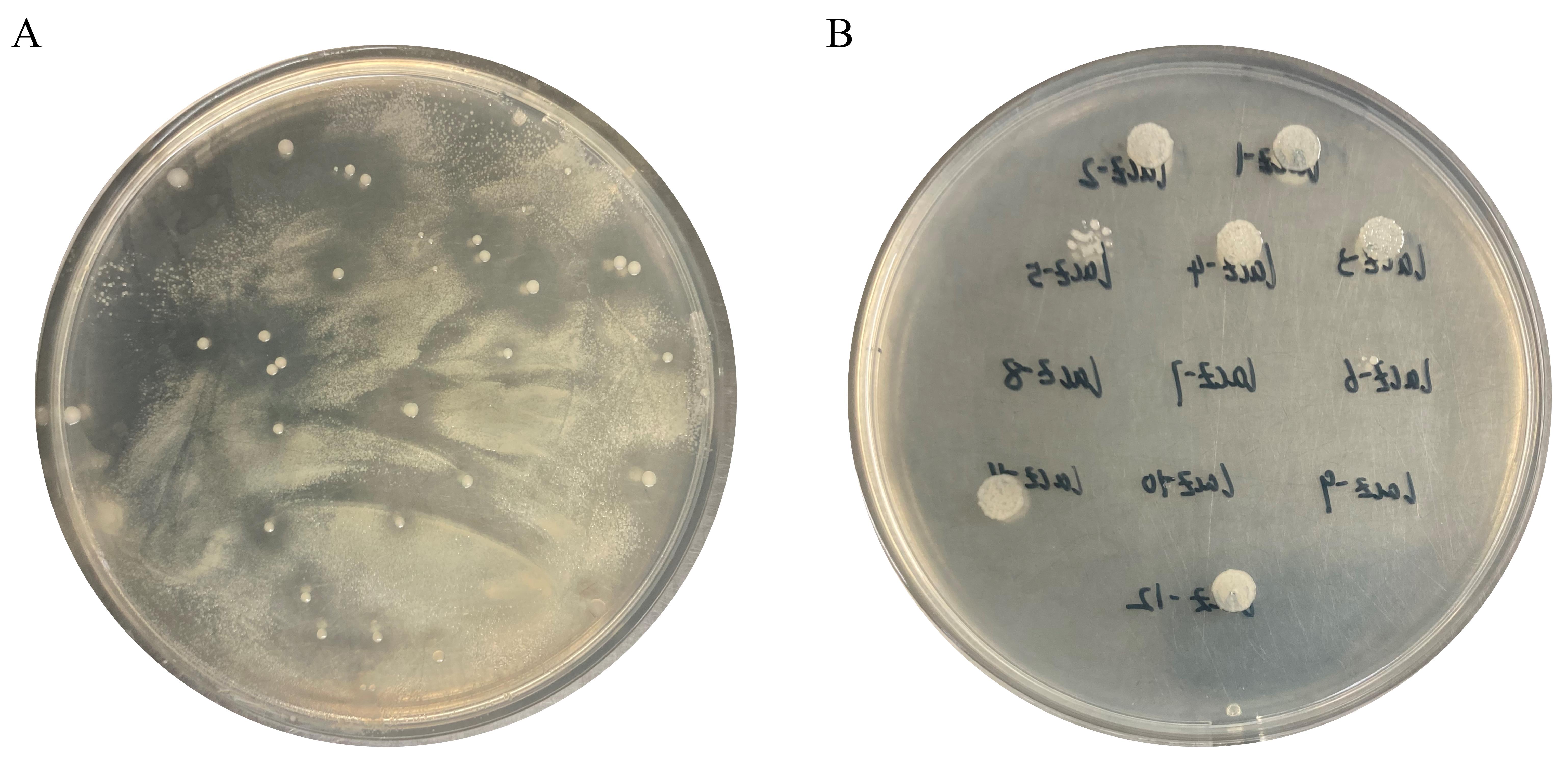
Figure 5. Colony of the first recombination exchange screening and resistance validation. A. First recombination exchange. B. Resistance validation.Confirm the first crossover strains through PCR using primers F7/R8 (use Tanon 1600 Gel Image System). The expected fragment size generated by PCR is 1,794 bp or 4,866 bp (Figure 6A).
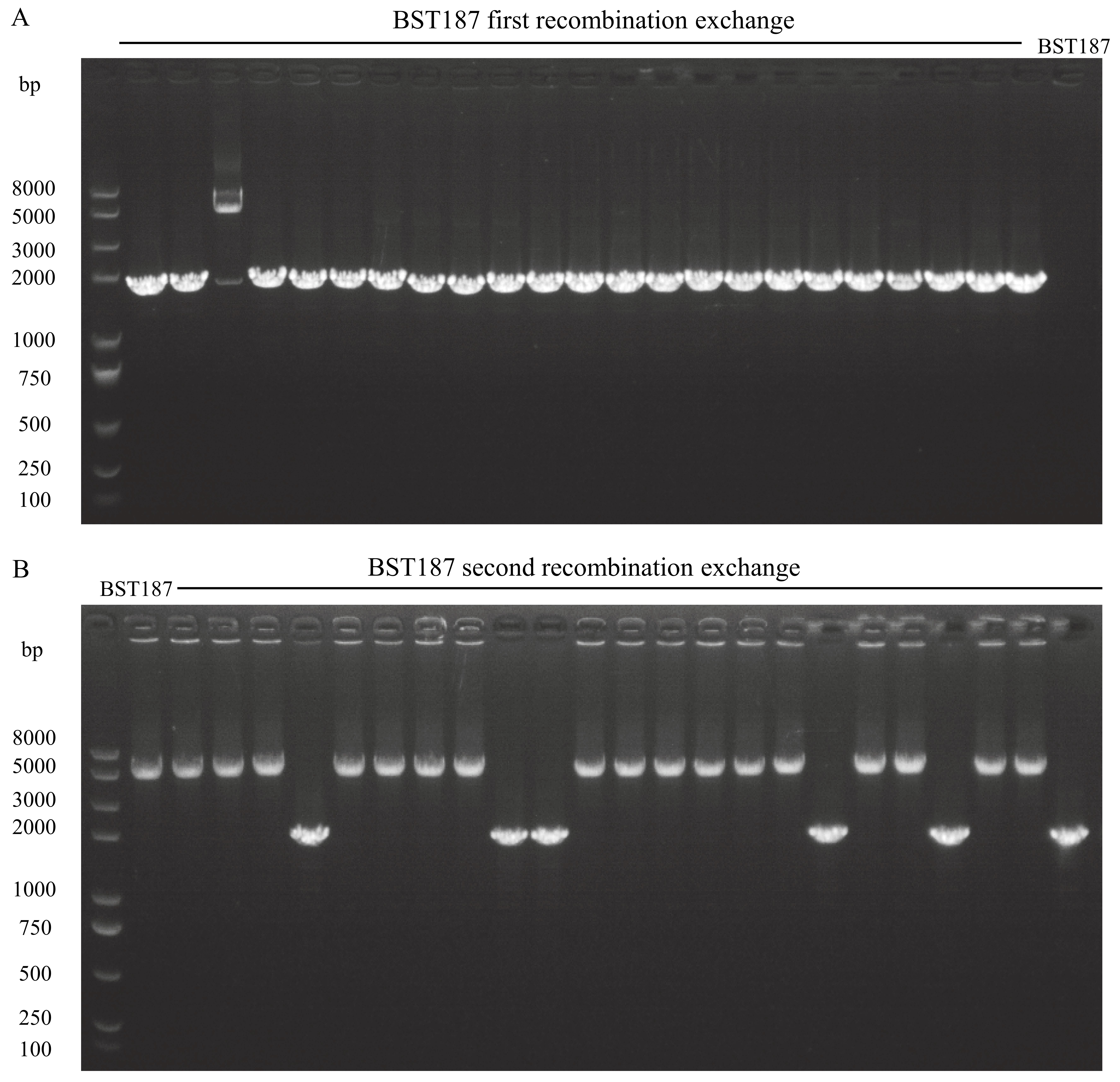
Figure 6. PCR amplification from BST187 that completed recombination exchange. A. First recombination exchange. B. Second recombination exchange.Second recombination exchange: Centrifuge the single-crossover strains at 1,878× g for 2 min and remove the supernatant.
Resuspend the cell pellets with 0.85% NaCl solution and for gradient dilution (10,000-fold). Plate the 100 µL dilutions on LB agar medium (15% sucrose) and incubate the plate at 28 °C for more than 24 h.
Pick single colonies into 500 µL of liquid LB (15% sucrose) and incubate on a rotary shaker (200 rpm) at 28 °C for 12 h. See General note 5.
Confirm the second recombination exchange strains through PCR using primers F8/R8 (use Tanon 1600 Gel Image System). The expected fragment size generated by PCR is 1887 bp or 4,971 bp (Figure 6B).
Aspirate 2.5 µL of the culture with a band size of 1,887 bp and dispense it onto LB agar medium (K+) at 28 °C for 24 h. A knockout strain (ΔlacZ) is obtained that was not grown on K+ LB agar medium (Figure 5B). See General note 6.
Data analysis
To verify the genome editing events, amplify the target region via colony PCR.
Editing efficiency = No. of correct mutants/No. of colonies tested.
Validation of protocol
For complete details on the use and execution of this protocol, please refer to Zhao et al. [13]. Fine-tuning gene expression of regulator AdmX for improved biosynthesis of andrimid in Erwinia persicina BST187. Applied Microbiology and Biotechnology (Figure 3).
General notes and troubleshooting
General notes
The T7 promoter does not work when used for the construction of overexpression cassette in E. persicina BST187.
Genes that can affect bacterial growth are prone to mutations when overexpressed with the J23119 promoter. It is recommended to use inducible or moderately strong promoter J23116.
Recombinant suicide vector (Replicon: R6K) is dependent on the λpir gene in competent cells.
E. coli S17-1 could grow on A+ plates and pick abnormally large colonies (Figure 5A).
BST187 will grow rapidly on sucrose plates and, after picking colonies, the plates will be placed at 4 °C.
Successful knockout of strains requires resistance verification in LB agar medium (K+) to avoid residual resistance fragments (Figure 5B).
Knockout efficiency is related to the target gene and, according to our experience, the editing efficiency is approximately 10%–80%.
DpnI is not usually necessary when gel purifying fragments.
Troubleshooting
Problem 1: After the first exchange, there was no growth of the target strain on LB agar medium (A++K+).
Possible cause(s): Instability of ampicillin causing the S17-1 strain to cover BST187.
Solution(s): Ready-to-use ampicillin plates.
Problem 2: After the second exchange, knockout strains contain residual resistance.
Possible cause(s): Incomplete suicide and sucrose selection is not entirely effective.
Solution(s): Selection of positive strains and repeat screening in 15% sucrose LB.
Acknowledgments
This work was financially supported by the Hundred Talents Program of the Chinese Academy of Sciences to LZ (E3J56201), the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-CXRC-027) and the international postdoctoral exchange fellowship program (Talent-Introduction Program) of the office of China postdoc council (YJ20210438).
We would like to acknowledge our original publication [13].
Competing interests
Authors have no competing interests.
References
- Aremu, B. R. and Babalola, O. O. (2015). Classification and Taxonomy of Vegetable Macergens. Front. Microbiol. 6: e01361.
- Gálvez, L., Gil-Serna, J., García-Díaz, M. and Palmero, D. (2015). First Report of a Garlic Bulb Rot Caused by Erwinia persicina in Europe. Plant Disease 99(5): 723–724.
- Cho, H., Park, J. Y., Kim, Y. K., Sohn, S. H., Park, D. S., Kwon, Y. S., Kim, C. W. and Back, C. G. (2019). Whole-Genome Sequence of Erwinia persicina B64, Which Causes Pink Soft Rot in Onions. Microbiol. Resour. Announce. 8(1): e01302–18.
- Kawaguchi, A., Abe, D., Saito, T., Nogata, Y., Nomiyama, K., Kohyama, N., Takahashi, A., Yoshioka, T., Ishikawa, N., Tomioka, K., et al. (2021). Pink seed of barley caused by Erwinia persicina. J. Gen. Plant Pathol. 87(2): 106–109.
- Li, L., Li, H. L., Shi, Y. X., Chai, A. L., Xie, X. W. and Li, B. J. (2021). First Report of Bacterial Leaf Spot of Cucurbita pepo Caused by Erwinia persicina in China. Plant Disease 105(5): 1558.
- Wang, J., Han, W., Pan, Y., Zhang, D., Zhao, D., Li, Q., Zhu, J. and Yang, Z. (2022). First Report of Erwinia persicina Causing Stalk Rot of Celery in China. Plant Disease 106(5): 1514.
- Wasendorf, C., Schmitz-Esser, S., Eischeid, C. J., Leyhe, M. J., Nelson, E. N., Rahic-Seggerman, F. M., Sullivan, K. E. and Peters, N. T. (2022). Genome analysis of Erwinia persicina reveals implications for soft rot pathogenicity in plants. Front. Microbiol. 13: e1001139.
- Yao, B., Huang, R., Zhang, Z. and Shi, S. (2022). Seed-Borne Erwinia persicina Affects the Growth and Physiology of Alfalfa (Medicago sativa L.). Front. Microbiol. 13: e891188.
- Kim, W. I., Choi, S. Y., Han, I., Cho, S. K., Lee, Y., Kim, S., Kang, B., Choi, O. and Kim, J. (2020). Inhibition of Salmonella enterica growth by competitive exclusion during early alfalfa sprout development using a seed-dwelling Erwinia persicina strain EUS78. Int. J. Food Microbiol. 312: 108374.
- Deroo, W., De Troyer, L., Dumoulin, F., De Saeger, S., De Boevre, M., Vandenabeele, S., De Gelder, L. and Audenaert, K. (2022). A Novel In Planta Enrichment Method Employing Fusarium graminearum-Infected Wheat Spikes to Select for Competitive Biocontrol Bacteria. Toxins 14(3): 222.
- Zhang, Y., Liu, X., Li, X., Zhao, L., Zhang, H., Jia, Q., Yao, B. and Zhang, Z. (2022). Physicochemical Properties and Antibiosis Activity of the Pink Pigment of Erwinia persicina Cp2. Agriculture 12(10): 1641.
- Cheng, T., Ge, T., Zhao, L., Hou, Y., Xia, J. and Zhao, L. (2023). Improved production of andrimid in Erwinia persicina BST187 strain by fermentation optimization. BMC Microbiol. 23(1): 263.
- Zhao, L., Ge, T., Cheng, T., Wang, Q., Cui, M., Yuan, H. and Zhao, L. (2023). Fine-tuning gene expression of regulator AdmX for improved biosynthesis of andrimid in Erwinia persicina BST187. Appl. Microbiol. Biotechnol. 107(22): 6775–6788.
- Gibson, D. G., Young, L., Chuang, R. Y., Venter, J. C., Hutchison, C. A. and Smith, H. O. (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6(5): 343–345.
Supplementary information
The following supporting information can be downloaded here:
- Table S1. Information primers used in this study
- Table S2. Ligation of homologous recombination
Article Information
Copyright
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Cheng, T., Ge, T., Zhao, X., Liu, Z. and Zhao, L. (2024). Efficient Genetic Transformation and Suicide Plasmid-mediated Genome Editing System for Non-model Microorganism Erwinia persicina. Bio-protocol 14(6): e4956. DOI: 10.21769/BioProtoc.4956.
Category
Microbiology > Microbial genetics > Genome editing
Molecular Biology > DNA > Transformation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.